

Abstract
Deubiquitinating enzymes (DUBs) are proteases that fulfill crucial roles in the ubiquitin (Ub) system, by deconjugation of Ub from its targets and disassembly of polyUb chains. The specificity of a DUB towards one of the polyUb chain linkages largely determines the ultimate signaling function. We present a novel set of diubiquitin FRET probes, comprising all seven isopeptide linkages, for the absolute quantification of chain cleavage specificity of DUBs by means of Michaelis–Menten kinetics. Each probe is equipped with a FRET pair consisting of Rhodamine110 and tetramethylrhodamine to allow the fully synthetic preparation of the probes by SPPS and NCL. Our synthetic strategy includes the introduction of N,N′‐Boc‐protected 5‐carboxyrhodamine as a convenient building block in peptide chemistry. We demonstrate the value of our probes by quantifying the linkage specificities of a panel of nine DUBs in a high‐throughput manner.
Keywords: deubiquitinating enzymes, FRET, native chemical ligation, solid-phase synthesis, ubiquitin conjugates
Ubiquitin (Ub), a 76 amino acid protein, is a post‐translational modifier that is crucial for a wide range of cellular processes, including protein degradation, trafficking, and signaling.1 Ub is generally attached via its C‐terminal carboxylate to the side‐chain amine of a lysine residue in the target protein, thereby forming an isopeptide bond. Target proteins are frequently modified with a polyUb chain, in which multiple Ub modules are successively linked at the N terminus (linear polyUb) or any of the seven internal lysine residues (isopeptide‐linked polyUb: K6, K11, K27, K29, K33, K48, and K63). The type of polyUb chain largely determines its signaling function.1
Ubiquitination is mediated by the concerted action of three enzymes, E1 (activating), E2 (conjugating), and E3 (ligase), the particular combination of which provides specificity for the protein target or polyUb chain topology. Removal of Ub from its targets and disassembly of polyUb chains are catalyzed by deubiquitinating enzymes (DUBs). About 100 human DUBs have been identified;2 some exhibit Ub linkage specificity. DUB action can rescue proteins from proteasomal degradation and alter Ub signaling functions through chain remodeling in a linkage‐specific manner.1 The synthesis of diubiquitin (diUb) has made it possible to study processing by DUBs.3 In order to determine specificity, a DUB can be incubated with either a native diUb molecule4 or with a diUb activity‐based probe5 of a given linkage. However current methods do not allow fast and absolute quantification of DUB linkage specificity, and furthermore cannot separate this specificity into binding affinity and catalytic turnover rate (K m and k cat, respectively, in Michaelis–Menten kinetics).
The application of FRET pairs has proved useful in the study of DUB activity, Ub chain conformation, and Ub‐interacting proteins.6 In order to investigate chain cleavage specificity across all isopeptide linkages, we developed a full chemical synthesis of all seven isopeptide‐linked diUb FRET pairs. These pairs carry a novel dye‐pair suitable for FRET and compatible with solid phase peptide synthesis (SPPS). We determined K m and k cat values of linkage‐specific DUBs that are used in Ub chain restriction analysis,7 in order to obtain insight into their catalytic action.
In the FRET‐based assay (Figure 1) the reagents consist of two Ub modules, one equipped with a donor fluorophore and the other with an acceptor; these are specifically linked by a native isopeptide bond to each of the seven lysine residues. We reasoned that the best position for fluorophore attachment would be the N termini of both Ub modules, because the distance between the N termini ranges from 30 to 50 Å, based on available crystallographic data (Table S1 in the Supporting Information), an ideal distance for FRET. Because the fluorophores need to be compatible with all synthetic steps (see below), we developed a new FRET pair by using 5‐carboxyrhodamine110 (Rho) as the donor and 5‐carboxytetramethylrhodamine (TAMRA) as the acceptor. Fluorescein, the more commonly used FRET donor, was initially tried but proved incompatible with the desulfurization step in the final synthesis step (see below) and was therefore replaced by Rho. Upon addition of a DUB, the diUb FRET pair is cleaved, thereby resulting in loss of the FRET signal and hence an increase in donor emission.
Figure 1.
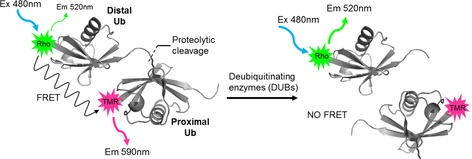
Principle of the FRET‐based diUb cleavage assay. Upon cleavage of the diUb FRET pair by a DUB, the FRET signal is lost.
A major problem in the use of Rho (but also TAMRA) in SPPS (Scheme 1 A) is that when Rho is attached to an amine in a globally side‐chain‐protected peptide, the 1‐carboxylate moiety of Rho is in an open conformation and can react upon further extension of the peptide chain (Scheme 1 A, 1→2). In addition, the coupling of Rho is generally difficult because of the poor solubility and intrinsic reactivity of the aniline moieties. We therefore prepared N,N′‐Boc‐protected Rho. When this molecule is coupled to a peptide, the dual Boc protection locks the 1‐carboxylate in the closed lactone form, thus making it unreactive (Scheme 1 A, 3→4). We modified the method reported by Grimm and Lavis8 to prepare N,N′‐Boc‐protected Rho 10 (Scheme 1). 5‐Carboxyfluorescein (5) was converted in four steps into ditriflate 8. Buchwald–Hartwig coupling with BocNH2 resulted in the formation of N,N′‐Boc‐protected Rho (9). Use of ethyl ester protection of the 5‐carboxylate allowed selective liberation of the 5‐carboxylate without affecting the Boc groups, thereby resulting in 10. In contrast to unprotected Rho, 10 is very soluble in organic solvents, can easily be coupled under standard peptide coupling conditions, and can be prepared on a multi‐gram scale.
Scheme 1.
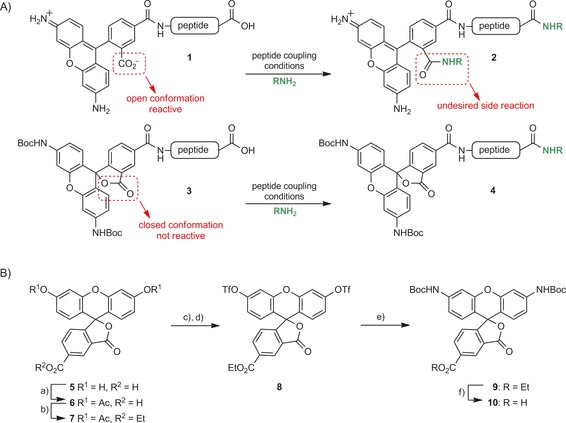
A) Problems encountered with unprotected Rho in peptide chemistry. B) Synthesis of N,N′‐Boc‐protected Rho. a) Ac2O, H2SO4, 120 °C (99 %); b) EtOH, EDC, CH2Cl2 (94 %); c) NaOEt, EtOH; d) Tf2O, pyridine, CH2Cl2 (57 %); e) BocNH2, Pd2dba3, Xantphos, Cs2CO3, dioxane, 100 °C (82 %); f) NaOH, THF (95 %).
The seven diUb FRET pairs 17 a–g were constructed by native chemical ligation (NCL) between Rho‐Ub‐thioester 14 and TAMRA‐Ub containing a γ‐thioLys building block3, 9 16 (Scheme 2). The individual Ub modules where synthesized by linear SPPS on hyper‐acid‐labile trityl resin.3 DiBoc‐protected Rho (10) was coupled to Ub1−75 (11) on resin to result in 12, which was subsequently cleaved from the resin under mild acidic conditions without affecting the global protection scheme. Methyl‐3‐(glycylthio)‐propionate was coupled to the liberated C‐terminal carboxylate to give 13. Global deprotection under strong acidic conditions followed by cation exchange and RP‐HPLC purification gave Rho‐Ub‐thioester 14. It is of note that the Boc groups on Rho are concomitantly removed during the global deprotection, thereby restoring its fluorescent properties. TAMRA‐Ub modules containing γ‐thioLys on each of the respective lysine positions (16 a–g) were prepared by coupling the 5‐carboxy isomer of TAMRA to the Ub1–76 polypeptides 15 a–g, followed by global deprotection and purification (Figure S1 in the Supporting Information). Methionine‐1 was replaced by the isostere norleucine to prevent oxidation of the thioether moiety. NCL reactions between 14 and 16 a–g, followed by desulfurization under radical conditions,10 purification by RP‐HPLC, and gel filtration gave the final seven diUb FRET pairs 17 a–g in good yield and purity.
Scheme 2.
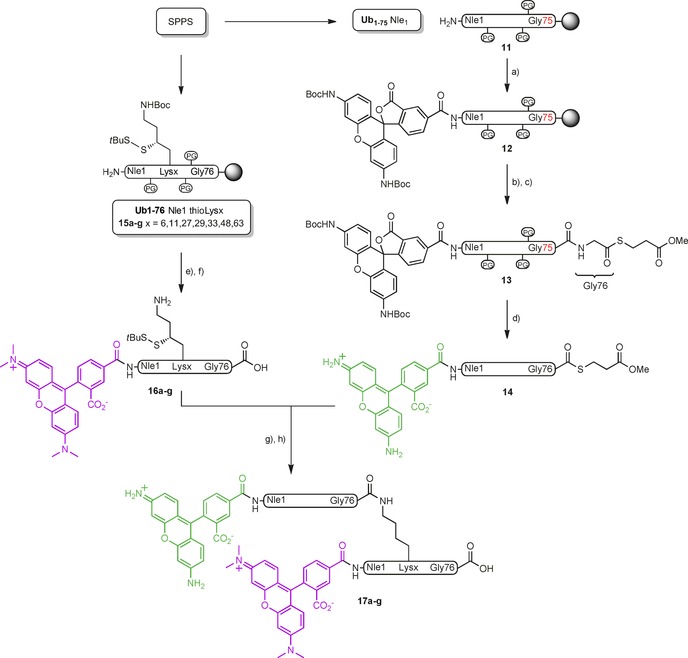
Synthesis of the seven isopeptide‐linked diUb FRET pairs. a) 10, PyBOP, DIPEA, NMP; b) 20 % HFIP/CH2Cl2; c) HCl⋅H‐Gly‐S(CH2)2CO2Me, EDC, HOBt, CH2Cl2; d) TFA/H2O/phenol/iPr3SiH (90.5:5:2.5:2); e) TAMRA, PyBOP, DIPEA, NMP; f) TFA/H2O/phenol/iPr3SiH (90.5:5:2.5:2); g) MPAA, 6 m Gnd⋅HCl, pH 7.2; h) TCEP, GSH, VA‐044, 6 m Gnd⋅HCl, pH 7.0.
The purities of 17 a–g were confirmed by LCMS analysis (Figure 2 A, B, and Supporting Information) and gel analysis (Figure 2 C). Upon excitation at 466 nm, the emission spectra of the diUb FRET pairs and Rho‐Ub and TAMRA‐Ub revealed that all seven molecules show a clear FRET signal (Figure 2 D). We performed fluorescence lifetime imaging microscopy (FLIM) to determine FRET efficiencies of all the FRET pairs (Figure 2 E, Table S3); these were found to be 0.45–0.60, depending on the linkage, thus demonstrating efficient FRET in all these molecules.
Figure 2.
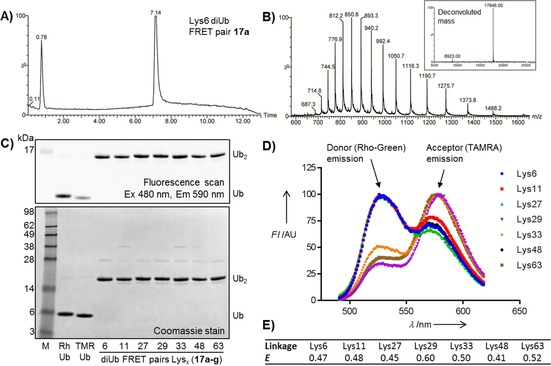
Characterization of diUb FRET pairs 17 a–g. A) Analytical HPLC and B) MS of Lys6‐linked diUb FRET pair 17 a. C) SDS‐PAGE analysis. D) Emission spectra recorded at λ ex=466 nm. E) FRET efficiencies (E) determined by FLIM.
DUB‐mediated cleavage of our new diUb FRET pairs was first assessed by incubation with USP7 and OTUD2, two well‐studied DUBs from the two largest DUB families and for which cleavage of unlabeled diUbs has been reported. Reactions were analyzed by SDS‐PAGE (Figures S2 and S3) which showed that the diUb selectivity of both DUBs was in good agreement with reported data.4a,4b We then incubated OTUD2 with Lys11‐ and Lys27‐linked diUb FRET pairs (17 b and 17 c, respectively) and the corresponding unlabeled diUbs. We also included a 1:1 mixture of the FRET pair and the unlabeled diUb for both linkages. SDS‐PAGE analysis (Figures 3 A, S4 and S5) showed that both the FRET pair and the unlabeled diUb substrates were equally processed, thus we concluded that the attached fluorophores do not affect DUB activity.
Figure 3.
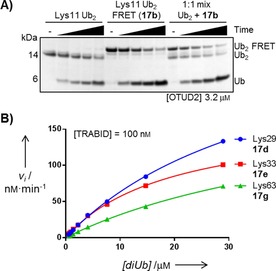
A) SDS‐PAGE analysis of Lys11‐linked diUb cleavage. OTUD2 was incubated with unlabeled diUb, FRET pair 17 b, or a 1:1 mixture; samples were taken after 10, 30, 60, and 180 min. B) Michaelis–Menten kinetics of TRABID for Lys29‐, Lys33‐ and Lys63‐linked diUb, as determined by the FRET assay.
We next applied our FRET reagents for the quantification of diUb linkage‐specificity for nine DUBs derived from three different DUB families; each was shown to display a distinct specificity (Table 1).4b We incubated the DUBs with all diUb FRET pairs at a fixed concentration (0.5 μm, to keep initial fluorescence constant for all samples) with an increasing concentration of the unlabeled diUb (0–28 μm). The enzyme concentration was chosen such that the reaction proceeded linearly for at least 20 min (Supporting Information). The total amount of processed diUb was then determined by monitoring the increase in donor fluorescence over time; from this the rates of initial velocity were calculated and fitted to the Michaelis–Menten equation (Figure 3 B and Supporting Information), from which K m and, k cat were determined.
Table 1.
Kinetic characterization of DUBs that are used in Ub chain restriction analysis7 for the diUb FRET pairs 17 a–g.[a]
| DUB | Linkage | K m [μm] | k cat [s−1] | k cat/K m [m −1 s−1] |
|---|---|---|---|---|
| AMSH | Lys63 | 45.4 | 0.0027 | 59 |
| AMSH*[b] | Lys63 | 2.4 | 0.17 | 70 048 |
| Cezanne | Lys11 | 19.4 | 1.5 | 78 818 |
| OTUB1 | Lys48 | ≫50 | n.d. | |
| OTUB1*[b] | Lys48 | 38.6 | 0.66 | 17 158 |
| OTUD1 | Lys63 | ≫50 | n.d. | 4020 |
| OTUD3 | Lys6 | ≫50 | n.d. | |
| Lys11 | 52.4 | 0.0085 | 162 | |
| Lys63 | 57.9 | 0.0061 | 105 | |
| TRABID | Lys29 | 40.1 | 0.053 | 1317 |
| Lys33 | 19.6 | 0.028 | 1431 | |
| Lys63 | 54.0 | 0.034 | 627 | |
| OTUD2 | Lys11 | 87.9 | 4.4 | 50 253 |
| Lys27 | ≫50 | n.d. | ||
| Lys29 | ≫50 | n.d. | ||
| vOTU | Lys6 | 3.6 | 0.87 | 242 031 |
| Lys11 | 3.4 | 0.22 | 63 110 | |
| Lys48 | 8.4 | 1.0 | 119 952 | |
| Lys63 | 1.4 | 0.091 | 66 266 | |
| USP21 | Lys6 | 2.1 | 0.12 | 60 633 |
| Lys11 | 1.4 | 0.087 | 62 735 | |
| Lys33 | 2.1 | 0.13 | 60 814 | |
| Lys48 | 1.7 | 0.067 | 39 181 | |
| Lys63 | 2.7 | 0.16 | 60 558 |
[a] Values in italics were obtained by extrapolation beyond the highest substrate concentration. [b] Activated versions of AMSH and OTUB1 were created by fusing them to their activators (STAM and UBE2D2, respectively).11
Table 1 shows the data for all combinations of DUB and diUb substrate for which activity could be measured. Overall, the individual diUb linkage types cleaved by each DUB were consistent with published qualitative data.4 As expected from earlier findings, the unspecific DUB USP21 showed similar activity towards most linkages.4a The virus‐derived DUB vOTU, which is also considered to be unspecific, showed some interesting results: the catalytic efficiency (k cat/K m) for Lys6 was two times higher than for Lys48, and four times higher than for Lys11 and Lys63 diUbs; this can largely be attributed to differences in k cat rather than K m. Another interesting result was for AMSH. In agreement with earlier findings, this DUB had absolute specificity for Lys63 diUb,4c although the overall efficiency was rather low. Remarkably, the recently reported fusion of AMSH with its natural activator STAM211 resulted in a more than 1000‐fold increase in catalytic efficiency, which can be attributed to increases in both affinity and catalytic turnover. Taken together, these data show that our quantitative assessment of the DUB linkage specificity is in accordance with reported data and that new insights can be obtained from the kinetic parameters.
In summary, the set of all seven isopeptide‐linked diUb FRET pairs allows absolute quantification of DUB linkage specificity. Our synthetic strategy, which includes a convenient N,N′‐Boc‐protected Rho building block, allows efficient preparation of these reagents in large quantities. The assay requires low amounts of material, can easily be automated, and can be used in high‐throughput small‐molecule screening or for the assessment of (di)Ub binding domains.6c Overall, we believe that our diUb FRET probes will be of great value in ongoing efforts to crack the ubiquitin code and that the FRET pair presented here will facilitate FRET‐pair synthesis in general.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dris El Atmioui and Henk Hilkman for SPPS and Prof. Dr. Kees Jalink for help with FLIM. This work was supported by the European Research Council (ERC grant no. 281699) and the Netherlands Organization for Scientific Research, (NWO VICI grant no. 724.013.002) to H.O. Work in the D.K. lab was funded by the Medical Research Council (U105192732), the European Research Council (Grant no. 309756), and the Lister Institute for Preventive Medicine, as well as the Marie Curie Initial Training Network “UPStream” (T.E.T.M.) and an EMBO Long‐Term Fellowship (J.N.P.).
P. P. Geurink, B. D. M. van Tol, D. van Dalen, P. J. G. Brundel, T. E. T. Mevissen, J. N. Pruneda, P. R. Elliott, G. B. A. van Tilburg, D. Komander, H. Ovaa, ChemBioChem 2016, 17, 816.
Contributor Information
Dr. Paul P. Geurink, Email: p.geurink@nki.nl
Prof. Dr. Huib Ovaa, Email: h.ovaa@nki.nl
References
- 1. Komander D., Rape M., Annu. Rev. Biochem. 2012, 81, 203–229. [DOI] [PubMed] [Google Scholar]
- 2. Reyes-Turcu F. E., Ventii K. H., Wilkinson K. D., Annu. Rev. Biochem. 2009, 78, 363–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. El Oualid F., Merkx R., Ekkebus R., Hameed D. S., Smit J. J., de Jong A., Hilkmann H., Sixma T. K., Ovaa H., Angew. Chem. Int. Ed. 2010, 49, 10149–10153; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 10347–10351. [Google Scholar]
- 4.
- 4a. Faesen A. C., Luna-Vargas M. P. A., Geurink P. P., Clerici M., Merkx R., van Dijk W. J., Hameed D. S., El Oualid F., Ovaa H., Sixma T. K., Chem. Biol. 2011, 18, 1550–1561; [DOI] [PubMed] [Google Scholar]
- 4b. Mevissen T. E. T., Hospenthal M. K., Geurink P. P., Elliott P. R., Akutsu M., Arnaudo N., Ekkebus R., Kulathu Y., Wauer T., El Oualid F., Freund S. M. V., Ovaa H., Komander D., Cell 2013, 154, 169–184; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Ritorto M. S., Ewan R., Perez-Oliva A. B., Knebel A., Buhrlage S. J., Wightman M., Kelly S. M., Wood N. T., Virdee S., Gray N. S., Morrice N. A., Alessi D. R., Trost M., Nat. Commun. 2014, 5, 4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Mulder M. P. C., El Oualid F., ter Beek J., Ovaa H., ChemBioChem 2014, 15, 946–949; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. McGouran J. F., Gaertner S. R., Altun M., Kramer H. B., Kessler B. M., Chem. Biol. 2013, 20, 1447–1455; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Li G., Liang Q., Gong P., Tencer A. H., Zhuang Z., Chem. Commun. 2014, 50, 216–218; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Haj-Yahya N., Hemantha H. P., Meledin R., Bondalapati S., Seenaiah M., Brik A., Org. Lett. 2014, 16, 540–543. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Ohayon S., Spasser L., Aharoni A., Brik A., J. Am. Chem. Soc. 2012, 134, 3281–3289; [DOI] [PubMed] [Google Scholar]
- 6b. Horton R. A., Strachan E. A., Vogel K. W., Riddle S. M., Anal. Biochem. 2007, 360, 138–143; [DOI] [PubMed] [Google Scholar]
- 6c. Ye Y., Blaser G., Horrocks M. H., Ruedas-Rama M. J., Ibrahim S., Zhukov A. A., Orte A., Klenerman D., Jackson S. E., Komander D., Nature 2012, 492, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hospenthal M. K., Mevissen T. E. T., Komander D., Nat. Protoc. 2015, 10, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grimm J. B., Lavis L. D., Org. Lett. 2011, 13, 6354–6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Kumar K. S. A., Haj-Yahya M., Olschewski D., Lashuel H. A., Brik A., Angew. Chem. Int. Ed. 2009, 48, 8090–8094; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 8234–8238; [Google Scholar]
- 9b. Pasunooti K. K., Yang R., Vedachalam S., Gorityala B. K., Liu C.-F., Liu X.-W., Bioorg. Med. Chem. Lett. 2009, 19, 6268–6271; [DOI] [PubMed] [Google Scholar]
- 9c. Merkx R., de Bruin G., Kruithof A., van den Bergh T., Snip E., Lutz M., El Oualid F., Ovaa H., Chem. Sci. 2013, 4, 4494–4498. [Google Scholar]
- 10. Wan Q., Danishefsky S. J., Angew. Chem. Int. Ed. 2007, 46, 9248–9252; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 9408–9412. [Google Scholar]
- 11. Michel M. A., Elliott P. R., Swatek K. N., Simicek M., Pruneda J. N., Wagstaff J. L., Freund S. M. V., Komander D., Mol. Cell 2015, 58, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary