

ABSTRACT
The Paneth cell is a unique intestinal epithelial cell that can sense the gut microbiome and secrete anti-microbial peptides, thereby playing critical roles in the maintenance of homeostasis at the intestinal-microbial interface. These roles in regulating innate immunity and intestinal microbial ecology are dependent on a functional autophagy pathway through ATG16L1. ATG16L1 is a regulator for autophagy and a risk gene for inflammatory bowel disease (IBD). We demonstrated that a low VDR/vitamin D receptor level in the intestine is associated with abnormal Paneth cells, impaired autophagy function, and imbalanced bacterial profile (dysbiosis), accompanied by a reduction of ATG16L1. We determined that VDR transcriptionally regulates ATG16L1 as a VDR target gene. Administration of the bacterial product butyrate increases intestinal VDR expression and suppresses inflammation in a colitis model. Thus, our study indicates that VDR may be a determinant of IBD risk through its actions on ATG16L1. These insights can be leveraged to define therapeutic targets for restoring Paneth cells and autophagy through VDR in chronic inflammation. It may also have applicability for infectious diseases and autoimmune diseases associated with skin or lung, where the host is in contact with bacteria.
KEYWORDS: ATG16L1, bacteria, dysbiosis, inflammation, nuclear receptor, Paneth cells, transcriptional factor, vitamin D, vitamin D receptor, vitamin D response element
Deletion of ATG16L1, encoding an important regulator for autophagy and an IBD risk gene, leads to abnormal Paneth cells. Intriguingly, our recent paper demonstrated that a low VDR protein level is associated with abnormal Paneth cells and impaired autophagy function, accompanied by a reduction of ATG16L1. We determined that VDR transcriptionally regulates ATG16L1 as a VDR target gene. Furthermore, we found that conditional removal of VDR in the intestinal epithelium made mice more susceptible to chemical induced-colitis. In the immune-based IL10−/− colitis mice, a significant decline in the expression of VDR was found in IL10−/− mice with colitis symptoms compared to mice without colitis symptoms, Intestinal VDR expression is decreased in IBD patients. Moreover, low levels of intestinal epithelial VDR correlate with reduced ATG16L1 and dysbiosis in experimental colitis models and intestinal tissue from patients with IBD. Administration of the bacterial product butyrate increases intestinal VDR expression and suppresses inflammation in a colitis model. Therefore, our study indicate that intestinal epithelial VDR may be a determinant of IBD risk through its actions on the autophagy regulatory gene ATG16L1, thus determining Paneth cell states and microbial assemblage in intestinal homeostasis.
Our study links dysbiosis, innate immune functions (Paneth cells), and genetic susceptibility (ATG16L1) through intestinal epithelial VDR. It fills an existing gap by characterizing the precise role of intestinal epithelial VDR in regulating intestinal homeostasis through alterations in intestinal autophagy and the microbiome. Our data suggest that decreased expression of intestinal epithelial VDR perturbs microbial homeostasis (fewer butyrate-producing bacteria) and VDRΔIEC (intestinal epithelial VDR conditional knockout) mice are susceptible to colitis. In an immune-based IL10−/− colitis model, we found significant reduction of VDR and ATG16L1. Administration of butyrate (a fermentation product of gut microbes) increases intestinal VDR expression and suppresses inflammation in a colitis model. These data bring up the possibility that microbial natural products can be used therapeutically to restore VDR-dependent functions in patients with IBD.
The lack of intestinal VDR is associated with chronic inflammation. Our previous study showed that VDR negatively regulates bacteria-induced NFKB activity in intestinal inflammation. Lack of VDR leads to a reduction of NFKBIA/IκBα, an endogenous inhibitor of NFKB activity. It is still not known whether intestinal VDR expression restores microbial homeostasis though specifically regulating Paneth cells and ATG16L1, or involves multiple pathways in colitis. This may occur through several unique mechanisms that include autophagy and NFKB. In the future, we will determine the effects and mechanisms necessary for restoration of intestinal VDR and host-microbial interactions in colitis.
Based on the current data, we propose a working model in Figure 1 that may provide a unifying hypothesis that can potentially account for defective autophagy, abnormal Paneth cells, and dysbiosis found in many patients with IBD and other intestinal disorders. As an upstream regulator, VDR regulates genetic susceptibility genes (ATG16L1), autophagy, Paneth cell function, and gut microbiota that are essential for the maintenance of intestinal homeostasis. If VDR is reduced, ATG16L1 is also reduced associated with abnormal Paneth cells, reduced autophagic acitivity, and dysbiosis, for the development of chronic states of mucosal inflammation. This knowledge can be immediately used to develop intestinal VDR as a clinical biomarker for identifying patients who might benefit from currently available interventions. This knowledge can also be exploited to define novel strategies to prevent and treat human IBD and intestinal disorders.
Figure 1.
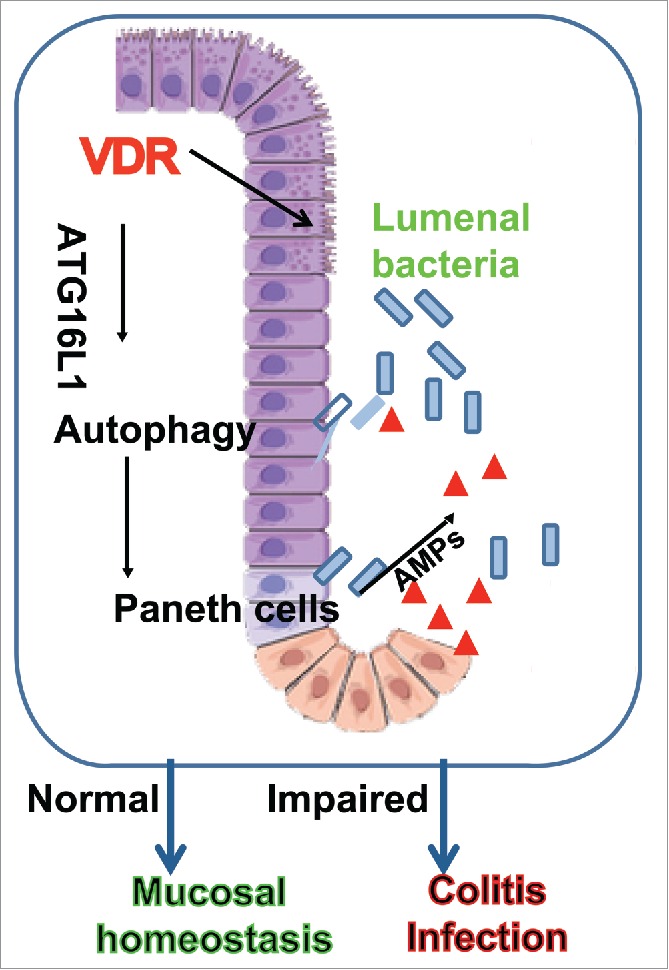
Working model of intestinal VDR in regulating the activity of autophagy, function of Paneth cells, and microbial homeostasis through ATG16L1. AMPs, antimicrobial peptides.
VDR is a nuclear receptor that mediates most known functions of 1,25-dihydroxyvitamin D (1,25[OH]2D3), the active form of vitamin D. Once VDR binds with 1,25(OH)2D3, VDR heterodimerizes with RXR (retinoid X receptor). Activated VDR then binds to the vitamin D-response element (VDRE) in the target gene promoter to regulate gene transcription. VDR targets include genes for CAMP/LL-37 (cathelicidin antimicrobial peptide), and DEFB/β defensin. We have determined that the genes encoding both tight junction protein CLDN2 (claudin 2) and the autophagy regulator ATG16L1 are novel VDR target genes. Indeed, ˜3% of the mouse and human genomes are regulated directly or indirectly by the vitamin D endocrine system, further suggesting the possibility of widespread effects of vitamin D and VDR in disease mechanisms.
Functionally, VDR and 1,25(OH)2D3 are known as key players in calcium homeostasis and in electrolyte and blood pressure regulation. Recent evidence demonstrates that vitamin D deficiency is a critical factor in the pathology of IBD and other diseases. Vitamin D and VDR appear to be important immunological regulators of the microbial community. Aberrant immune responses and dysbiosis (imbalanced bacteria profile) are implicated in various diseases. Our current studies use a tissue-specific VDR knockout model to understand the pathogenesis of IBD. These findings are important not only for a better understanding of intestinal diseases, but may also have applicability for infectious diseases and autoimmune diseases associated with skin or lung, where the host is in contact with bacteria. Vitamin D protects the host from Mycobacterium tuberculosis in lungs. Vitamin D may benefit the gut microbiome and improve glucose homeostasis in diabetes. Vitamin D3 and its analog can increase autophagy in immune cells. Vitamin D3 signaling is able to regulate autophagy at different steps, including induction, nucleation, elongation, closure, and maturation. Further studies are needed to understand whether the role of vitamin D3-VDR in regulating autophagy is tissue specific. Insights gained from understanding how the vitamin D3-VDR pathway is integrally involved in regulating autophagy may serve as a paradigm for understanding the nature of host defense signals in infectious diseases and chronic inflammation.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.