

ABSTRACT
Pseudomonas aeruginosa can establish life-long chronic infection in patients with cystic fibrosis by generating genetic loss-of-function mutations, which enhance fitness of the bacterium in the airways. However, the precise role of the pathoadaptive mutations in persistence in chronic airways infection remains largely unknown. Here we demonstrate that pyocyanin, a well-described P. aeruginosa virulence factor that plays an important role in the initial infection, promotes autophagy in bronchial epithelial cells. Disruption of phzM, which is required for pyocyanin biosynthesis, leads to a significant reduction in autophagy in Beas-2B cells and lung tissues. Pyocyanin-induced autophagy is mediated by the EIF2AK4/GCN2-EIF2S1/eIF2α-ATF4 pathway. Interestingly, rats infected with the phzMΔ mutant strain have high mortality rate and numbers of colony-forming units, compared to those infected with wild-type (WT) P. aeruginosa PA14 strain, during chronic P. aeruginosa infection. In addition, the phzMΔ mutant strain induces more extensive alveolar wall thickening than the WT strain in the pulmonary airways of rats. As autophagy plays an essential role in suppressing bacterial burden, our findings provide a detailed understanding of why reduction of pyocyanin production in P. aeruginosa in chronic airways infections has been associated with better host adaptation and worse outcomes in cystic fibrosis.
KEYWORDS: autophagy, EIF2AK4/GCN2, lung, pathoadaptive mutations, Pseudomonas aeruginosa, pyocyanin
Introduction
Pseudomonas aeruginosa is a common opportunistic pathogen found throughout the environment. P. aeruginosa causes infection in patients with chronic obstructive pulmonary disease and cystic fibrosis (CF).1 In CF disease, P. aeruginosa is the most important pathogen associated with mortality. P. aeruginosa initiates infection by secreting a large arsenal of virulence factors, such as proteases, lipases, phospholipases, and pyocyanin (PYO).2 After acute infection, P. aeruginosa can establish life-long chronic infection and persistent colonization in the airways of patients by evading immune system detection.3,4 P. aeruginosa adapts by loss-of-function mutations, i.e., the genetic properties of late P. aeruginosa isolates differ greatly from the properties of the initially acquired strains during the course of chronic infections.3,5,6 Mutations in many virulence genes required for the initiation of acute infection have been detected at the late stage of chronic infection, indicating that reduced virulence of the late strains probably leads to more persistent phenotypes.5,7,8 However, the precise role of the pathoadaptive mutations in the persistence in chronic airways infection remains largely unknown.
Autophagy is an evolutionarily conserved catabolic process that involves the nonspecific bulk degradation of cytoplasmic components.9 From an evolutionary perspective, autophagy plays a prominent role in resistance to bacterial, viral and protozoan infections in metazoan organisms by capturing and degrading the invading microbes.10-12 Although P. aeruginosa is traditionally considered as an extracellular bacterium,13 the bacterium can enter and reside within bronchial epithelial cells and mast cells.14,15 Recent studies have demonstrated that P. aeruginosa induces autophagy in macrophages, mast cells, and bronchial epithelial cells.15-17 Moreover, autophagy contributes to the clearance of intracellular P. aeruginosa in bronchial epithelial and mast cells as well as from the lungs of infected mice in vivo.15,16 P. aeruginosa-induced autophagy in alveolar macrophages is mediated by the classical BECN1/Beclin-1-ATG5-ATG7 autophagy pathway.16 Recently, Jabir et al.17 has revealed that P. aeruginosa infection induces autophagy via the TLR4 (toll-like receptor 4) and its adaptor TICAM1/TRIF (toll-like receptor adaptor molecule 1) in bone-marrow-derived macrophages.
In this report, we identified that PYO, a major component secreted by P. aeruginosa PA14,2,18 induced autophagy in airway epithelial cells. Furthermore, signaling through the EIF2AK4/GCN2 (eukaryotic translation initiation factor 2 α kinase 4)-EIF2S1/eIF2α (eukaryotic translation initiation factor 2 subunit α)-ATF4 (activating transcription factor 4) pathway was required for PYO-mediated autophagy during P. aeruginosa PA14 infection. When comparing the wild-type and pyocyanin-deficient strains of P. aeruginosa, we found that lack of pyocyanin caused a higher mortality rate and induced more extensive alveolar wall thickening in the pulmonary airways of rats.
Results
PYO secreted by P. aeruginosa promotes autophagy in bronchial epithelial cells
Recent studies have revealed that P. aeruginosa infection induces autophagy in bronchial and alveolar epithelial cell lines.15,16 Upon the induction of autophagy, MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 β) is cleaved at the carboxyl terminus and conjugated to phosphatidylethanolamine.19 This conjugated form of LC3B (LC3B-II) becomes associated with phagophore (the precursor to the autophagosome) membranes. The conversion of cytosolic LC3B (LC3B-I) to phagophore-associated LC3B-II is a marker for autophagy. To examine autophagosome formation after P. aeruginosa PA14 infection, Beas-2B human lung bronchial epithelial cells stably expressing GFP-LC3B were used. We observed that the numbers of GFP-LC3B puncta were markedly increased in Beas-2B cells at 8 h after P. aeruginosa PA14 infection (Fig. 1A), indicating that LC3B is activated and localized to autophagosomes. Next, we investigated whether component(s) secreted by P. aeruginosa PA14 could induce autophagy in Beas-2B cells. After P. aeruginosa PA14 cultures were grown to early log phase, the supernatant fractions were collected and extracted with chloroform. The crude extract was isolated by silica gel G column chromatography and Sephadex LH-20 column chromatography. An active candidate that significantly induced autophagy in Beas-2B cells was isolated by activity-guided isolation. Mass spectrometry and nuclear magnetic resonance analysis identified it as PYO (Fig. 1B and C).
Figure 1.

Pyocyanin produced by P. aeruginosa induces formation of autophagosomes. (A) Beas-2B cells stably expressing GFP-LC3B were infected with wild-type (WT) P. aeruginosa PA14, or the phzMΔ mutant, or treated with pyocyanin (PYO, 0.1 or 0.5 mM) for 8 h. The puncta in each cell were counted (right panel). Values are from 100 cells/sample. Data are expressed as mean ± SD of 3 independent experiments. a, P < 0.01 vs. vehicle; b, P < 0.05 vs. WT; c, P < 0.05 vs. phzMΔ. (B and C) A candidate compound was obtained by activity-guided isolation, and further identified as pyocyanin by mass spectra (B), and 13C-NMR (C).
PYO is an important virulence factor in P. aeruginosa.2,18 Two previous studies demonstrate that the concentrations of PYO in the sputum from patients infected with P. aeruginosa are up to 130 and 80 μM, respectively.20,21 As PYO induces apoptosis in human peripheral blood neutrophils,22 we first determined the effect of PYO on cytotoxicity in Beas-2B cells using the 3-(4,5 -dimethylthiazol-2-yl)-5- (3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H- tetrazolium inner salt (MTS) assay. We found that only 0.8 mM of PYO significantly inhibited cell proliferation (Fig. S1). Next, we tested the effect of PYO (0.01 to 0.5 mM) on autophagy in Beas-2B cells. We found that the numbers of GFP-LC3B puncta were markedly increased in Beas-2B cells at 8 h after treatment of PYO at the concentrations of 0.05, 0.1, and 0.5 mM (Fig. 1A and Fig. S2A). Meanwhile, we monitored the levels of the endogenous lipidated LC3B-II form by western blotting. As expected, we observed a dramatic increase in the protein levels of LC3B-II in Beas-2B cells infected with treatment of PYO or P. aeruginosa PA14 (Fig. 2A and Fig. S2B). Preincubation with bafilomycin A1, a lysosomal proton pump inhibitor,23 increased the accumulation of LC3B-II. In addition, we determined the protein levels of SQSTM1/p62. SQSTM1 is incorporated into phagophores and thus becomes sequestered within the completed autophagosomes and degraded in autolysosomes; thus, decreased SQSTM1 levels are positively correlated with autophagy activation.23 We found that treatment of PYO or P. aeruginosa PA14 infection led to a decrease in SQSTM1 protein levels (Fig. 2B and Fig. S2D).
Figure 2.
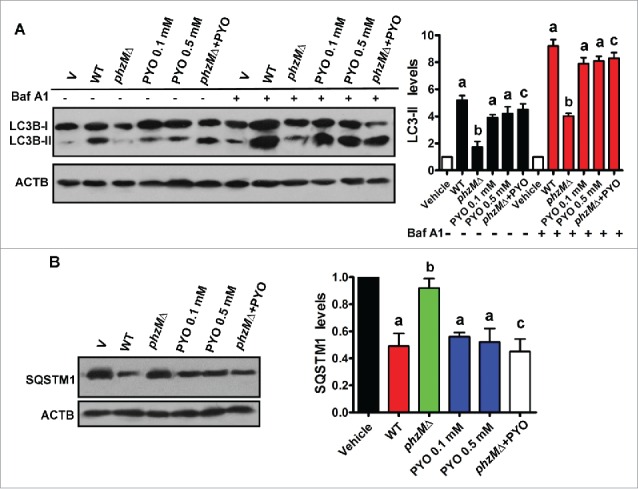
Pyocyanin is involved in autophagy in Beas-2B cells after P. aeruginosa infection. (A) After Beas-2B cells were infected with wild-type (WT) P. aeruginosa PA14, or the phzMΔ mutant in the presence or absence of pyocyanin (PYO, 0.1 mM), or treated with pyocyanin (PYO, 0.1 and 0.5 mM) for 8 h, the protein levels of LC3B were determined by western blotting. The blot is representative of 3 experiments. The right panel shows quantification of the ratio of LC3B-II to ACTB. Data are expressed as mean ± SD of 3 independent experiments. Baf A1, bafilomycin A1 (100 nM). a, P < 0.05 vs. vehicle (V); b, P < 0.05 vs. WT; c, P < 0.05 vs. phzMΔ. (B) Immunoblot analysis of SQSTM1 in Beas-2B cells. The right panel shows quantification of the ratio of SQSTM1 to ACTB. a, P< 0.05 vs. vehicle (V); b, P < 0.05 vs. WT; c, P < 0.05 vs. phzMΔ.
In P. aeruginosa PA14, PYO is synthesized from its precursor, phenazine-1-carboxylic acid, a process that is mediated by phenazine-specific methytransferase, PhzM.18 To further confirm the effect of PYO on autophagy, we deleted phzM in P. aeruginosa PA14 (Fig. S3). The disruption of phzM led to an approximately 23-fold decrease in the production of PYO (Fig. S4). Both the numbers of GFP-LC3B puncta and the protein levels of LC3B-II were significantly decreased in cells infected with the phzMΔ mutant, compared with those infected with the wild-type (WT) PA14 strain (Fig. 1A and Fig. 2A). In contrast, the protein levels of SQSTM1 in cells infected the phzMΔ mutant were higher than those infected with WT PA14 strain (Fig. 2B). However, application of PYO (0.1 mM) markedly increased the numbers of GFP-LC3B puncta and the protein levels of LC3B-II, and reduced the protein levels of SQSTM1 in cells infected with the phzMΔ mutant (Fig. 1A, and Fig. 2A and B).
It has been reported that the autophagy gene atg5 is essential for P. aeruginosa-induced autophagy.15,16 In order to examine the role of PYO in P. aeruginosa-induced autophagy, endogenous ATG5 expression was significantly ablated by siRNA in Beas-2B cells (Fig. S5A). As expected, knockdown of ATG5 markedly reduced the numbers of GFP-LC3B puncta in Beas-2B cells after P. aeruginosa PA14 infection or PYO treatment (0.1 mM) (Fig. S6). Taken together, these results suggest that PYO is a major component that induces autophagy in bronchial epithelial cells during P. aeruginosa PA14 infection.
PYO induces autophagy in lung tissue of rats
To investigate whether PYO induces autophagy in vivo, we first examined the lung tissues of rats, using transmission electron microscopy. Autophagosomes are double-membrane-bound vesicles characteristic of autophagy (Fig. 3A). The number of autophagosomes in the lungs of rats inoculated intratracheally with PYO was markedly higher than that inoculated intratracheally with vehicle (saline) (Fig. 3B). Meanwhile, deletion of phzM led to a significant decrease in the number of autophagosomes in the lungs of rats after P. aeruginosa PA14 infection. Likewise, rats inoculated intratracheally with PYO exhibited higher protein levels of LC3B-II (Fig. 3C), but had lower SQSTM1 protein levels (Fig. 3D), than those inoculated with vehicle. Furthermore, LC3B-II protein levels (Fig. 3E) were much lower, and SQSTM1 protein levels were higher (Fig. 3F) in the lung tissues from rats inoculated with the phzMΔ mutant than those from rats inoculated with the WT strain. Thus, PYO elicits autophagy in rat lung tissue.
Figure 3.
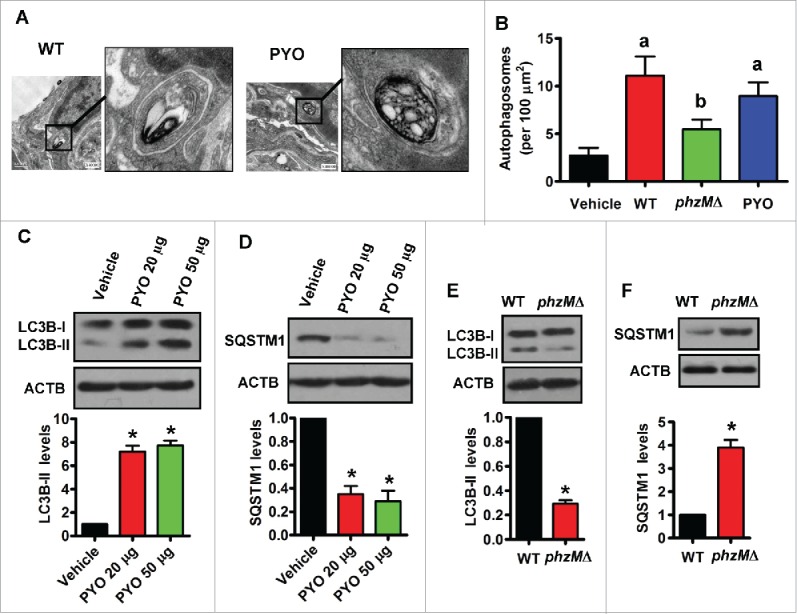
Pyocyanin induces autophagy in lung tissues. (A) After rats were inoculated intratracheally with wild-type (WT) P. aeruginosa or pyocyanin (50 μg), the lung tissues were collected and fixed, and processed for transmission electron microscopy study. Representative images of the lung tissues from rats treated with WT P. aeruginosa (left) and pyocyanin (PYO) (right) are shown. (B) The numbers of autophagosomes were counted. Data are expressed as mean ± SD of 3 independent experiments. a, P < 0.05 vs. vehicle; b, P < 0.05 vs. WT. (C and D) After rats were inoculated intratracheally with pyocyanin, the protein levels of LC3B (C) and SQSTM1 (D) were determined in lung tissues by western blotting. The blot is representative of 3 experiments. The lower panels show quantification of the ratio of LC3B-II or SQSTM1 to ACTB. *, P < 0.05 vs. vehicle. (E and F) After rats were inoculated intratracheally with P. aeruginosa, the protein levels of LC3B (E) and SQSTM1 (F) were determined in lung tissues. The blot is representative of 3 experiments. The lower panels show quantification of the ratio of LC3B-II or SQSTM1 to ACTB. *, P< 0.05 vs. WT. Bars, 0.5 μm.
PYO activates the EIF2AK4-EIF2S1-ATF4 pathway
It is well established that PYO has a profound impact on airway epithelial cells. PYO causes ciliary dyskinesia, induces and releases many proinflammatory cytokines, decreases glutathione contents, and inhibits catalase activity.18 However, PYO-mediated signaling pathways in airway epithelial cells remain largely unknown. It has been reported that PYO elicits activation of the proinflammatory transcription factor NFKB1/NF-κB (nuclear factor of kappa light polypeptide gene enhancer in B-cells 1) in a CF airway epithelial cell line, CF15.24 To clarify whether P. aeruginosa PA14 induces autophagy through NFKB1 signaling, we tested 2 specific inhibitors of NFKB, dimethylfumarate (10 μM)25 and parthenolide (0.5 μM).26 However, we found that both the inhibitors failed to suppress autophagy in Beas-2B cells exposed to PYO (Fig. S7). Furthermore, PYO can activate MAP2K/MEK-MAPK1/ERK2-MAPK3/ERK1 signaling (known as the mitogen-activated protein kinase pathway), via oxidative stress.27 We thus tested 2 specific MAP2K inhibitors U0126 (10 μM) and PD98059 (10 μM), and found that neither of the inhibitors affected PYO-induced autophagy (Fig. S7). These results suggest that the activation of either NFKB or MAPK is unlikely to be involved in the induction of autophagy by PYO.
It has been shown that PYO can oxidize NAD(P)H to produce reactive oxygen species, a process that consumes oxygen.18 As the CF airways are relatively hypoxic,28 we thus tested the effect of PYO on autophagy at low oxygen concentrations of 12% and 15%. We found that PYO (0.1 mM) could induce autophagy in alveolar epithelial cells at the oxygen concentrations of 12% and 15%, similar to that observed in normoxia (21% of oxygen) (Fig. S8). It has been shown that mutations in phzM result in accumulation of 1-hydroxyphenazine,29 which affects respiratory epithelia through generation of reactive oxygen. However, we found that 1-hydroxyphenazine (0.05 and 0.1 mM) did not induce autophagy (Fig. S2). Thus, oxidative stress is not the primary mediator of PYO-induced autophagy.
Recent studies have demonstrated that Shigella infection or IFNG/INFγ (interferon, gamma) treatment activates EIF2AK4, a kinase of EIF2S1, resulting in the activation of autophagy in HeLa cells and human kidney epithelial cells, respectively.30,31 These observations prompted us to investigate whether P. aeruginosa PA14 has an impact on autophagy mediated by the EIF2AK4-EIF2S1 pathway. First, we detected phosphorylation of EIF2AK4 at residue Thr898, indicative of its activation.32 We found that application of PYO led to an increase in phosphorylation of EIF2AK4 in Beas-2B cells (Fig. 4A). This was accompanied by an increase in phosphorylation levels of its substrate, EIF2S1 (Fig. 4B). Consistent with these results, the phosphorylation levels of EIF2AK4 and EIF2S1 in Beas-2B cells infected with the phzMΔ mutant were significantly lower than those infected with the WT PA14 strain (Fig. 4A and B).
Figure 4.
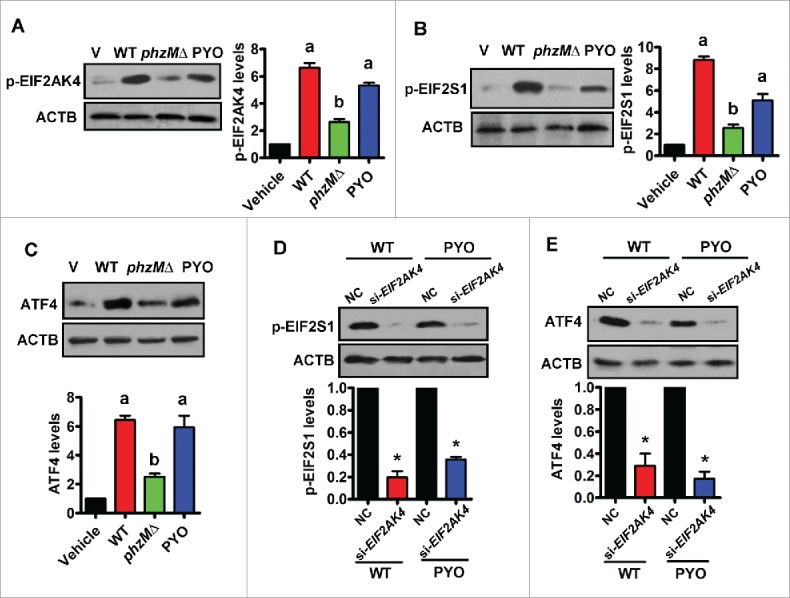
Pyocyanin activates the EIF2AK4-EIF2S1-ATF4 pathway in Beas-2B cells. (A and B) Beas-2B bronchial epithelial cells were infected with wild-type (WT) P. aeruginosa PA14, or the phzMΔ mutant, or treated with pyocyanin (PYO, 0.1 mM) for 8 h. The phosphorylation (p-) levels of EIF2AK4 (A) and EIF2S1 (B) were determined by western blotting. The blot is representative of 3 experiments. The right panels show quantification of phosphorylated EIF2AK4 or EIF2S1. All results are standardized to ACTB. a, P < 0.05 vs. vehicle (V); b, P < 0.05 vs. WT. (C) The protein levels of ATF4 were determined by western blotting. The lower panel shows quantification of ATF4. a, P < 0.05 vs. vehicle (V); b, P < 0.05 vs. WT. (D and E) Knockdown of EIF2AK4 by RNAi inhibited protein levels of phosphorylated EIF2S1 (D) and ATF4 (E) in Beas-2B cells infected with WT PA14 or treated by PYO. The lower panels show quantification of proteins. *, P < 0.05 vs. negative control (NC).
In general, induction of the EIF2AK4-EIF2S1 axis elicits the reduction of general protein synthesis and induces translation of specific mRNAs, such as ATF4.33 We found that exogenous application of PYO augmented the protein levels of ATF4 in Beas-2B cells (Fig. 4C). The protein levels of ATF4 in Beas-2B cells exposed to the phzMΔ mutant were significantly lower than those exposed to the WT PA14 strain. However, addition of PYO restored the protein levels of ATF4 in cells exposed to the phzMΔ mutant (Fig. 4C). As a stress-dependent transcription factor, ATF4 together with its target, DDIT3/CHOP (DNA damage inducible transcript 3), is required for the transcriptional induction of a set of autophagy genes.34 We found that the expression of DDIT3 was significantly upregulated in Beas-2B cells treated with PYO (Fig. S9A and B). Likewise, the expression of DDIT3 in Beas-2B cells exposed to the phzMΔ mutant was significantly lower than those exposed to the WT PA14 strain. Finally, we determined the mRNA of 3 autophagy genes (ATG16L1, ATG7, and ATG10), which are targets of ATF4 and/or DDIT3.34 As show in Figure S9C, exogenous application of PYO markedly upregulated the mRNA levels of ATG16L1, ATG7, and ATG10 in Beas-2B cells. As expected, the expression of these autophagy genes in cells exposed to the phzMΔ mutant was significantly lower than those exposed to the WT PA14 strain.
In order to examine whether EIF2AK4 functions upstream of EIF2S1 and ATF4, endogenous EIF2AK4 expression was significantly ablated by siRNA in Beas-2B cells (Fig. S5B). Knockdown of EIF2AK4 markedly blocked the phosphorylation levels of EIF2S1 and protein levels of ATF4 in Beas-2B cells infected with WT PA14 or treated by PYO (Fig. 4D and E), indicating that EIF2S1 phosphorylation and ATF4 protein expression are EIF2AK4-dependent.
Next, we tested whether PYO activated the EIF2AK4-ATF4 signaling in vivo. We found that rats inoculated intratracheally with PYO exhibited increased phosphorylation levels of EIF2AK4 and EIF2S1 as well as protein levels of ATF4 in the lung tissues, compared with those inoculated with vehicle (Fig. 5A and B). Furthermore, the phosphorylation levels of EIF2AK4 and EIF2S1, and the protein levels of ATF4 in the lung tissues of rats infected with the WT PA14 were significantly higher than those in rats infected with the phzMΔ mutant (Fig. 5C and D). Taken together, these results suggest that PYO mediates activation of the EIF2AK4-EIF2S1-ATF4 pathway during P. aeruginosa PA14 infection.
Figure 5.
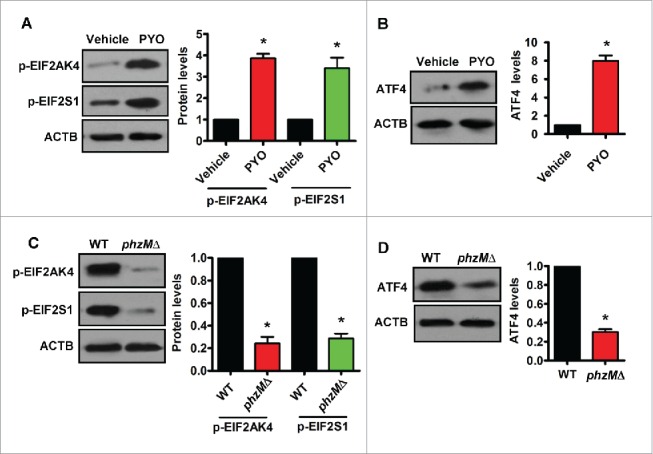
Pyocyanin activates the EIF2AK4-EIF2S1-ATF4 pathway in the lung of rats. (A and B) After rats were inoculated intratracheally with pyocyanin (PYO, 50 μg) for 8 h, the phosphorylation (p-) levels of EIF2AK4 and EIF2S1 (A), and protein levels of ATF4 (B) in the lung were determined by western blotting. The blot is representative of 3 experiments. The right panels show quantification of protein levels. All results are standardized to the levels of ACTB. *, P < 0.05 vs. vehicle. (C and D) After rats inoculated intratracheally with the wild-type (WT) P. aeruginosa PA14 or phzMΔ mutant for 8 h, the phosphorylation levels of EIF2AK4 and EIF2S1 (C), and protein levels of ATF4 (D) in the lung were determined by western blotting. The right panels show quantification of protein levels. *, P < 0.05 vs. WT.
The EIF2AK4-ATF4 pathway is required for PYO-induced autophagy
To address the role of EIF2AK4-ATF4 signaling in autophagy during P. aeruginosa PA14 infection, endogenous EIF2AK4 or ATF4 expression was significantly ablated by siRNA in Beas-2B cells (Fig. S5B and C). We found that knockdown of EIF2AK4 or ATF4 by RNAi significantly suppressed the numbers of GFP-LC3B puncta as well as the protein levels of LC3B-II in Beas-2B cells infected with WT P. aeruginosa PA14 (Fig. 6A and B). Likewise, an increase in the numbers of GFP-LC3B puncta and LC3B-II levels induced by PYO was also reduced in Beas-2B cells subjected to EIF2AK4 or ATF4 RNAi. In addition, knockdown of EIF2AK4 or ATF4 led to the accumulation of SQSTM1 in cells infected with WT PA14 or treated with PYO (Fig. 6C). Thus, the EIF2AK4-ATF4 pathway is involved in PYO-induced autophagy.
Figure 6.
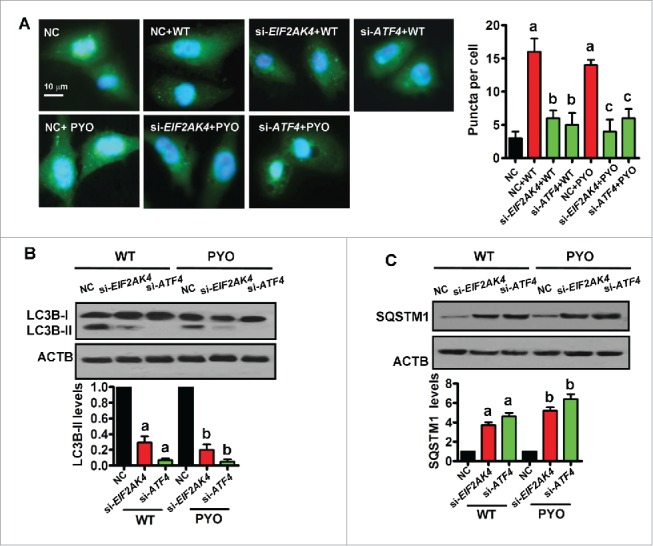
The EIF2AK4-ATF4 pathway is required for in pyocyanin-induced autophagy. (A) Knockdown of EIF2AK4 or ATF4 by RNAi inhibited the numbers of GFP-LC3B puncta in Beas-2B cells infected with WT P. aeruginosa PA14 or treated with pyocyanin (PYO, 0.1 mM). The puncta in each cell were counted (right panel). Data are expressed as mean ± SD of 3 independent experiments. a, P < 0.05 vs. negative control (NC); b, P < 0.05 vs. NC + WT ; c, P < 0.05 vs. NC + PYO. (B and C) Immunoblot analysis of LC3B and SQSTM1 in cells. The blot is representative of 3 experiments. The lower panels show quantification of the ratio of LC3B-II or SQSTM1 to ACTB. a, P < 0.05 vs. NC + WT; b, P < 0.05 vs. NC + PYO.
PYO contributes to inflammation and inhibits bacterial clearance during acute P. aeruginosa infection
PYO induces bronchopneumonia with an influx of neutrophils during the early stage of P. aeruginosa infection.35,36 To investigate the role of PYO in the inflammatory response, rats were inoculated intratracheally with the WT and mutant strains of P. aeruginosa or pyocyanin, and alveolar tissues were examined using hematoxylin and eosin staining. It should be noted that the deletion of phzM does not influence growth of P. aeruginosa PA14 in Luria-Bertani (LB) medium supplemented with 10% fetal calf serum.37 At the chosen endpoint of 16 h, airway inflammatory infiltrates classified by a semiquantitative histology score were measured. Inoculation with PYO alone induced neutrophil influx and led to an increase in histology score (Fig. 7A). Furthermore, histology of the lungs infected with the WT PA14 strain demonstrated more severe acute pneumonia than that infected with the phzMΔ mutant. In addition, we determined the colony-forming unit (CFU) after infection with the bacterium. The viable counts of the WT PA14 strain were 20-fold higher than those of the phzMΔ mutant (Fig. 7B). These results suggest that PYO mediates the pathology of acute P. aeruginosa pneumonia.
Figure 7.
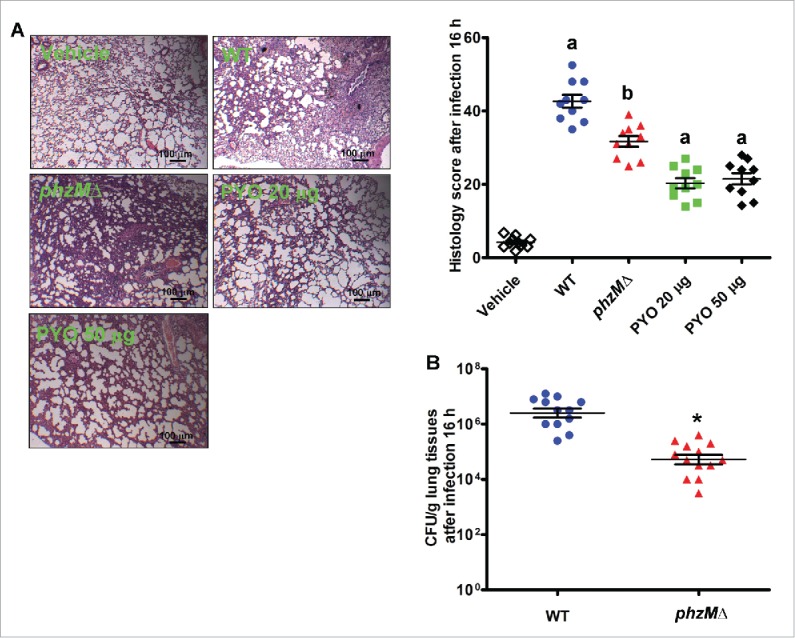
PYO causes severe pneumonia and contributes to high CFU in rats during acute P. aeruginosa infection. (A) After rats were inoculated intratracheally with P. aeruginosa or pyocyanin (20 or 50 μg) for 16 h, alveolar tissues were collected and stained by hematoxylin and eosin (left panel). The airway inflammation, classified by the histology score, was measured (right panel) (n = 10). a, P < 0.05 vs. vehicle; b, P < 0.05 vs. WT. (B) CFUs of the wild-type (WT) and the phzMΔ mutant strains (n = 10). *, P < 0.05 vs. WT.
Loss of PYO aggravates lung damage, development of animal death and inhibits bacterial clearance during chronic P. aeruginosa infection
Next, we evaluated the role of PYO in chronic P. aeruginosa infection. We found that all the animals survived after inoculation of PYO. Interestingly, the survival rate of animals infected with the WT PA14 strain was significantly higher than that of animals infected with the phzMΔ mutant (Fig. 8A). At the chosen endpoint of 15 d, we examined the CFUs and pathologic changes in all the surviving rats. Compared with 16 h of infection, viable counts of bacteria were substantially decreased 15 d after inoculation (Fig. 8B). Interestingly, viable counts of the WT PA14 strain were 62-fold lower than those of the phzMΔ mutant. PYO treatment markedly reduced viable counts of the bacterium in rats inoculated with phzMΔ mutant strain. Unexpectedly, the thickness of the alveolar septum was increased by 43% in rats inoculated with the phzMΔ mutant, compared with those inoculated with the WT P. aeruginosa PA14 (Fig. 8C).
Figure 8.
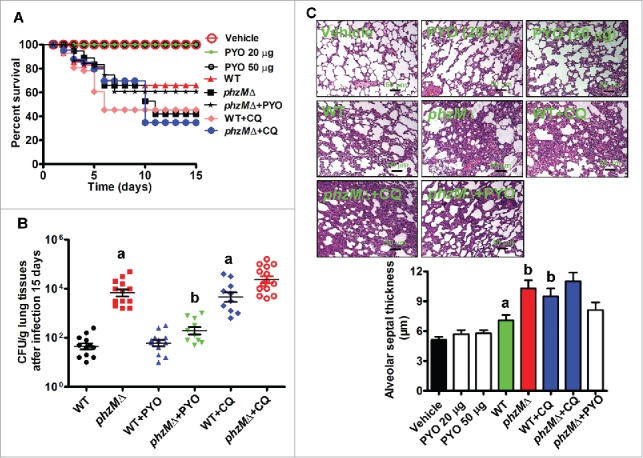
Rats infected with the phzMΔ mutant have a high mortality rate and CFU counts, and exhibit severe lung damage during chronic P. aeruginosa infection. (A) After rats were inoculated intratracheally with pyocyanin alone (PYO, 20 or 50 μg), P. aeruginosa in the presence or absence of PYO (50 μg) or chloroquine (CQ, 60 mg/kg body weight), the survival rates of rats were determined (n = 14). P< 0.05, phzMΔ, or wild type (WT) +CQ vs. WT. (B) After 15 d of inoculation, tissues were collected in the surviving rats. CFUs of the WT and phzMΔ mutant strains were determined. a, P < 0.05 vs. WT; b, P < 0.05 vs. phzMΔ. (C) The alveolar tissues were stained by hematoxylin and eosin (upper panel). The alveolar septal thickness was measured (lower panel). a, P < 0.05 vs. vehicle; b, P < 0.05 vs. WT.
To further assess the impact of autophagy inhibition on chronic lung infection, chloroquine, an autophagy inhibitor,19,23 was used in our experiment. After inoculation with the WT PA14 or phzMΔ mutant strains, rats were injected with chloroquine (60 mg/kg/d) for 15 d or an equivalent volume of saline as control. Chloroquine treatment significantly accelerated mortality in animals infected with the WT PA14 strain (Fig. 8A). Furthermore, chloroquine treatment led to a substantial increase in the viable counts of bacteria and the thickness of the alveolar septum in rats inoculated with the WT PA14 strain (Fig. 8B and C). In contrast, the mortality rate, the thickness of the alveolar septum, and the viable counts of bacteria were not altered by chloroquine in rats inoculated with the phzMΔ mutant. Taken together, these results indicate that loss of PYO results in more severe pathological damage and worse prognosis during chronic P. aeruginosa infection, which is associated with reduced autophagic activity.
Discussion
Our findings provide the first evidence that PYO, an important virulence factor,18 is involved in P. aeruginosa PA14-induced autophagy in bronchial epithelial cells. Our results demonstrate that during P. aeruginosa lung infection, PYO triggers autophagy via the EIF2AK4-EIF2S1 signaling pathway. The molecular mechanisms by which the EIF2AK4-EIF2S1 signaling pathway promotes autophagy involve the upregulation of ATF4.34 Consistent with this idea, our results show that knockdown of ATF4 by RNAi significantly suppresses autophagy induced by P. aeruginosa PA14 infection or PYO treatment. ATF4 in turn upregulates the expression of the transcription factor DDIT3. ATF4 and DDIT3 induce the expression of a set of autophagy genes, which are involved in the formation, elongation and function of the autophagosome.34 However, the signaling pathways that act upstream of EIF2AK4 to regulate PYO-mediated autophagy in bronchial epithelial cells remain unknown. It should be noted that the phzMΔ mutant also induced autophagy to some extent although less effectively than WT does, suggesting that PYO is not the only factor that induces autophagy in lung bronchial epithelial cells. In bone-marrow-derived macrophages, the TLR4-TICAM1 signaling is required for P. aeruginosa-induced autophagy.17 The fact that LPS induces autophagy through the TLR4-TICAM1 signaling indicates that LPS is one of the components that mediates P. aeruginosa-induced autophagy via TLR4 signaling in macrophages.38 However, there is currently no evidence that PYO can activate TLR4-TICAM1 signaling. Therefore, it is not clear whether PYO promotes autophagy via TLR4.
As a membrane-permeable toxin, PYO is involved in the acute infection of P. aeruginosa.18,35 In vitro studies have demonstrated that PYO inhibits DUOX (dual oxidase) activity by competing for intracellular NADPH in airway epithelial cells.39 One of the airway antimicrobial defense systems is the hydrogen peroxide-thiocyanate-lactoperoxidase system, which is essential for airway epithelium defense against pathogens. As DUOXs serve as the major source of hydrogen peroxide needed to the activity of the defense system, inhibition of DUOXs can enhance bacterial survival.39 It has been shown that the viable counts of the PYO-deficient phzM- and phzS (flavin-containing monooxygenase)- mutant strains are much lower than that of WT P. aeruginosa PAO1 strain in an acute mouse pneumonia model of infection.35 Meanwhile, the WT PAO1 strain causes more severe pneumonia than the PYO-deficient strains in mice. Our study confirms these observations in rats infected with the WT and PYO-deficient strains of P. aeruginosa PA14. These results indicate that PYO confers a survival advantage for P. aeruginosa and contributes to the bacterial virulence at the initial infection.
Physiologically relevant concentrations of PYO can promote neutrophil apoptosis, thus inhibiting host defense and favoring bacterial persistence.22 However, it has been shown that transposon inactivation of the pilY1 (type 4 fimbrial biogenesis protein PilY1) gene markedly reduces the secretion of quinolones and PYO, rendering the bacterial cells of the P. aeruginosa strain TBCF10839 more resistant to killing by polymorphonuclear neutrophils, which is an important host defense against P. aeruginosa infection.40 As autophagy can promote clearance of P. aeruginosa by host cells,15,16 PYO appears to be a 2-edged sword for the bacterium during infection of a host. A similar case has been seen in the pathogenic bacterium Streptococcus pneumoniae, where pneumolysin, a virulence factor produced by the pathogen, can form transmembrane pores in cell membranes, thus contributing substantially to the development of invasive pneumococcal disease.41 However, TLR4 in mammalian cells can recognize this toxin to initiate innate immune responses against S. pneumoniae. The phenomenon that the host is capable of perceiving and responding to a bacterial toxin seems to be a common consequence of an evolutionary arms race between pathogens and hosts.
P. aeruginosa can persist continuously in the lungs of patients with CF for years due to adaptive evolution mediated by genetic variation.3-6 Many genes used by P. aeruginosa to invade and injure the host are mutated, resulting in changes or loss of protein functions, during the chronic infection.4,5 Loss-of-function mutations in virulence factors and regulators probably help the bacterium lower innate immune system detection, as many virulence factors are ligands for the immune system. For example, alterations in lipopolysaccharide structure of P. aeruginosa late strains lead to lower leukocyte recruitment in the lungs than the early strains.3 However, the precise role of the pathoadaptative mutations associated with the persistent lifestyle remains largely unknown. Using PCR-based signature-tagged mutagenesis screening, Bianconi et al.6 have revealed that compared to the PAO1293 WT strains, the PA4842 signature-tagged mutagenesis mutant lacking PYO secretion causes a significant increase in mortality rate of mice after 14 d of infection, which is probably due to a higher CFU level. Our data demonstrate that loss of PYO production or treatment with chloroquine, an autophagy inhibitor, increases the CFU counts and the mortality rate in rats during chronic infection with P. aeruginosa. As an essential component of innate immunity, autophagy is involved in eliminating and clearing pathogens. Thus, reduction of PYO production in P. aeruginosa probably is a strategy to minimize provoking host immune system by reducing autophagic responses. However, the increased CFU counts in rats infected with the phzMΔ mutant are not merely due to reduced autophagy. For example, the reduced secretion of PYO in P. aeruginosa confers resistance to killing by polymorphonuclear neutrophils.40 These results suggest that the reduced proinflammatory response in animals infected with the phzMΔ mutants probably results in a less effective clearance of P. aeruginosa by the innate immune system independent of autophagy.
Interestingly, reduced PYO production in chronic infections has been observed in the clinical isolates of P. aeruginosa from CF patients.4,42 The antineutrophil cytoplasmic antibody-positive strains, which have much lower levels of PYO than the antineutrophil cytoplasmic antibody-negative strains,42 significantly correlate with extensive lung damage and poor prognosis in CF patients.43,44 These results contradict the observation by Hunter et al.21 that PYO concentrations in the CF lung negatively correlate with lung function. Animal models of chronic lung infection use the P. aeruginosa strains with a gene mutation that only influences PYO production or secretion. In contrast, the clinical isolates of P. aeruginosa from CF patients typically will undergo genetic adaptations, leading to the accumulation of numerous mutations. The genetic heterogeneity in these isolates can overwhelm the effect of a single factor (e.g., PYO) on lung function and disease severity, thus providing a plausible explanation for these paradoxical observations in CF patients.
In light of the fact that autophagy is involved in host defense by augmenting bacterial clearance, the reduction of PYO production by P. aeruginosa, which is common in the course of chronic pulmonary infection, probably represents an adaptive mechanism of bacterial survival and colonization. Thus, our findings provide a striking example of the pathoadaptative mutations associated with persistent infection.
Materials and methods
Reagents and antibodies
Bafilomycin A1 (Santa Cruz Biotechnology, sc-201550) and 1-hydroxyphenazine (Tokyo Chemical Industry, H0289) were dissolved in DMSO. For western blotting, the following primary antibodies were used: anti-phospho-EIF2S1 (rabbit polyclonal antibody, 1:1000 dilution; Cell Signaling Technology, 3597s), anti-phospho-EIF2AK4 (mouse monoclonal antibody, 1:1000 dilution; Cell Signaling Technology, 3301), anti-ATF4 (rabbit polyclonal antibody, 1: 1000 dilution; Santa Cruz Biotechnology, sc-200), anti-LC3B (rabbit polyclonal antibody, 1:3000 dilution; Sigma-Aldrich, L7543), anti-SQSTM1 (rabbit polyclonal antibody, 1:3000; Santa Cruz Biotechnology, sc-25575), anti-DDIT3 (mouse monoclonal antibody, 1:1000; Santa Cruz Biotechnology, sc-7351), and anti-ACTB antibodies (rabbit polyclonal antibody, 1:3000 dilution; Sigma-Aldrich, A2066).
Animals
SD rats were obtained from the Animal Center, Kunming Medical University, China, and used between the ages of 3 to 4 mo. Animals were kept under a constant 12-h light-dark cycle and were allowed to eat and drink ad libitum. The investigation conforms to the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health. The protocol of the experiments was approved by the Animal Care and Use Committee of Yunnan University.
Cell culture
The human lung bronchial epithelial line Beas-2B cells (a gift from Dr. YG Huang, the Third Affiliated Hospital, Kunming Medical University, China) were grown in RPMI-1640 medium (Invitrogen, 11875–093) containing L-glutamine, glucose, NaHCO3, 10% fetal bovine serum (Invitrogen, 10099–141) and 1% penicillin-streptomycin and maintained in a humidified, 5% CO2:95% air incubator at 37°C. To test the effects of PYO on autophagy at low oxygen concentrations, Beas-2B cells were exposed to 10%, 15%, and 21% (normoxia as a control) supplied with 5% CO2 for 8 h.
Transfection
The vector expressing GFP-LC3B (a gift from Dr. Yoshinori Ohsumi, School of Life Science, The Graduate University for Advanced Studies, Japan) was transfected into Beas-2B cells using Lipofectamine 2000 reagent in Opti-MEM medium (Invitrogen, 11668–019 and 31985–070, respectively) according to the manufacturer's specifications. In control experiments, the expression vectors were replaced with empty vectors. Stable Beas-2B cells that express GFP-LC3B were produced by positive colony selection using G418 (Sigma-Aldrich, A1720) at a concentration of 500 μg/ml.
GFP-LC3B assay
After Beas-2B cells that express GFP-LC3B were grown to 70% to 80% confluence on poly-L-lysine coated glass coverslips, cells were exposed to 50 μl of P. aeruginosa suspensions containing 106 CFU/ml (multiplicity of infection of 10:1) or different concentrations of PYO. In some experiments, cells were preincubated for 1 h with 10 μM dimethylfumarate (Sigma-Aldrich, 50744), 0.5 μM parthenolide (Sigma-Aldrich, P0667), 10 μM U0126 (Sigma-Aldrich, U120), and 10 μM PD98059 (Sigma-Aldrich, P215), respectively, before treatment with PYO (0.1 mM). After 8 h of bacterial infection or PYO treatment, supernatant fractions were removed and cells were fixed in 4% paraformaldehyde and examined by a Zeiss Axioskop 2 plus fluorescence microscope (Carl Zeiss, Jena, Germany). The mean number of LC3B puncta per cell was counted, and at least 100 cells per slide were examined.
Isolation and identification of active compound
P. aeruginosa PA14 was grown in 5 L of LB medium (Sigma-Aldrich, L9234) on a rotary shaker at 37°C for 24 h, fermentation supernatant was collected and condensed. The supernatant fraction was extracted with an equal volume of chloroform. The chloroform extract was concentrated to produce a crude extract (8.6 g). Column chromatography was performed on a silica gel G column eluting with chloroform:acetone (50:1 to 1:1, v/v) providing fractions A1-A20. Subsequently, these fractions were tested for their abilities to induce autophagy by counting the number of LC3B puncta in Beas-2B cells expressing GFP-LC3B. The active fraction A5 (1.83 g) was further separated and purified on a Sephadex LH-20 column (600 g) eluting with acetone to yield fractions A5–1-A5–10. Bioassay showed that only the fraction A5–2 could induce autophagy. Then the active fraction A5–2 (923 mg) was further purified on Sephadex LH-20 (200 g) eluting with acetone to obtain a single candidate active compound (326 mg). An NMR experiment was carried out on Bruker Avance III-600 NMR spectrometers with TMS as internal standard (Bruker Corporation, Fällanden, Switzerland). ESI-MS profiles were recorded on a Finnigan LCQ-Advantage mass spectrometer (Thermo Electron Corporation, San Jose, CA). The concentration of pyocyanin is determined based upon the absorbance at 520 nm in acidic solution.45
Cytotoxicity assay
The cytotoxic effect of PYO on cells was measured using the MTS assay (Promega, G5421). Briefly, the 1×105 Beas-2B cells/well were placed into 96-well plates at 37°C overnight. Then PYO were added to the plates at the final concentrations of 0.1 to 0.8 mM. After 12 or 24 h of incubation, 20 μl of MTS solution was added to all the wells and incubated for a further 2 h at 37°C. The absorbance at OD490 nm was measured using a microplate reader (Molecular Devices, Sunnyvale, CA).
Deletion of phzM in P. aeruginosa PA14
The P. aeruginosa PA14 phzM knockout vector was constructed by using the gene replacement vector pEX18tc vector. Briefly, A 559-bp fragment upstream of phzM and a 511-bp fragment downstream of phzM were amplified from P. aeruginosa PA14 genomic DNA using primers phzM-KpnI-F (KO1) (5′-GGG GTA CCG AAA TTT CGC GTT ACA TAT G-3′) , phzM-Gmrup-R (KO2) (5′-TCA GAG CGC TTT TGA AGC TAA ATC GCT TTT ATT CTC TCT CGT TAC AC-3′), and phzM-Gmrdown-F (KO3) (5′-AGG AAC TTC AAG ATC CCC AAT TCG GTG ATC GAG CGG ACC ATC T-3′) and phzM-HindIII-R (KO4) (5′-CCC AAG CTT AAC GCG CTC AAC CAA CTG G-3′), respectively. A 1053-bp fragment carrying the open reading frame of the Gentamicin resistance gene (Gmr) was amplified with primers Gm-F (5′-CGA ATT AGC TTC AAA AGC GCT CTG A-3′) and Gm-R (5′-CGA ATT GGG GAT CTT GAA GTT CCT-3′) from plasmid pPS856. Equal amounts of 3 fragments were mixed for 3 PCR cycles without added primers. Primers phzM-KpnI-F and phzM-HindIII-R were added at the end of third cycle, followed by 30 cycles run to harvest fragment phzMup-Gmr-phzMdown. The fragment was cloned into the plasmid pMD19T vector by T/A cloning, the plasmid pMD19T-phzMup-Gmr-phzMdown were digested with KpnI/HindIII and the fragment phzMup-Gmr-phzMdown inserted into pEX18tc vector. The final plasmid pEX18tc-phzM-Gmr (500 ng) was mixed with 100 μl of PA14 cells and transferred to a 2-mm gap width electroporation cuvette. After applying a pulse (settings: 6ms; 2.5 kV on a BTX BCM 399 electroporation system), the cells were transferred to a polystyrene tube and shaken for 1 h at 37°C. The entire mixture was then plated on LB plates containing 100 μg/ml gentamicin (Gm100) and 10% sucrose (Shanghai Sangon Biotechnology, A502792). The plates were incubated at 37°C until colonies appeared. The colonies on the LB + Gm100 + 10% sucrose plates were then transferred to LB + tetracycline (50 μg/ml) (Tc50) and LB + Gm100 plates, respectively. Colonies growing on the LB-Gm, but not on the LB-Tc plates were then identified by colony PCR using primers out-phzM-F (5′-GTA TGC CGG AGA AAC TTT TC-3′) , out-phzM-R (5′-CCA CCG CCG AAC TCT ATC-3′), and in-phzM-F (5′-ATT TGA TAC AAG TTG TTA CCG G-3′), in-phzM-R (5′-GCG ACA GCA GGT AGA TAT CG-3′).
Western blotting
For detection of protein expression in the lung tissues, the samples were homogenized in liquid nitrogen. Then the homogenate was lysed on ice for 30 min in lysis buffer (BioTeke Corporation, PP1901). For detection of protein expression in Beas-2B cells, cells were lysed on ice for 30 min in lysis buffer. The lysates (25 μg) of total protein were loaded per well and separated on a 10% SDS polyacrylamide gel. Proteins were then transferred to immobilon-PSQ transfer PVDF membrane (Millipore, IPVH00010), and probed with the primary antibodies. The secondary antibody was a peroxidase-coupled anti-rabbit or mouse IgG (1:5000 dilution; Abmart Incorporation, M21002 and M21001, respectively). Proteins were detected by enhanced chemoluminescence (ECL Prime; GE Healthcare, RPN2232). The membranes were exposed to Kodak X-OMAT film (Kodak, Xiamen, China), and the film was developed.
Quantitative real-time PCR
Total RNA was isolated from cells and lung tissues with TRIzol Reagent (Invitrogen, 15596–018). Random-primed cDNAs were generated by reverse transcription of the total RNA samples with SuperScript II (Takara Biomedical Technology, RR047A). A real time-PCR analysis was conducted using the Roche LightCycler 480®System (Roche Applied Science, Penzberg, Germany) using SYBR®Premix-Ex TagTM GC (Takara Biomedical Technology, RR071A). The primers used for PCR were as follows: EIF2AK4, 5′-AAT GCC CAC CTA CCT ATC C-3′ (F) 5′-GCT TGT TAT GCT CGC TGA-3′ (R); ATF4, 5′-CCA ACA ACA GCA AGG AGG AT-3′ (F), 5′-GTG TCA TCC AAC GTG GTC AG-3′ (R); DDIT3, 5′- AAC GGC TCA AGC AGG AAA TCG-3′ (F); 5′- TCT GGG AAA GGT GGG TAG TGT G −3′(R); ATG16L1, 5′-AGA AGA AGC ACA TGG GCT CC-3′ (F), 5′-CAG GGA GGG GTC TGT AGT TC-3′ (R); ATG7, 5′-CAC TGT GAG TCG TCC AGG AC-3′ (F), 5′-CGC TCA TGT CCC AGA TCT CA-3′ (R); ATG10, 5′-CCA AGA GTT TAC CTG GCC AG-3′ (F), 5′-CCT GGG TTA AAG CCA ACC TC-3′ (R); ACTB, 5′-TCC CTG GAG AAG AGC TAC GA-3′ (F), 5′-AGC ACT GTG TTG GCG TAC AG-3′ (R).
RNA interference
All chemically synthesized siRNAs were obtained from GenePharma Corporation. To knock down the expression of EIF2AK4 or ATF4 by RNA interference, Beas-2B cells were transfected at 50% confluence with 100 nM of EIF2AK4- or ATF4-specific siRNAs in Opti-MEM medium using Lipofectamine 2000 transfection agent. Gene silencing efficiency was determined by quantitative real-time PCR 72 h post-transfection. The following siRNAs were used (sequence of the sense strand): EIF2AK4, 5′-GGG AAA UGU AUU GGC AGU GUU-3′ (F), 5′- CAC UGC CAA UAC AUU UCC CUU-3′(R); ATF4, 5′-UCA UCU AAG AGA CCU AGG CTT −3′(F), 5′-GCC UAG GUC UCU UAG AUG ATT-3′ (R); ATG5, 5′-CCA UCA AUC GGA AAC UCA UTT-3′ (F), 5′-AUG AGU UUC CGA UUG AUG GTT-3′ and negative control 5′-UUC UCC GAA CGU GUC ACG UTT-3′ (F) 5′-ACG UGA CAC GUU CGG AGA ATT-3′ (R).
Transmission electron microscopy
The lung tissues were fixed in 2.5% glutaraldehyde, postfixed in 1% osmium tetroxide, dehydrated with ethanol, and embedded in epoxy resin for thin sectioning, sliced on grid, followed by standard staining in uranium and lead salts. Thin sections were observed in a JEM-1011 transmission electron microscope (Japan Electron Optics Laboratory, Tokyo, Japan). Autophagosomes were identified based on the appearance of their characteristic double membrane, and heterogeneous contents.
Pathohistology for acute lung infection
For the vial count and inflammation assays during acute infection, animals were inoculated by 20 or 50 μg of PYO, or 1×106 CFU of P. aeruginosa PA14 strains for 16 h. The right lower lobes were immediately removed and quickly frozen in liquid nitrogen for the determination of protein expression. The remaining part of the lung tissues was fixed in formalin, dehydrated in graded ethanol, embedded in paraffin, and stained with hematoxylin and eosin. Inflammation was determined using a semiquantitative pathohistological score as described previously.46 The score of airway inflammation was classified into the following categories: almost not visible (0 to 5); slight (6 to 20); moderate (21 to 40); severe/profound inflammation (41 to 60). The remaining tissues were homogenized in sterile phosphate-buffered saline (PBS) buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4). For counting bacterial CFU, 10 μL of the homogenate was spread over LB agar plates, and incubated for 24 h at 37°C.
Bacterial growth in agarose beads
P. aeruginosa-laden agarose beads were prepared as previously described.47 Briefly, bacteria were grown to an optical density at 600 nm of 1. A 5-ml aliquot of the bacterial broth was mixed with 50 ml of 2% agarose (Sigma-Aldrich, A9045) in PBS, pH7.4. The agarose-broth mixture was added to heavy mineral oil equilibrated at 50°C to 55°C, rapidly stirred with a magnetic stirring bar for 6 min at room temperature, followed by cooling for 15 min. The agarose beads were washed once with 0.5% deoxycholic acid, sodium salt (Sigma-Aldrich, D6750) in PBS, once with 0.25% deoxycholic acid, sodium salt in PBS, and 4 times with PBS. The bead slurry was allowed to settle so that 75% of the final volume consisted of beads. The number of bacteria in the beads was determined by homogenizing the bacterium-bead suspension and plating 10-fold serial dilutions on LB agar plates. Bead diameter was measured using an inverted light microscope with the software package Image ProPlus (Media Cybernetics, Baltimore, MD).
Chronic lung infection for morphometry and survival assays
Rats were inoculated with P. aeruginosa-laden agarose beads, as previously described.47 Animals were anesthetized using 10% chloral hydrate (0.25 ml/100 g body weight). The ventral cervical region was surgically prepared, a 0.5-cm skin incision was made at the thoracic inlet, and the trachea was visualized by blunt dissection. A 27 G 1-in over-the-needle intravenous catheter (angiocatheter) was used to cannulate the trachea. The average 0.05-ml bolus contained 1.0 × 106 CFUs/rats. Meanwhile, some of the rats were also inoculated with 20 or 50 μg of PYO at 1, 4, 7, and 10 d. At the same time, some of animals received daily doses of 60 mg/kg body weight chloroquine (Sigma-Aldrich, C6628) through intraperitoneal injection for 5 d.
The alveolar septal thickness was determined by linear intercept counting as described previously.48 Briefly, images of the lung were viewed at 400× magnification through a superimposed test grids. The shortest distance across the alveolar septum in any direction was determined at the point where an airspace septum intersected a test line (between alveolar lumen to the left and alveolar tissue to the right). This yielded "orthogonal intercepts" of the alveolar wall, and the arithmetic mean (la) of the intercepts was calculated. The mean thickness (Ta) of alveolar septae was determined by using the following equation: Ta=(π/4) × la.
Statistical analysis
Data from experiments are expressed as mean ± SD. Differences in survival rates were analyzed using the log-rank test. A significant difference was determined by a one-way ANOVA test followed by a Student-Newman-Keuls test. Values of P < 0.05 were considered significant. Data were analyzed using SPSS11.0 software (SPSS Inc.).
Supplementary Material
Abbreviations
- ATG
autophagy related
- ATF4
activating transcription factor 4
- CF
cystic fibrosis
- CFU
colony-forming unit
- DDIT3/CHOP
DNA damage inducible transcript 3
- DUOX
dual oxidase
- EIF2S1/eIF2α
eukaryotic translation initiation factor 2 subunit α
- EIF2AK4/GCN2
eukaryotic translation initiation factor 2 α kinase 4
- GFP
green fluorescent protein
- LB
Luria-Bertani
- MAP1LC3B/LC3B
microtubule associated protein 1 light chain 3 β
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt
- NFKB1/NF-κB
nuclear factor of kappa light polypeptide gene enhancer in B-cells 1
- phzM
phenazine-specific methytransferase
- PYO
pyocyanin
- SQSTM1/p62
sequestrosome 1
- TLR4
toll-like receptor 4
- TICAM1/TRIF
toll-like receptor adaptor molecule 1
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Dr. Z An (University of Texas Health Science Center at Houston) for critical reading of this manuscript. We thank Drs. YG Huang and Y Ohsumi for Beas-2B cells and GFP-LC3B vector. We are grateful to A/Prof. J-L Song (Kunming Medical University, China) for her technical assistance with transmission electron microscopy.
Funding
This work was supported in part by a grant from the National Natural Science Foundation of China (81471916) and a grant from Yunnan Department of Science and Technology (2013HB087).
References
- [1].Cohen TS, Prince A. Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat Med 2012; 18:509-19; PMID:22481418; http://dx.doi.org/ 10.1038/nm.2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sadikot RT, Blackwell TS, Christman JW, Prince AS. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med 2005; 171:1209-23; PMID:15695491; http://dx.doi.org/ 10.1164/rccm.200408-1044SO [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cigana C, Curcuru L, Leone MR, Ierano T, Lore NI, Bianconi I, Silipo A, Cozzolino F, Lanzetta R, Molinaro A, et al.. Pseudomonas aeruginosa exploits lipid A and muropeptides modification as a strategy to lower innate immunity during cystic fibrosis lung infection. PLoS One 2009; 4:e8439; PMID:20037649; http://dx.doi.org/ 10.1371/journal.pone.0008439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bragonzi A, Paroni M, Nonis A, Cramer N, Montanari S, Rejman J, Di Serio C, Doring G, Tummler B. Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am J Respir Crit Care Med 2009; 180:138-45; PMID:19423715; http://dx.doi.org/ 10.1164/rccm.200812-1943OC [DOI] [PubMed] [Google Scholar]
- [5].Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D'Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, et al.. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 2006; 103:8487-92; PMID:16687478; http://dx.doi.org/ 10.1073/pnas.0602138103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bianconi I, Milani A, Cigana C, Paroni M, Levesque RC, Bertoni G, Bragonzi A. Positive signature-tagged mutagenesis in Pseudomonas aeruginosa: tracking patho-adaptive mutations promoting airways chronic infection. PLoS Pathog 2011; 7:e1001270; PMID:21304889; http://dx.doi.org/ 10.1371/journal.ppat.1001270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ciofu O, Mandsberg LF, Bjarnsholt T, Wassermann T, Hoiby N. Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology 2010; 156:1108-19; PMID:20019078; http://dx.doi.org/ 10.1099/mic.0.033993-0 [DOI] [PubMed] [Google Scholar]
- [8].Hogardt M, Heesemann J. Microevolution of Pseudomonas aeruginosa to a chronic pathogen of the cystic fibrosis lung. Curr Top Microbiol Immunol 2013; 358:91-118; PMID:22311171 [DOI] [PubMed] [Google Scholar]
- [9].Melendez A, Neufeld TP. The cell biology of autophagy in metazoans: a developing story. Development 2008; 135:2347-60; PMID:18567846; http://dx.doi.org/ 10.1242/dev.016105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, et al.. Autophagy defends cells against invading group A Streptococcus. Science 2004; 306:1037-40; PMID:15528445; http://dx.doi.org/ 10.1126/science.1103966 [DOI] [PubMed] [Google Scholar]
- [11].Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119:753-66; PMID:15607973; http://dx.doi.org/ 10.1016/j.cell.2004.11.038 [DOI] [PubMed] [Google Scholar]
- [12].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/ 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cornelis GR. Type III secretion: a bacterial device for close combat with cells of their eukaryotic host. Philos Trans R Soc Lond B Biol Sci 2000; 355:681-93; PMID:10874740; http://dx.doi.org/ 10.1098/rstb.2000.0608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pier GB, Grout M, Zaidi TS. Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aeruginosa from the lung. Proc Natl Acad Sci U S A 1997; 94:12088-93; PMID:9342367; http://dx.doi.org/ 10.1073/pnas.94.22.12088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Junkins RD, Shen A, Rosen K, McCormick C, Lin TJ. Autophagy enhances bacterial clearance during P. aeruginosa lung infection. PLoS One 2013; 8:e72263; PMID:24015228; http://dx.doi.org/ 10.1371/journal.pone.0072263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yuan K, Huang C, Fox J, Laturnus D, Carlson E, Zhang B, Yin Q, Gao H, Wu M. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J Cell Sci 2012; 125:507-15; PMID:22302984; http://dx.doi.org/ 10.1242/jcs.094573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jabir MS, Ritchie ND, Li D, Bayes HK, Tourlomousis P, Puleston D, Lupton A, Hopkins L, Simon AK, Bryant C, et al.. Caspase-1 cleavage of the TLR adaptor TRIF inhibits autophagy and β-interferon production during Pseudomonas aeruginosa infection. Cell Host Microbe 2014; 15:214-27; PMID:24528867; http://dx.doi.org/ 10.1016/j.chom.2014.01.010 [DOI] [PubMed] [Google Scholar]
- [18].Rada B, Leto TL. Pyocyanin effects on respiratory epithelium: relevance in Pseudomonas aeruginosa airway infections. Trends Microbiol 2013; 21:73-81; PMID:23140890; http://dx.doi.org/ 10.1016/j.tim.2012.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151-75; PMID:18188003; http://dx.doi.org/ 10.4161/auto.5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wilson R, Sykes DA, Watson D, Rutman A, Taylor GW, Cole PJ. Measurement of Pseudomonas aeruginosa phenazine pigments in sputum and assessment of their contribution to sputum sol toxicity for respiratory epithelium. Infect Immun 1988; 56:2515-7; PMID:3137173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hunter RC, Klepac-Ceraj V, Lorenzi MM, Grotzinger H, Martin TR, Newman DK. Phenazine content in the cystic fibrosis respiratory tract negatively correlates with lung function and microbial complexity. Am J Respir Cell Mol Biol 2012; 47:738-45; PMID:22865623; http://dx.doi.org/ 10.1165/rcmb.2012-0088OC [DOI] [PubMed] [Google Scholar]
- [22].Usher LR, Lawson RA, Geary I, Taylor CJ, Bingle CD, Taylor GW, Whyte MK. Induction of neutrophil apoptosis by the Pseudomonas aeruginosa exotoxin pyocyanin: a potential mechanism of persistent infection. J Immunol 2002; 168:1861-8; PMID:11823520; http://dx.doi.org/ 10.4049/jimmunol.168.4.1861 [DOI] [PubMed] [Google Scholar]
- [23].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schwarzer C, Fu Z, Fischer H, Machen TE. Redox-independent activation of NF-kappaB by Pseudomonas aeruginosa pyocyanin in a cystic fibrosis airway epithelial cell line. J Biol Chem 2008; 283:27144-53; PMID:18682396; http://dx.doi.org/ 10.1074/jbc.M709693200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Seidel P, Merfort I, Hughes JM, Oliver BG, Tamm M, Roth M. Dimethylfumarate inhibits NF-{kappa}B function at multiple levels to limit airway smooth muscle cell cytokine secretion. Am J Physiol Lung Cell Mol Physiol 2009; 297:L326-39; PMID:19465513; http://dx.doi.org/ 10.1152/ajplung.90624.2008 [DOI] [PubMed] [Google Scholar]
- [26].Nehra R, Riggins RB, Shajahan AN, Zwart A, Crawford AC, Clarke R. BCL2 and CASP8 regulation by NF-kappaB differentially affect mitochondrial function and cell fate in antiestrogen-sensitive and -resistant breast cancer cells. FASEB J 2010; 24:2040-55; PMID:20154269; http://dx.doi.org/ 10.1096/fj.09-138305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rada B, Gardina P, Myers TG, Leto TL. Reactive oxygen species mediate inflammatory cytokine release and EGFR-dependent mucin secretion in airway epithelial cells exposed to Pseudomonas pyocyanin. Mucosal Immunol 2011; 4:158-71; PMID:20962773; http://dx.doi.org/ 10.1038/mi.2010.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J 2004; 23:146-58; PMID:14738247; http://dx.doi.org/ 10.1183/09031936.03.00057003 [DOI] [PubMed] [Google Scholar]
- [29].Parsons JF, Greenhagen BT, Shi K, Calabrese K, Robinson H, Ladner JE. Structural and functional analysis of the pyocyanin biosynthetic protein PhzM from Pseudomonas aeruginosa. Biochemistry 2007; 46:1821-8; PMID:17253782; http://dx.doi.org/ 10.1021/bi6024403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LA, Yang C, Emili A, Philpott DJ, Girardin SE. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 2012; 11:563-75; PMID:22704617; http://dx.doi.org/ 10.1016/j.chom.2012.04.012 [DOI] [PubMed] [Google Scholar]
- [31].Fougeray S, Mami I, Bertho G, Beaune P, Thervet E, Pallet N. Tryptophan depletion and the kinase GCN2 mediate IFN-gamma-induced autophagy. J Immunol 2012; 189:2954-64; PMID:22896630; http://dx.doi.org/ 10.4049/jimmunol.1201214 [DOI] [PubMed] [Google Scholar]
- [32].Visweswaraiah J, Lageix S, Castilho BA, Izotova L, Kinzy TG, Hinnebusch AG, Sattlegger E. Evidence that eukaryotic translation elongation factor 1A (eEF1A) binds the Gcn2 protein C terminus and inhibits Gcn2 activity. J Biol Chem 2011; 286:36568-79; PMID:21849502; http://dx.doi.org/ 10.1074/jbc.M111.248898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bunpo P, Cundiff JK, Reinert RB, Wek RC, Aldrich CJ, Anthony TG. The eIF2 kinase GCN2 is essential for the murine immune system to adapt to amino acid deprivation by asparaginase. J Nutr 2010; 140:2020-7; PMID:20861212; http://dx.doi.org/ 10.3945/jn.110.129197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 2013; 41:7683-99; PMID:23804767; http://dx.doi.org/ 10.1093/nar/gkt563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lau GW, Ran H, Kong F, Hassett DJ, Mavrodi D. Pseudomonas aeruginosa pyocyanin is critical for lung infection in mice. Infect Immun 2004; 72:4275-8; PMID:15213173; http://dx.doi.org/ 10.1128/IAI.72.7.4275-4278.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Look DC, Stoll LL, Romig SA, Humlicek A, Britigan BE, Denning GM. Pyocyanin and its precursor phenazine-1-carboxylic acid increase IL-8 and intercellular adhesion molecule-1 expression in human airway epithelial cells by oxidant-dependent mechanisms. J Immunol 2005; 175:4017-23; PMID:16148150; http://dx.doi.org/ 10.4049/jimmunol.175.6.4017 [DOI] [PubMed] [Google Scholar]
- [37].Allen L, Dockrell DH, Pattery T, Lee DG, Cornelis P, Hellewell PG, Whyte MK. Pyocyanin production by Pseudomonas aeruginosa induces neutrophil apoptosis and impairs neutrophil-mediated host defenses in vivo. J Immunol 2005; 174:3643-9; PMID:15749902; http://dx.doi.org/ 10.4049/jimmunol.174.6.3643 [DOI] [PubMed] [Google Scholar]
- [38].Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007; 27:135-44; PMID:17658277; http://dx.doi.org/ 10.1016/j.immuni.2007.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rada B, Lekstrom K, Damian S, Dupuy C, Leto TL. The Pseudomonas toxin pyocyanin inhibits the dual oxidase-based antimicrobial system as it imposes oxidative stress on airway epithelial cells. J Immunol 2008; 181:4883-93; PMID:18802092; http://dx.doi.org/ 10.4049/jimmunol.181.7.4883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bohn YS, Brandes G, Rakhimova E, Horatzek S, Salunkhe P, Munder A, van Barneveld A, Jordan D, Bredenbruch F, Haussler S, et al.. Multiple roles of Pseudomonas aeruginosa TBCF10839 PilY1 in motility, transport and infection. Mol Microbiol 2009; 71:730-47; PMID:19054330; http://dx.doi.org/ 10.1111/j.1365-2958.2008.06559.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A 2003; 100:1966-71; PMID:12569171; http://dx.doi.org/ 10.1073/pnas.0435928100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Carlsson M, Shukla S, Petersson AC, Segelmark M, Hellmark T. Pseudomonas aeruginosa in cystic fibrosis: pyocyanin negative strains are associated with BPI-ANCA and progressive lung disease. J Cyst Fibros 2011; 10:265-71; PMID:21463973; http://dx.doi.org/ 10.1016/j.jcf.2011.03.004 [DOI] [PubMed] [Google Scholar]
- [43].Dorlochter L, Carlsson M, Olafsdottir EJ, Roksund OD, Rosendahl K, Fluge G. Anti-neutrophil cytoplasmatic antibodies and lung disease in cystic fibrosis. J Cyst Fibros 2004; 3:179-83; PMID:15463905; http://dx.doi.org/ 10.1016/j.jcf.2004.04.005 [DOI] [PubMed] [Google Scholar]
- [44].Carlsson M, Eriksson L, Pressler T, Kornfalt R, Mared L, Meyer P, Wiik A, Wieslander J, Segelmark M. Autoantibody response to BPI predict disease severity and outcome in cystic fibrosis. J Cyst Fibros 2007; 6:228-33; PMID:17166780; http://dx.doi.org/ 10.1016/j.jcf.2006.10.005 [DOI] [PubMed] [Google Scholar]
- [45].Kong KF, Jayawardena SR, Indulkar SD, Del Puerto A, Koh CL, Hoiby N, Mathee K. Pseudomonas aeruginosa AmpR is a global transcriptional factor that regulates expression of AmpC and PoxB β-lactamases, proteases, quorum sensing, and other virulence factors. Antimicrob Agents Chemother 2005; 49:4567-75; PMID:16251297; http://dx.doi.org/ 10.1128/AAC.49.11.4567-4575.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Munder A, Wolbeling F, Kerber-Momot T, Wedekind D, Baumann U, Gulbins E, Tummler B. Acute intratracheal Pseudomonas aeruginosa infection in cystic fibrosis mice is age-independent. Respir Res 2011; 12:148; PMID:22059807; http://dx.doi.org/ 10.1186/1465-9921-12-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].van Heeckeren AM, Schluchter MD. Murine models of chronic Pseudomonas aeruginosa lung infection. Lab Anim 2002; 36:291-312; PMID:12144741; http://dx.doi.org/ 10.1258/002367702320162405 [DOI] [PubMed] [Google Scholar]
- [48].Tanaka R, Al-Jamal R, Ludwig MS. Maturational changes in extracellular matrix and lung tissue mechanics. J Appl Physiol (1985) 2001; 91:2314-21; PMID:11641376 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.