

ABSTRACT
As a central node of the macroautophagy/autophagy process, the BECN1/Beclin1-PIK3C3/VPS34 complex participates in different steps of autophagy by interacting with multiple molecules. The ATG14-associated PIK3C3 complex is involved in autophagy initiation, whereas the UVRAG-associated complex mainly modulates autophagosome maturation and endosome fusion. However, the molecular mechanism that coordinates the sequential execution of the autophagy program remains unknown. We have recently discovered that a Golgi-resident protein, PAQR3, regulates autophagy initiation as it preferentially facilitates the formation of the ATG14-linked PIK3C3 complex instead of the UVRAG-associated complex. Upon glucose starvation, AMPK directly phosphorylates T32 of PAQR3, which is crucial for the activation of the ATG14-associated class III PtdIns3K. Furthermore, Paqr3-deleted mice have a deficiency in exercise-induced autophagy as well as behavioral disorders. Thus, this work not only uncovers the regulatory mechanism of PAQR3 on autophagy initiation, but also provides a potential candidate therapeutic target for neurodegenerative diseases.
KEYWORDS: AMPK, ATG14, autophagy, Beclin 1, class III PtdIns3K, glucose starvation, nutrient sensing, PAQR3
As a member of the progestin and adipoQ receptors (PAQR) superfamily, PAQR3 is a 7-transmembrane protein in the Golgi apparatus. By sequestering critical signaling molecules to the Golgi apparatus, PAQR3 acts as a negative regulator for multiple signaling pathways, such as RAS-RAF-MAP2K/MEK-MAPK/ERK and PI3K-AKT cascades. However, it remains poorly understood whether PAQR3 can also positively regulate signal transduction for certain biological pathways. In a recent study, we reported an interesting discovery that under glucose starvation conditions, PAQR3 is detached from the Golgi apparatus into punctiform structures, which are colocalized with the autophagosome marker LC3. These data suggest that there might exist a functional connection between PAQR3 and autophagy. Consistently, autophagic activity is significantly blunted in Paqr3-deficient HeLa or MEF cells under various autophagy-promoting conditions, such as glucose starvation, amino acid starvation, incubation in Hank's balanced salt solution or following rapamycin treatment. Furthermore, chloroquine-induced LC3-II accumulation is also reduced by Paqr3 deletion in MEFs, indicating that PAQR3 might directly affect autophagosome formation, but not LC3-II degradation. Two major upstream autophagy-regulatory signaling pathways, involving AMPK and MTOR, are not modulated by PAQR3 under glucose starvation. The candidate target for PAQR3 in autophagy regulation was later narrowed down to class III PtdIns3K, which modulates PtdIns3P generation in the initiation phase of autophagy. We found that PAQR3 is essential for glucose starvation-induced PtdIns3P generation, as the punctiform distribution pattern of its major downstream effectors ZFYVE1/DFCP1 and WIPI1 are dramatically blocked in PAQR3 knockdown or Paqr3 knockout cells upon glucose starvation. Consistent with these results, Paqr3 deletion significantly attenuates the activity of the ATG14-associated class III PtdIns3K, which is critical for autophagy initiation and autophagosome formation. However, the UVRAG-associated PIK3C3 complex involved in autophagosome maturation and endocytic fusion is not affected by PAQR3. Remarkably, we discovered that the NH2-terminal 21–60 amino acids of PAQR3 are not only indispensable for its regulation of class III PtdIns3K activity, but also critical for the constitution of the ATG14-linked PIK3C3 complex. Furthermore, PAQR3 was identified as a constitutive binding partner of the ATG14-linked PIK3C3 complex regardless of autophagy-promoting signals. However, PAQR3 could not interact with UVRAG. As a scaffold protein, PAQR3 could form a ternary complex with BECN1 and ATG14 via 2 distinct structural motifs, thus promoting the formation of the ATG14-linked PIK3C3 complex.
As PAQR3 regulates autophagic activity more dramatically under glucose starvation compared to energy-rich conditions, we postulated that PAQR3 might receive signals from glucose starvation to improve its regulatory effect on autophagy. Using a method based on Phos-tag gels in which phosphorylated proteins are super-shifted away from the unphosphorylated proteins, we found that PAQR3 is phosphorylated by AMPK upon glucose starvation in an ATG14-dependent manner. Since the NH2-terminal 71 amino acids of PAQR3 might be directly phosphorylated by AMPK because of its cytosolic orientation in topology, we investigated the potential phosphorylation site within this region. We found that a T32A mutation of PAQR3 completely abolishes AMPK-mediated phosphorylation. Furthermore, mass spectrometry provided direct evidence that T32 of PAQR3 is phosphorylated by AMPK in vitro. Although PAQR3 T32 phosphorylation does not affect the interaction between PAQR3 and the ATG14-linked PIK3C3 complex, it is pivotal for autophagy initiation and ATG14-linked PIK3C3 activity upon glucose starvation.
In 8-wk-old mice, exercise-induced autophagy in both liver and skeletal muscle is dramatically attenuated by Paqr3 deletion. Remarkably, the aged Paqr3 knockout mice display multiple neurodegenerative phenotypes, such as impaired stability on the accelerating rotarod, abnormal limb clasping, ataxic walking pattern, and weakened grip strength. Besides, both the formation and activity of the ATG14-associated class III PtdIns3K complex are compromised by Paqr3 deletion in vivo. Therefore, PAQR3 could be regarded as a potential candidate drug target for neurodegenerative diseases.
In summary, PAQR3 participates in autophagy initiation via 2 layers of regulation (Fig. 1). First, PAQR3 acts as a scaffold to enhance the relative abundance of the ATG14-linked PIK3C3 complex, so that a higher PtdIns3P generation capacity is acquired before the arrival of starvation signals. This regulation process can be recognized as a cellular self-protecting mechanism to prepare for the possible arrival of stress conditions by ensuring sufficient reserve of pre-autophagy class III PtdIns3K. For the second layer of regulation, AMPK phosphorylates PAQR3 at T32 upon glucose starvation, switching on powerful PtdIns3P production and autophagosome formation. This mode of the regulatory mechanism thus ensures a prompt and effective autophagy initiation upon glucose starvation.
Figure 1.
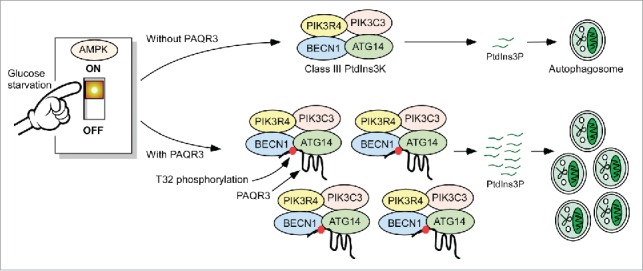
A schematic model of PAQR3 regulation of autophagy in response to glucose starvation. See text for details.
Notably, PAQR3 acts as a dual-modulator for both class I PI3K and class III PtdIns3K, but the regulation modes are different. PAQR3 promotes the formation and activity of the ATG14-linked class III PtdIns3K. However, PAQR3 reduces class I PI3K activity via blocking the interaction between PIK3CA/p110α and PIK3R/p85. Why are there 2 opposite modes of regulation? One possible explanation is that upon growth factor stimulation, PAQR3 keeps class I PI3K signaling in check to prevent cells from overactivation so as to resist abnormal cell proliferation. Under starvation conditions, PAQR3 adequately and swiftly drives autophagy initiation to maintain cell survival. Therefore, PAQR3 exquisitely balances 2 classes of lipid kinases to sustain cellular homeostasis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.