

ABSTRACT
MIF (macrophage migration inhibitory factor [glycosylation-inhibiting factor]) is a pro-inflammatory cytokine expressed in multiple cells types, including macrophages. MIF plays a pathogenic role in a number of inflammatory diseases and has been linked to tumor progression in some cancers. Previous work has demonstrated that loss of autophagy in macrophages enhances secretion of IL1 family cytokines. Here, we demonstrate that loss of autophagy, by pharmacological inhibition or siRNA silencing of Atg5, enhances MIF secretion by monocytes and macrophages. We further demonstrate that this is dependent on mitochondrial reactive oxygen species (ROS). Induction of autophagy with MTOR inhibitors had no effect on MIF secretion, but amino acid starvation increased secretion. This was unaffected by Atg5 siRNA but was again dependent on mitochondrial ROS. Our data demonstrate that autophagic regulation of mitochondrial ROS plays a pivotal role in the regulation of inflammatory cytokine secretion in macrophages, with potential implications for the pathogenesis of inflammatory diseases and cancers.
KEYWORDS: autophagosome, cytokines, inflammation, LPS, macrophage, reactive oxygen species, starvation
Introduction
MIF (macrophage migration inhibitory factor [glycosylation-inhibiting factor]) was first discovered in the 1960s, making it one of the first cytokines to be identified.1,2 However, human MIF cDNA was not cloned until 1989 and the first mif−/− mice not reported until 1999.3,4 Since this time there has been significant scientific interest in MIF, which functions not only as a pro-inflammatory protein, but also as a stress and growth factor, and is released by cells of the anterior pituitary gland, similar to a hormone.5,6
Most immune cells produce MIF. It is constitutively expressed and stored within intracellular vesicles, so de novo synthesis is not required for secretion. Macrophages secrete MIF following treatment with pro-inflammatory stimuli, including lipopolysaccharide (LPS), TNF (tumor necrosis factor) and IFNG (interferon, gamma),7 and it can act in an autocrine or paracrine manner to stimulate the release of other pro-inflammatory mediators, enhance phagocytosis and increase production of reactive oxygen species (ROS) and nitric oxide (NO).7-10 MIF has been demonstrated to be an important protective factor in the immune response to Gram-negative bacteria, including Escherichia coli and Klebsiella pneumonia.11 However, hypersecretion of MIF is associated with pathogenesis in a number of autoimmune conditions, including rheumatoid arthritis, atherosclerosis and systemic lupus erythematosus.12,13
Previous reports have demonstrated that loss of autophagy in macrophages and dendritic cells (DCs) leads to increased secretion of pro-inflammatory cytokines, including IL6, type I IFN and the IL1 (interleukin 1) family members IL1A, IL1B and IL18.14-18 Moreover, through effects on IL1B, loss of autophagy leads to increased IL23 secretion by macrophages and DCs, and supernatant fractions from these LPS-treated autophagy-defective cells, high in IL1B and IL23, enhance the secretion of IL17, IL22 and IFNG by innate γδ T cells.19 Similarly, mice with deletion of the autophagy gene Atg5 in myeloid cells produce more IL1A and IL17 in response to infection with Mycobacterium tuberculosis.20 Conversely, induction of autophagy suppresses the secretion of IL1B and IL23, both in vitro and in vivo.15,19 Together, these findings indicate that autophagy is a process that can directly influence multiple cytokines.
A role for MIF in regulating autophagy has been highlighted by a number of recent studies, although with seemingly conflicting results. MIF has been shown to induce autophagy in cardiomyocytes,21,22 endothelial cells23 and a human hepatoma cell line,24 while other work has suggested an inhibitory role for MIF on autophagy in cardiomyocytes and breast cancer cell lines.25-27 Intriguingly, one study demonstrated that amino acid starvation, which induces autophagy, also induces MIF secretion,24 but the effect of autophagy on the release of MIF remains poorly understood. However, as MIF is constitutively expressed in cells, autophagy might be a key regulator of its turnover and secretion. To determine whether autophagy acts as a regulator of MIF, we looked at the effect of pharmacological inhibitors of autophagy and siRNA against Atg5 on the secretion of MIF by human and mouse macrophages. We found that loss of autophagy in these cells resulted in greater secretion of MIF, both basally and in response to stimuli. Intriguingly, amino acid starvation also induced MIF secretion in a ROS-dependent manner, but pharmacological induction of autophagy had no effect on MIF secretion.
Results
Loss of autophagy increases MIF secretion by macrophages
A number of studies have demonstrated a role for autophagy in the regulation of pro-inflammatory cytokines.14,15,17,19 Here, we looked at the effect of autophagy inhibition on the secretion of MIF in response to the TLR4 (toll like receptor 4) agonist LPS or heat-killed Escherichia coli (HK E. coli). Treatment of human phorbol 12-myristate 13-acetate (PMA)-differentiated THP-1 cells with LPS or HK E. coli in the presence of the class III phosphatidylinositol 3-kinase (PtdIns3K)/autophagy inhibitor 3-methyladenine (3-MA) resulted in an increase in MIF secretion (Fig. 1A, B). On its own, LPS induced MIF secretion by THP-1 but HK E. coli did not, while 3-MA alone also did not induce significant MIF release (Fig. 1A, B). However, 3-MA significantly increased MIF secretion in response to HK E. coli in undifferentiated monocytic THP-1 cells (Fig. 1C). Moreover, as previously reported,15 3-MA also increased IL1B secretion, but inhibited TNF secretion (Fig. 1D, E). Neither LPS nor 3-MA, either separately or together, had any effect on levels of Mif mRNA in RAW264.7 cells (Fig. S1A). Moreover, treatment with LPS + 3-MA for 6 h had no significant effect on cell viability (Fig. S1B).
Figure 1.
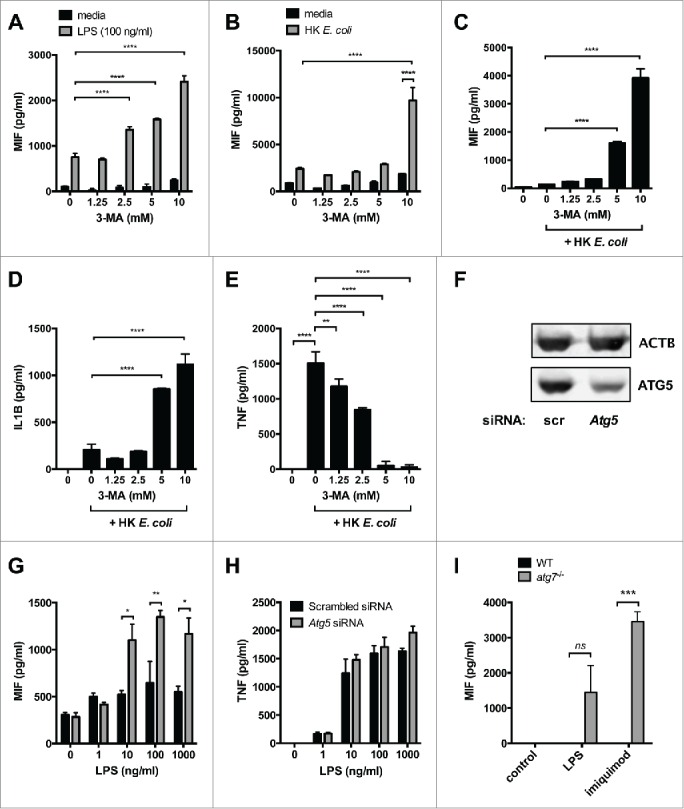
Loss of autophagy enhances MIF secretion by human and mouse macrophages. (A and B) PMA-differentiated THP-1 cells were treated with 3-methyladenine (3-MA) in the presence or absence of (A) LPS (100 ng/ml) or (B) heat-killed Escherichia coli (HK E. coli, multiplicity of infection 1:1) for 6 h and MIF measured in the culture medium by ELISA. (C-E) Undifferentiated THP-1 cells were treated with 3-MA and HK E. coli for 6 h and secreted (A) MIF, (B) IL1B and (C) TNF measured by ELISA. (F) Western blot analysis of ACTB/β-actin and ATG5 protein in lysates from RAW264.7 cells transfected with siRNA against Atg5 or nontargeting (scrambled, scr) siRNA for 48 h. (G and H) RAW264.7 cells were transfected with scrambled or Atg5 siRNA for 48 h then treated with LPS for 6 h. Secreted (G) MIF and (H) TNF were measured by ELISA. Bars represent means ± SEM of triplicates, representative of at least 3 experiments. (I) Bone marrow-derived macrophages from atg7wt/wt× LysMcre (WT) and atg7fl/fl× LysMcre (atg7−/−) mice were treated with LPS (100 ng/ml) or imiquimod (10 μg/ml) for 6 h and secreted MIF measured by ELISA. Bars represent means ± SEM for 3 mice. *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001.
To further confirm the role of autophagy in MIF secretion, we transfected mouse RAW264.7 macrophages with siRNA against Atg5 and compared MIF secretion to cells transfected with control, nontargeting (scrambled) siRNA. Loss of ATG5 protein was confirmed by western blot (Fig. 1F) and inhibitory effects on autophagosome formation confirmed by confocal microscopy in GFP-LC3+ macrophages (Fig. S1C). Cells transfected with Atg5 siRNA secreted higher levels of MIF, both basally and in response to LPS, although LPS itself had no effect on MIF release in these cells (Fig. 1G). Loss of Atg5 had no effect on TNF in these cells (Fig. 1H). Similarly, increases in MIF secretion by RAW264.7 cells transfected with siRNA against Atg5 were seen in response to the TLR7 agonist imiquimod, while TNF secretion was again unaffected (Fig. S1D and E).
Finally, we looked at MIF secretion by bone marrow-derived macrophages from atg7fl/fl×LysMcre mice treated with LPS or imiquimod. Unlike the cell lines, these macrophages did not secrete detectable levels of MIF in the absence of stimulation (Fig. 1I). Moreover, cells from Atg7 competent mice (atg7wt/wt×LysMcre) did not produce MIF in response to overnight treatment with LPS or imiquimod. However, cells from atg7fl/fl×LysMcre mice secreted MIF in response to both LPS and imiquimod, although this was not statistically significant for LPS (Fig. 1I). Taken together, these data indicate that loss of autophagy enhances the secretion of MIF by monocytes/macrophages in response to inflammatory stimuli.
Increased MIF secretion in autophagy-deficient cells is dependent on mitochondrial ROS
Previous studies have demonstrated that increased IL1 family cytokine secretion in autophagy-deficient cells is dependent on ROS.15,17 Similarly, ROS inhibitors can inhibit starvation-induced MIF secretion.24 Thus, to determine whether MIF secretion in autophagy-deficient macrophages was dependent on ROS, we treated human PMA-differentiated THP-1 cells with LPS in the presence or absence of 3-MA (10 mM) and the ROS scavenger N-acetyl-L-cysteine (NAC, 25 mM). As before, 3-MA increased LPS-induced MIF secretion in a dose-dependent manner and this effect was inhibited by treatment with NAC (Fig. 2A). Accordingly, 3-MA-induced MIF secretion was completely abrogated by NAC at 3 different concentrations in mouse immortalized bone marrow-derived macrophages (iBMM, Fig. 2B). We confirmed these effects using another ROS inhibitor, (2R,4R)-APDC, which also inhibited 3-MA-induced MIF secretion in iBMM (Fig. 2C).
Figure 2.
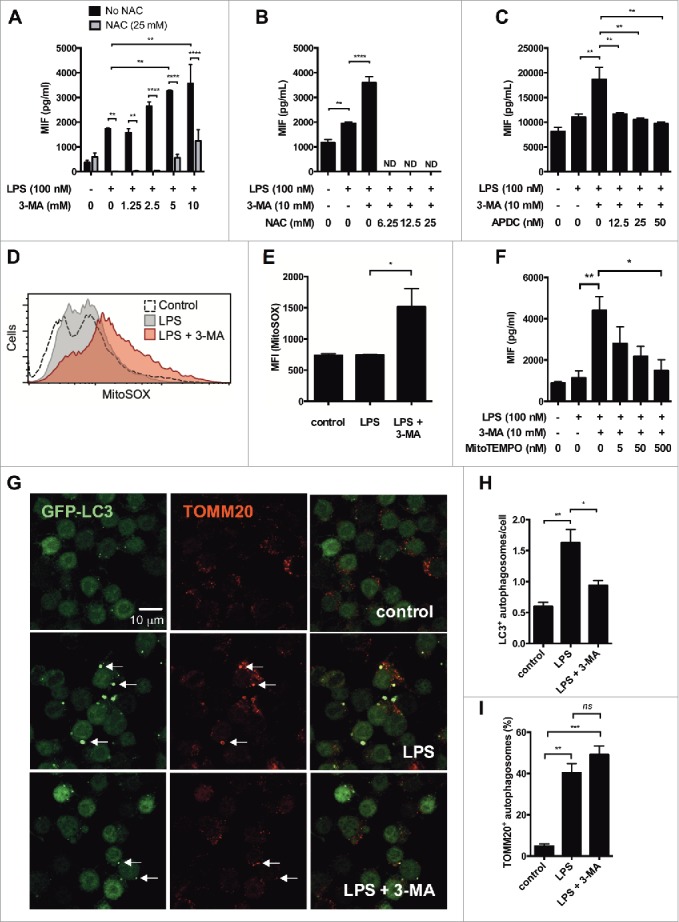
MIF secretion in autophagy-deficient cells is dependent on mitochondrial reactive oxygen species (ROS). (A) PMA-differentiated THP-1 cells were treated with LPS (100 ng/ml) and different concentrations of 3-methyladenine (3-MA) in the presence or absence of the ROS inhibitor N-acetyl-L-cysteine (NAC, 25 mM) for 6 h. Secreted MIF was measured by ELISA. (B and C) Immortalized bone marrow-derived macrophages (iBMM) were treated with LPS (100 ng/ml) and 3-MA (10 mM) in the presence or absence of different concentrations of (B) NAC or (C) the ROS inhibitor (2R,4R)-APDC (APDC). Bars represent means ± SEM of triplicates, representative of at least 3 experiments. (D) RAW264.7 cells were treated with LPS (100 ng/ml) or LPS + 3-MA (10 mM) for 6 h, then incubated with MitoSOX Red to measure mitochondrial superoxide production by FACS. The geometric mean fluorescence index (MFI) is shown in (D and E). (F) iBMM were treated with LPS + 3-MA ± MitoTEMPO for 6 h at the concentrations stated and MIF secretion measured by ELISA. (G) iBMM stably transfected with GFP-LC3 were treated with LPS (100 ng/ml) or LPS + 3-MA (10 mM) for 6 h, fixed and stained with antibody against TOMM20. Autophagosome formation (H) and percentage of TOMM20+ autophagosomes (I) were analyzed. Bars represent means ± SEM of 3 separate experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001.
Studies have highlighted a specific role for mitochondrial ROS in the secretion of IL1B by autophagy-deficient macrophages.28,29 To examine the production of mitochondrial superoxide by macrophages we used MitoSOX Red, which is oxidized by superoxide but not by other ROS or reactive nitrogen species. Upon oxidation, MitoSOX Red is highly fluorescent, allowing for analysis of superoxide in live cells. Treatment of RAW 264.7 cells with LPS for 6 h had no effect on superoxide production, but treatment with LPS + 3-MA significantly increased the percentage of cells positive for MitoSOX (Fig. 2D, E, Fig S2), demonstrating that loss of autophagy leads to an accumulation of mitochondrial ROS in the cytosol. Moreover, increased MIF secretion in response to LPS + 3-MA was inhibited by the mitochondrial ROS inhibitor MitoTEMPO (Fig. 2F).
These data suggest that loss of mitophagy is responsible for the ROS-dependent MIF secretion observed. To test this further, we looked at mitophagy in iBMM stably transfected with GFP-LC3 (GFP-LC3 iBMM) by confocal microscopy, staining cells with antibody against TOMM20 (translocase of outer mitochondrial membrane 20). Treatment of cells with LPS increased both autophagosome formation and the percentage of TOMM20+ autophagosomes, indicating a significant increase in mitophagy (Fig. 2G-I). Addition of 3-MA had no effect on the percentage of TOMM20+ autophagosomes, but did significantly inhibit autophagosome formation (Fig. 2G–I). These data indicate that 3-MA inhibits autophagy globally, but that its effect on mitophagy is most pertinent to the effects observed on MIF secretion.
Amino acid starvation, but not pharmacological induction of autophagy, increases MIF secretion in a mitochondrial ROS-dependent manner
A previous study has reported that amino acid starvation increases MIF secretion in Hu-H7 cells.24 These data might indicate that induction of autophagy could increase MIF secretion. To investigate this further, we first looked to confirm these findings in macrophages. We determined the effect of amino acid starvation on MIF secretion by PMA-differentiated THP-1 cells over time. Cells were cultured in Hank's balanced salt solution (HBSS), depriving them of amino acids, for up to 6 h, and secreted MIF measured by ELISA. At 2, 4 and 6 h, cells cultured in HBSS secreted significantly higher levels of MIF than those cultured in complete medium (Fig. 3A) confirming that amino acid starvation can increase MIF release. The same effect was also observed in RAW264.7 cells at 2, 4 and 6 h (Fig. 3B). IL1B, TNF and CCL2/MCP-1 (chemokine [C-C motif] ligand 2) were not detected in supernatant fractions from starved macrophages (data not shown). Moreover, starvation did not affect cell viability over 6 h (Fig. S1B). To determine whether this increased secretion was dependent on ROS, we treated THP-1 cells cultured in HBSS for 6 h with NAC (25 mM). Starvation in HBSS for 6 h significantly increased MIF secretion in these cells and this effect was inhibited by NAC (Fig. 3C). Enhanced secretion of MIF in response to starvation was not altered in mouse cybb/nox2−/− BMM, suggesting that NADPH oxidase-derived ROS are not responsible for this effect (Fig. 3D). Furthermore, starvation over a 6-h period significantly enhanced MitoSOX fluorescence in RAW264.7 cells (Fig. 3E-G) and MitoTEMPO inhibited starvation-induced MIF secretion (Fig. 3H). These data suggest that the observed increases in MIF in response to starvation are dependent on mitochondrial ROS.
Figure 3.
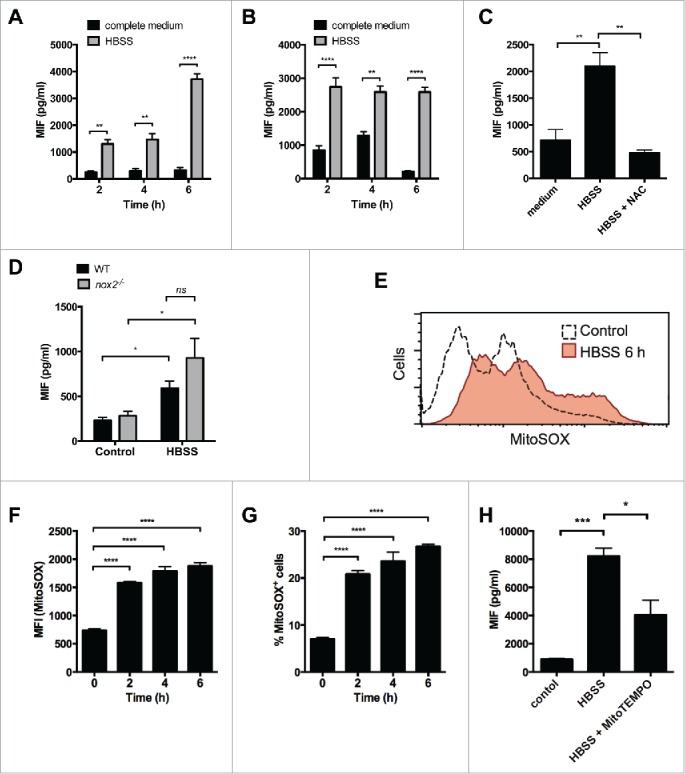
Amino acid starvation induces MIF secretion by macrophages. (A) PMA-differentiated THP-1 cells or (B) RAW264.7 cells were starved in Hank's balanced salt solution for the times stated and secreted MIF measured by ELISA. (C) PMA-differentiated THP-1 cells were starved in Hank's balanced salt solution (HBSS) for 6 h in the presence or absence of the ROS inhibitor N-acetyl-L-cysteine (NAC, 25 mM) and secreted MIF measured by ELISA. (D) Primary BMM from WT and cybb−/− mice (n = 3) were treated with HBSS for 6 h and MIF secretion measured by ELISA. (E) FACS analysis (geometric mean) of MitoSOX in RAW264.7 cells treated with HBSS for 6 h. Geometric mean fluorescence for MitoSOX (F) and percentage of MitoSOX+ RAW264.7 cells (G) after treatment with HBSS over time. (H) iBMM were treated with HBSS or HBSS + MitoTEMPO (5 nM) for 6 h. MIF secretion was measured by ELISA. Bars represent means ± SEM of 3 separate experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001.
Next, we investigated the effect of starvation in autophagy-deficient cells, to determine whether loss of autophagy altered the secretion of MIF in response to starvation. RAW264.7 cells were transfected with siRNA against Atg5, or with nontargeting scrambled siRNA and cultured in complete medium or HBSS for 6 h. Starvation increased MIF secretion in cells transfected either with scrambled siRNA or Atg5 siRNA (Fig. 4A), suggesting that MIF secretion in response to starvation is not mediated via effects on autophagy induction.
Figure 4.
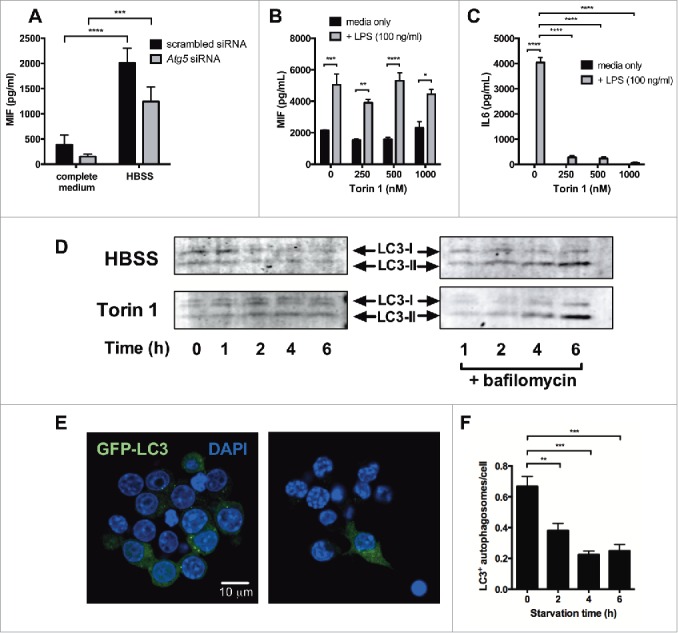
MIF secretion in starved cells is not dependent on autophagy induction. (A) RAW264.7 cells were transfected with nontargeting (scrambled) or Atg5 siRNA for 48 h and starved in HBSS for 6 h. Secreted MIF was measured by ELISA. (B and C) PMA-differentiated THP-1 cells were treated with different doses of the MTOR inhibitor Torin 1 in the presence or absence of LPS (100 ng/ml). Secretion of (B) MIF and (C) IL6 was measured by ELISA. (D) Western blot analysis of LC3-I and LC3-II in lysates from RAW264.7 cells starved in HBSS or treated with Torin 1 for the times indicated. Experiments were done in the presence or absence of bafilomycin A1. Blot shown is representative of 3 separate experiments. (E) Immortalized bone marrow-derived macrophages (iBMM) stably expressing GFP-LC3 (green) were cultured in complete medium or HBSS for 6 h, fixed and analyzed for autophagosome formation by confocal microscopy. (F) Bar graph showing the number of GFP-LC3+ autophagosomes per cell. Bars represent means ± SEM from triplicates, representative of at least 3 separate experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001.
To investigate this further, we treated human and mouse macrophages with the MTOR inhibitors/autophagy inducers Torin 1, rapamycin and AZD8055, none of which had any effect on MIF secretion (Fig 4B, Fig. S3); however, Torin 1 did inhibit LPS-induced IL6 secretion (Fig. 4C), a finding consistent with a previous report demonstrating a role for autophagy in the regulation of this cytokine.14 In addition, while we did see IL1B in autophagosomes, we did not observe colocalization of MIF with LC3 in LPS-treated GFP-LC3+ macrophages (Fig. S4), suggesting that autophagosomes are not directly involved in MIF secretion, as has been proposed for IL1B.30,31
We hypothesized that starvation over time might induce rapid turnover of autophagosomes, potentially leading to a net loss of autophagy. To test this, we first looked at intracellular levels of LC3-I and LC3-II in starved RAW264.7 cells by western blot. Over time, total levels of both LC3-I and LC3-II decreased with starvation, suggesting that the protein was steadily depleted in starved cells (Fig. 4D). The addition of bafilomycin A1, however, increased levels of LC3-II over time (Fig. 4D), confirming that autophagosome turnover did occur. In contrast, LC3 levels in macrophages treated with the MTOR inhibitor Torin 1 did not decrease over time, even though conversion of LC3-I to LC3-II could be seen in the presence of bafilomycin A1 (Fig. 4D). This would suggest that, in contrast to amino acid starvation, pharmacological induction of autophagy does not deplete LC3 protein levels over 6 h. To investigate the effect of starvation on autophagosomes further, we starved GFP-LC3 iBMM in HBSS. After 2, 4 and 6 h, the number of LC3+ autophagosomes per cell was significantly decreased in the starved cells (Fig. 4E, F), again suggesting that starvation ultimately depletes autophagosomes. Together, these data suggest that while amino acid starvation does increase MIF secretion by macrophages, this is not dependent on the induction of autophagy.
Discussion
This is the first study to demonstrate that inhibition of autophagy leads to increased secretion of MIF. Our data show that this is dependent on ROS, which have previously been shown to induce MIF secretion.24 Similarly, previous studies have demonstrated that increased secretion of IL1A, IL1B and IL18 in autophagy-deficient macrophages and DCs is linked with increased ROS.15,17,28 The source of this ROS may be mitochondria; previous studies have demonstrated that mitochondrial, but not NADPH-dependent, ROS are responsible for increased IL1 secretion by macrophages and DCs in which autophagy is inhibited.15,28 Moreover, Zhou et al.29 demonstrated that blocking autophagy in LPS-stimulated macrophages leads to an accumulation of ROS-producing mitochondria, driving NLRP3 inflammasome activation and IL1B secretion. Likewise, in this study, we demonstrated that inhibition of autophagy leads to an accumulation mitochondrial ROS in macrophages treated with LPS, whereas LPS treatment significantly increased mitophagy. In addition, increased secretion of IL1A and IL1B in autophagy-deficient macrophages and DCs can directly lead to increased IL23 secretion by the same cells.19 However, MIF secretion in autophagy-deficient cells did not appear to be dependent on IL1, as neutralizing antibodies against IL1A and IL1B had no effect in mouse bone marrow-derived macrophages (data not shown). Also, MIF secretion in autophagy-deficient macrophages was increased in the presence of imiquimod, which does not induce IL1B secretion in autophagy-impaired macrophages.15 Moreover, IL1B is not secreted by RAW cells following inhibition of autophagy, as these cells do not express the inflammasome component PYCARD/ASC (PYD and CARD domain containing).32
A previous study has suggested that starvation-induced autophagy increases MIF secretion in human Hu-H7 hepatoma cells.24 Our data suggest that, in macrophages, while amino acid starvation does increase MIF secretion, this may not be dependent on the induction of autophagy, but rather on the depletion of autophagosomes over time due to rapid turnover, potentially coupled with a loss of de novo synthesis of autophagy proteins, such as LC3. Loss of LC3 protein over time was not seen in cells treated with the MTOR inhibitor Torin 1, which, along with other MTOR inhibitors, had no effect on MIF secretion. Complementary to the study by Chuang et al.,24 starvation-induced MIF secretion was dependent on mitochondrial ROS. Thus, we hypothesize that accumulation of ROS from damaged mitochondria, combined with a decrease in autophagy over time, leads to increased MIF secretion in amino acid-starved cells.
The secretion of MIF is still not well understood. One study demonstrated a role for the membrane- trafficking molecule USO1/p115/TAP (USO1 vesicle transport factor).33 Phosphorylated USO1 is found in the cytosol, whereas the unphosphorylated molecule locates on membranes of the Golgi complex and transcytotic vesicles.34 This study demonstrated interaction between MIF and USO1, as well as colocalization within vesicles of human peripheral blood mononuclear cells (PBMC). Moreover, USO1 was required for the secretion of MIF in response to LPS and was released alongside MIF. In contrast to these findings, we did not observe colocalization between MIF and USO1 nor any effect of autophagy inhibition or starvation on levels of USO1 protein or release from cells (data not shown). This would suggest that USO1 is not involved in the secretion of MIF in autophagy-deficient or starved cells. Another study suggested a role for the ATP-binding cassette transporter ABCA1 in unconventional secretion of MIF.35 However, treatment of cells with the inhibitors glyburide or cyclosporine A35,36 did not inhibit MIF secretion in response to 3-MA or starvation (data not shown), suggesting that this is not the mechanism through which MIF is secreted here. Finally, MIF can be released by necrotic cells, much like a classical danger-associated molecular pattern molecule.37 However, neither 3-MA nor starvation induced significant cell death in our experiments, suggesting this is also not the mechanism of release. Thus, further work is needed to uncover the potentially novel mechanism of MIF secretion under these conditions and how this is specifically driven by mitochondrial ROS.
As well as being affected by loss of autophagy, as our study demonstrates, MIF has itself been shown to regulate autophagy. Studies have shown that MIF increases autophagy in cardiomyocytes, endothelial cells and Hu-H7 hepatoma cells, although other work suggests the opposite effect in cardiomyocytes and breast cancer cells. Further work is needed to reconcile these apparently conflicting data regarding the effects of MIF upon autophagy. Given the role of autophagy in regulating pro-inflammatory cytokine secretion in macrophages, it would be of considerable interest to determine the effects of MIF on autophagy in these cells. This is potentially complicated by the fact that MIF is constitutively expressed and released by macrophages, which may mean that its effects are highly dependent on concentration, or may be more easily studied in MIF-deficient cells. Previous studies have demonstrated that both IL1A and IL1B induce autophagy in macrophages, which in turn limits their secretion.15,17,38 This would suggest a bidirectional feedback mechanism for release of these pro-inflammatory cytokines. A role for MIF in promoting or inhibiting the autophagy-dependent regulation of IL1A and IL1B would be a novel and important finding in the context of inflammation.
MIF is a key protein involved in inflammatory responses, particularly to Gram-negative bacteria. Moreover, MIF is implicated in the pathology of autoimmune diseases, including rheumatoid arthritis, systemic lupus erythematosus and atherosclerosis12,13,39 and is linked with cancer-associated inflammation, promoting tumor growth in a number of different cancers.40 Clinically, the role for MIF in reducing glucocorticoid sensitivity is particularly important, given the wide use of these drugs in the treatment of inflammatory diseases. Moreover, a growing body of evidence has highlighted significant links between autophagy and the regulation of inflammation and autoimmune disease. In particular, polymorphisms in autophagy genes have been linked to Crohn disease and systemic lupus erythematosus.41 Thus, dysfunction of autophagy is directly linked to inflammatory disease and our data here, along with previous reports on other cytokines, highlight a potentially important contributory mechanism to this. Our data establish a role for autophagy in regulating the secretion of MIF by macrophages. Further characterization of this, including analysis of MIF in patients with known polymorphisms in autophagy genes, may reveal the potential to therapeutically regulate multiple pro-inflammatory cytokines by targeting autophagy in macrophages.
Materials and methods
Cells and culture conditions
Human leukemic THP-1 monocytic cells were cultured in complete RPMI: RPMI 1640 medium (Gibco, 11875–093) supplemented with 10% fetal bovine serum (JRH Biosciences, 12103), 2 nM L-glutamine, 50 U/ml penicillin and 50 μg/ml streptomycin (Gibco, 10378–106). Cells were differentiated with PMA (5 ng/ml, Sigma, P8139) for 24 h, after which the PMA was removed and replaced with fresh complete RPMI. Cells were cultured for a further 24 h before final stimulation. Mouse immortalized bone marrow-derived macrophages (iBMM) and RAW264.7 cells were cultured in complete RPMI. iBMM stably expressing GFP-LC315 were cultured as for normal iBMM, but with the addition of puromycin dihydrochloride (10 μg/ml; Sigma, P9620) to the culture medium. Cells were cultured at 0.5–1 × 106 cells/ml. Bone marrow-derived macrophages from female atg7fl/fl × LysMcre and atg7wt/wt × LysMcre mice and cybb−/− mice (obtained from Professor Christopher Sobey, Monash University) were cultured in complete RPMI with 15% culture supernatant from MCSF-expressing L929 cells (L-cell conditioned medium) for 7 d.
Reagents
3-methyladenine (Sigma, M9281), freshly prepared for each experiment, was dissolved in phosphate-buffered saline (PBS; 8 g/l NaCl, 0.2 g/l KCl, 1.44 g/l Na2HPO4, 0.24 g/l KH2PO4) at 50°C at a stock concentration of 100 mM. N-acetyl-L-cysteine (NAC; Sigma, A7250) was dissolved in complete DMEM at a stock concentration of 500 mM and pH-balanced with sodium bicarbonate (Sigma, S5761). (2R, 4R)-APDC (P8765), bafilomycin A1 (B1793) and MitoTEMPO (SML0737) were from Sigma, AZD8055 from Synkinase (SYN-1166), Torin 1 from Cayman chemicals (10997) and rapamycin from Adipogen (AG-CN2-0025).
siRNA transfection
Mouse iBMM and RAW264.7 cells were transfected with siRNA as previously described.42 Briefly, cells were resuspended in 100 μl BTXpress electroporation buffer (BTX Harvard Apparatus, 45–0803) with 400 nM Atg5 or nontargeting (scrambled) ON-TARGETplus siRNA (SMARTpool, GE Healthcare Dharmacon) and electroporated using an Amaxa Nucleofector device (Lonza, Victoria, Australia). Cells were cultured at 5 × 105 cells/ml, medium replaced after 24 h and incubated for a further 24 h prior to final stimulation. Knockdown of ATG5 protein was confirmed for each experiment by western blot with antibody against ATG5 (Abcam, ab108327).
Enzyme-linked immunosorbent assay (ELISA)
Secreted cytokines were measured by ELISA of culture supernatants according to the manufacturers' protocols (R&D Systems for human MIF and mouse MIF, DY289 and DY1978; Biolegend for human IL6 and mouse TNF, 431302 and 430902).
Western blot
Cells were washed twice with PBS and lysed with 2% Igepal CA-130 (Sigma, I8896) in Tris buffer, as previously described,15 or in RIPA buffer (Cell Signaling Technology, 9806). Lysates were boiled with Laemmli buffer (under reducing conditions), loaded and separated on 4–12% NuPAGE® bis-tris gels (Novex, NP0322PK2) and transferred to PVDF membranes. The membranes were blocked with 0.5% teleostean gelatin (Sigma, G7765)-0.1% casein (Sigma, C7078) in PBS for 1 h and stained with primary antibodies overnight at 4°C (1:1,000 dilution in blocking buffer). The membranes were then probed with fluorescent secondary antibodies (diluted 1:10,000 in TBS-T) for 60 min at room temperature and analyzed on a LI-COR Odyssey fluorescence imaging system (LI-COR Biosciences, Lincoln, NE, USA). Adobe Photoshop was used for final image preparation.
Confocal microscopy
GFP-LC3 iBMM were cultured on glass coverslips, amino acid starved in HBSS (Sigma, H6648) for 6 h, and fixed in 2% paraformaldehyde in PBS. The cells were permeabilized with 0.1% Triton X-100 (Sigma, X-100) in PBS for 10 min at room temperature, then blocked with 0.5% teleostean gelatin-0.1% casein in PBS with 5% goat serum (Sigma, G6767) for 1 h at room temperature. The cells were stained with antibody against MIF (R&D Systems, MAB1978; 5 μg/ml in blocking buffer), IL1B (Abcam, ab9722; 2.5 μg/ml) or TOMM20 (Santa Cruz Biotechnology, sc-17764; 200 ng/ml) for 1 h, then with Alexa Fluor 568 anti-mouse Alexa Fluor 568- (Abcam, ab175473; 1:1000 in PBS) and/or anti-rabbit Alexa Fluor 637-conjugated secondary antibody (Abcam, ab150079; 1:1,000 in PBS) for 1 h at room temperature. Nuclei were stained with DAPI (2-[4-Amidinophenyl]-6-indolecarbamidine dihydrochloride; Sigma, D9542; 5 μg/ml) for 5 min. Coverslips were mounted on glass slides and cells analyzed on a Nikon C1 confocal microscope. For analysis of GFP-LC3-positive autophagosomes, at least 100 cells per treatment were analyzed.
Mitochondrial ROS assay
To measure the production of mitochondrial ROS, MitoSOX Red (Molecular Probes, M36008) was used, per the manufacturer's protocol. Briefly, RAW264.7 cells were grown in complete medium on CELLview 4 chamber culture dishes (Greiner Bio One, 627870) and treated with LPS (Sigma, L2630, 100 ng/ml) or LPS + 3-MA (10 mM) for 5 h. Cells were treated with MitoSOX Red (2.5 μM) for 10 min at 37°C, then washed 3 times with HBSS, followed by incubation in complete medium. Live cells were analyzed on a Nikon C1 confocal microscope with a 60× water immersion lens; 3–4 images were taken and an average of 386 cells (250–509) counted per sample. For flow cytometry, cells were prepared and treated as above, then analyzed on a FACS Canto II (BD Biosciences, North Ryde, NSW, Australia).
Propidium iodide incorporation assay
RAW264.7 cells were treated with LPS (100 ng/ml), LPS + 3-MA (10 MM), HBSS or Torin 1 (1 μM) for 6 h. The cells were loaded with 5 μg/ml propidium iodide (PI; Sigma, P4170) for 5 min and analyzed by flow cytometry on a FACS Canto II.
Quantitative PCR
RAW cells were treated with LPS (100 ng/ml), 3-MA (10 mM) or HBSS for 6 h. Total RNA was extracted from cells using a TRI reagent (Sigma, T9424) according to the manufacturer's protocol. Extracted RNA samples (0.5 µg) were reverse transcribed using Superscript III reverse transcriptase (Invitrogen, 18080044). PCR amplification was performed on a Rotor-Gene 3000 (Corbett Research, Mortlake, NSW, Australia). Primers for murine Mif and Rn18s/18S rRNA were used for quantitative PCR, as previously described.43 Relative quantification of target mRNA expression was calculated and normalized to Rn18s rRNA.
Statistics
Results were expressed as means ± standard error of the mean (SEM). Statistical differences were determined using one-way ANOVA or 2-way ANOVA followed by Tukey's post-hoc test.
Supplementary Material
Abbreviations
- 3-MA
3-methyladenine
- ACTB/β-actin
actin, β
- ATG
autophagy related
- BMM
bone marrow-derived macrophage
- cybb/nox2
cytochrome b-245, β polypeptide
- DC
dendritic cell
- GFP
green fluorescent protein
- HBSS
Hank's balanced salt solution
- HK
heat killed
- iBMM
immortalized bone marrow-derived macrophage
- IFN
interferon
- IL
interleukin
- LC3
microtubule-associated protein 1 light chain 3
- LPS
lipopolysaccharide
- LysMcre
lysozyme-M cre
- 3-MA
3-methyladenine
- MIF
macrophage migration inhibitory factor (glycosylation-inhibiting factor)
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NAC
N-acetyl L-cysteine
- NADPH
nicotinamide adenine dinucleotide phosphate-oxidase
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- PMA
phorbol 12-myristate 13-acetate
- ROS
reactive oxygen species
- TNF
tumor necrosis factor
- TOMM20
translocase of outer mitochondrial membrane 20
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Professor Christopher Sobey, Monash University for providing the cybb−/− mice and Camden Lo at Monash Micro Imaging for his invaluable assistance with confocal microscopy.
Funding
This work was supported by a Project Grant (1068040) from the National Health & Medical Research Council (NHMRC) of Australia and in part by a Rebecca L. Cooper Medical Research Foundation Grant.
References
- [1].Bloom BR, Bennett B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science 1966; 153:80-2; PMID:5938421; http://dx.doi.org/ 10.1126/science.153.3731.80 [DOI] [PubMed] [Google Scholar]
- [2].David JR. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc Natl Acad Sci USA 1966; 56:72-7; PMID:5229858; http://dx.doi.org/ 10.1073/pnas.56.1.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bozza M, Satoskar AR, Lin G, Lu B, Humbles AA, Gerard C, David JR. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med 1999; 189:341-6; PMID:9892616; http://dx.doi.org/ 10.1084/jem.189.2.341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Weiser WY, Temple PA, Witek-Giannotti JS, Remold HG, Clark SC, David JR. Molecular cloning of a cDNA encoding a human macrophage migration inhibitory factor. Proc Natl Acad Sci USA 1989; 86:7522-6; PMID:2552447; http://dx.doi.org/ 10.1073/pnas.86.19.7522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature 1993; 365:756-9; PMID:8413654; http://dx.doi.org/ 10.1038/365756a0 [DOI] [PubMed] [Google Scholar]
- [6].Lan HY. Role of macrophage migration inhibition factor in kidney disease. Nephron Exp Nephrol 2008; 109:e79–83; PMID:18663334; http://dx.doi.org/ 10.1159/000145463 [DOI] [PubMed] [Google Scholar]
- [7].Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med 1994; 179:1895-902; PMID:8195715; http://dx.doi.org/ 10.1084/jem.179.6.1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF). Biochemistry 1994; 33:14144-55; PMID:7947826; http://dx.doi.org/ 10.1021/bi00251a025 [DOI] [PubMed] [Google Scholar]
- [9].Cunha FQ, Moss DW, Leal LM, Moncada S, Liew FY. Induction of macrophage parasiticidal activity by Staphylococcus aureus and exotoxins through the nitric oxide synthesis pathway. Immunology 1993; 78:563-7; PMID:8495974 [PMC free article] [PubMed] [Google Scholar]
- [10].Onodera S, Suzuki K, Matsuno T, Kaneda K, Takagi M, Nishihira J. Macrophage migration inhibitory factor induces phagocytosis of foreign particles by macrophages in autocrine and paracrine fashion. Immunology 1997; 92:131-7; PMID:9370935; http://dx.doi.org/ 10.1046/j.1365-2567.1997.00311.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Roger T, Delaloye J, Chanson AL, Giddey M, Le Roy D, Calandra T. Macrophage migration inhibitory factor deficiency is associated with impaired killing of gram-negative bacteria by macrophages and increased susceptibility to Klebsiella pneumoniae sepsis. J Infect Dis 2013; 207:331-9; PMID:23125447; http://dx.doi.org/ 10.1093/infdis/jis673 [DOI] [PubMed] [Google Scholar]
- [12].Foote A, Briganti EM, Kipen Y, Santos L, Leech M, Morand EF. Macrophage migration inhibitory factor in systemic lupus erythematosus. J Rheumatol 2004; 31:268-73; PMID:14760795 [PubMed] [Google Scholar]
- [13].Morand EF, Leech M, Bernhagen J. MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat Rev Drug Discov 2006; 5:399-410; PMID:16628200; http://dx.doi.org/ 10.1038/nrd2029 [DOI] [PubMed] [Google Scholar]
- [14].Buffen K, Oosting M, Mennens S, Anand PK, Plantinga TS, Sturm P, van de Veerdonk FL, van der Meer JW, Xavier RJ, Kanneganti TD, et al.. Autophagy modulates Borrelia burgdorferi-induced production of interleukin-1beta (IL-1beta). J Biol Chem 2013; 288:8658-66; PMID:23386602; http://dx.doi.org/ 10.1074/jbc.M112.412841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, et al.. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 2011; 286:9587-97; PMID:21228274; http://dx.doi.org/ 10.1074/jbc.M110.202911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, Sasai M, Latz E, Brinkmann MM, Iwasaki A, et al.. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity 2012; 37:986-97; PMID:23219390; http://dx.doi.org/ 10.1016/j.immuni.2012.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al.. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456:264-8; PMID:18849965; http://dx.doi.org/ 10.1038/nature07383 [DOI] [PubMed] [Google Scholar]
- [18].Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci USA 2009; 106:2770-5; PMID:19196953; http://dx.doi.org/ 10.1073/pnas.0807694106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Peral de Castro C, Jones SA, Ni Cheallaigh C, Hearnden CA, Williams L, Winter J, Lavelle EC, Mills KH, Harris J. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J Immunol 2012; 189:4144-53; http://dx.doi.org/ 10.4049/jimmunol.1201946 [DOI] [PubMed] [Google Scholar]
- [20].Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, Delgado-Vargas M, Timmins GS, Bhattacharya D, Yang H, et al.. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci USA 2012; 109:E3168-76; PMID:23093667; http://dx.doi.org/ 10.1073/pnas.1210500109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xu X, Hua Y, Nair S, Bucala R, Ren J. Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy. Hypertension 2014; 63:490-9; PMID:24366076; http://dx.doi.org/ 10.1161/HYPERTENSIONAHA.113.02219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Xu X, Pacheco BD, Leng L, Bucala R, Ren J. Macrophage migration inhibitory factor plays a permissive role in the maintenance of cardiac contractile function under starvation through regulation of autophagy. Cardiovasc Res 2013; 99:412-21; PMID:23674514; http://dx.doi.org/ 10.1093/cvr/cvt116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen HR, Chuang YC, Chao CH, Yeh TM. Macrophage migration inhibitory factor induces vascular leakage via autophagy. Biol Open 2015; 4:244-52; PMID:25617421; http://dx.doi.org/ 10.1242/bio.201410322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chuang YC, Su WH, Lei HY, Lin YS, Liu HS, Chang CP, Yeh TM. Macrophage migration inhibitory factor induces autophagy via reactive oxygen species generation. PloS One 2012; 7:e37613; PMID:22629429; http://dx.doi.org/ 10.1371/journal.pone.0037613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu Y, Zhao L, Ju Y, Li W, Zhang M, Jiao Y, Zhang J, Wang S, Wang Y, Zhao M, et al.. A novel androstenedione derivative induces ROS-mediated autophagy and attenuates drug resistance in osteosarcoma by inhibiting macrophage migration inhibitory factor (MIF). Cell Death Dis 2014; 5:e1361; PMID:25101674; http://dx.doi.org/ 10.1038/cddis.2014.300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wu MY, Fu J, Xu J, O'Malley BW, Wu RC. Steroid receptor coactivator 3 regulates autophagy in breast cancer cells through macrophage migration inhibitory factor. Cell Res 2012; 22:1003-21; PMID:22430150; http://dx.doi.org/ 10.1038/cr.2012.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xu X, Ren J. Macrophage migration inhibitory factor (MIF) knockout preserves cardiac homeostasis through alleviating Akt-mediated myocardial autophagy suppression in high-fat diet-induced obesity. Int J Obes 2015; 39:387-96; PMID:25248618; http://dx.doi.org/ 10.1038/ijo.2014.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al.. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12:222-30; PMID:21151103; http://dx.doi.org/ 10.1038/ni.1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469:221-5; PMID:21124315; http://dx.doi.org/ 10.1038/nature09663 [DOI] [PubMed] [Google Scholar]
- [30].Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J 2011; 30:4701-11; PMID:22068051; http://dx.doi.org/ 10.1038/emboj.2011.398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang M, Kenny S, Ge L, Xu K, Schekman R. Translocation of interleukin-1beta into a vesicle intermediate in autophagy-mediated secretion. Elife 2015; 4:e11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pelegrin P, Barroso-Gutierrez C, Surprenant A. P2´7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J Immunol 2008; 180:7147-57; http://dx.doi.org/ 10.4049/jimmunol.180.11.7147 [DOI] [PubMed] [Google Scholar]
- [33].Merk M, Baugh J, Zierow S, Leng L, Pal U, Lee SJ, Ebert AD, Mizue Y, Trent JO, Mitchell R, et al.. The Golgi-associated protein p115 mediates the secretion of macrophage migration inhibitory factor. J Immunol 2009; 182:6896-906; http://dx.doi.org/ 10.4049/jimmunol.0803710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sohda M, Misumi Y, Yano A, Takami N, Ikehara Y. Phosphorylation of the vesicle docking protein p115 regulates its association with the Golgi membrane. J Biol Chem 1998; 273:5385-8; PMID:9478999; http://dx.doi.org/ 10.1074/jbc.273.9.5385 [DOI] [PubMed] [Google Scholar]
- [35].Flieger O, Engling A, Bucala R, Lue H, Nickel W, Bernhagen J. Regulated secretion of macrophage migration inhibitory factor is mediated by a non-classical pathway involving an ABC transporter. FEBS Lett 2003; 551:78-86; PMID:12965208; http://dx.doi.org/ 10.1016/S0014-5793(03)00900-1 [DOI] [PubMed] [Google Scholar]
- [36].Nagao K, Maeda M, Manucat NB, Ueda K. Cyclosporine A and PSC833 inhibit ABCA1 function via direct binding. Biochim Biophys Acta 2013; 1831:398-406; PMID:23153588; http://dx.doi.org/ 10.1016/j.bbalip.2012.11.002 [DOI] [PubMed] [Google Scholar]
- [37].Arndt U, Wennemuth G, Barth P, Nain M, Al-Abed Y, Meinhardt A, Gemsa D, Bacher M. Release of macrophage migration inhibitory factor and CXCL8/interleukin-8 from lung epithelial cells rendered necrotic by influenza A virus infection. J Virol 2002; 76:9298-306; PMID:12186913; http://dx.doi.org/ 10.1128/JVI.76.18.9298-9306.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shi CS, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem 2008; 283:33175-82; PMID:18772134; http://dx.doi.org/ 10.1074/jbc.M804478200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lang T, Foote A, Lee J, Morand EF, Harris J. MIF: Implications in the patho-aetiology of systemic lupus erythematosus. Front Immunol 2015; 6:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Candido J, Hagemann T. Cancer-related inflammation. J Clin Immunol 2013; 33(Suppl 1):S79-84; PMID:23225204; http://dx.doi.org/ 10.1007/s10875-012-9847-0 [DOI] [PubMed] [Google Scholar]
- [41].Jones SA, Mills KH, Harris J. Autophagy and inflammatory diseases. Immunol Cell Biol 2013; 91:250-8; PMID:23318657; http://dx.doi.org/ 10.1038/icb.2012.82 [DOI] [PubMed] [Google Scholar]
- [42].Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, Deretic V. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 2007; 27:505-17; PMID:17892853; http://dx.doi.org/ 10.1016/j.immuni.2007.07.022 [DOI] [PubMed] [Google Scholar]
- [43].Gu R, Santos LL, Ngo D, Fan H, Singh PP, Fingerle-Rowson G, Bucala R, Xu J, Quinn JM, Morand EF. Macrophage migration inhibitory factor is essential for osteoclastogenic mechanisms in vitro and in vivo mouse model of arthritis. Cytokine 2015; 72:135-45; PMID:25647268; http://dx.doi.org/ 10.1016/j.cyto.2014.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.