

ABSTRACT
Mitochondrial morphology is dynamically remodeled by fusion and fission in cells, and dysregulation of this process is closely implicated in tumorigenesis. However, the mechanism by which mitochondrial dynamics influence cancer cell survival is considerably less clear, especially in hepatocellular carcinoma (HCC). In this study, we systematically investigated the alteration of mitochondrial dynamics and its functional role in the regulation of autophagy and HCC cell survival. Furthermore, the underlying molecular mechanisms and therapeutic application were explored in depth. Mitochondrial fission was frequently upregulated in HCC tissues mainly due to an elevated expression ratio of DNM1L to MFN1, which significantly contributed to poor prognosis of HCC patients. Increased mitochondrial fission by forced expression of DNM1L or knockdown of MFN1 promoted the survival of HCC cells both in vitro and in vivo mainly by facilitating autophagy and inhibiting mitochondria-dependent apoptosis. We further demonstrated that the survival-promoting role of increased mitochondrial fission was mediated via elevated ROS production and subsequent activation of AKT, which facilitated MDM2-mediated TP53 degradation, and NFKBIA- and IKK-mediated transcriptional activity of NFKB in HCC cells. Also, a crosstalk between TP53 and NFKB pathways was involved in the regulation of mitochondrial fission-mediated cell survival. Moreover, treatment with mitochondrial division inhibitor-1 significantly suppressed tumor growth in an in vivo xenograft nude mice model. Our findings demonstrate that increased mitochondrial fission plays a critical role in regulation of HCC cell survival, which provides a strong evidence for this process as drug target in HCC treatment.
KEYWORDS: apoptosis, autophagy, cell survival, liver cancer, mitochondrial dynamics
Introduction
Mitochondria are highly dynamic, constantly changing their morphology to satisfy the variable need of cells and adapt to the cellular environment. Morphologically, the mitochondrial network exists as mixed structures of long interconnected tubules with short isolated dot-like spheres, which is precisely regulated by 2 opposing processes: fusion and fission.1 Highly conserved dynamin-related GTPases have been identified as the primary regulators of mitochondrial dynamics.1 Notably, DNM1L (dynamin 1-like) and FIS1 (fission, mitochondrial 1) are essential for mitochondrial fission, whereas MFN1 (mitofusin 1) and MFN2 and OPA1 (optic atrophy 1 [autosomal dominant]) are required for mitochondrial fusion. The dynamic nature of mitochondrial network is thought to be important for intermixing of DNA and proteins between mitochondria and for rapid repair of damaged mitochondria.2
Recently, cumulative evidence is beginning to reveal the close links between cancers and unbalanced mitochondrial dynamics. Several studies have reported that the expression of mitochondrial dynamic proteins such as DNM1L, MFN1 and MFN2 is dysregulated in human cancers of lung, bladder and breast.3-5 Moreover, a significant association has been observed between the clinical prognosis of lung cancer patients and abnormalities in proteins that govern mitochondrial dynamics.6 Importantly, several studies have shown that the disruption of the mitochondrial network exhibits a considerable effect on the apoptosis of cancer cells, including lung, bladder and colon cancers.3,4,7 However, to date, the molecular mechanisms by which dysregulated mitochondrial dynamics affect cancer cell survival remain largely to be elucidated and represent a major research challenge.
Autophagy, a lysosome-dependent proteolytic pathway, enables cells to sequester portions of cytosol or damaged and surplus organelles into autophagosomes and degrade them in autolysosomes.8 More and more studies support that autophagy is also a critical adaptive effect for tumor cells to survive different stresses by eliminating damaged mitochondria, controlling reactive oxygen species production, and reducing apoptosis.9,10 Recently, mitochondrial fission has been linked to both mitophagy (mitochondrial autophagy) and global autophagy in mouse embryonic fibroblasts, human fibroblasts and cardiac cells.11-13 However, it is still unclear whether mitochondrial fission induces autophagy, as well as what their combined effects may be on apoptosis in cancer cells.
In most mammalian cells, mitochondria are a major source of reactive oxygen species (ROS), which are generally elevated in cancer cells and functionally act as essential signaling molecules in the regulation of the autophagy and tumor development.14,15 A previous study has shown that the sustained fragmentation of mitochondria in human immortalized fibroblasts is associated with increased ROS production,13 which is also observed in a lung cancer cell model with increased DNM1L induced by cytoplasmic irradiation.16 However, we still do not know precisely how mitochondrial dynamics can be integrated into those ROS-related signaling pathways involved in cell survival.
In the present study, we systematically investigated the alteration of mitochondrial dynamics in HCC cells and their functional roles in the regulation of autophagy and cell survival. More importantly, the underlying molecular mechanisms and therapeutic application were explored in depth. Our study facilitates our understanding of the pathological roles played by mitochondrial dynamics and provides a strong evidence for a novel strategy targeting proteins of the mitochondrial fission machinery, in HCC treatment.
Results
Mitochondrial fission is frequently upregulated in HCC cells and significantly contributes to poor patient prognosis
To investigate the alterations of mitochondrial dynamics in human HCC cells, we examined mitochondrial morphology in paired HCC tissues using transmission electron microscopy (TEM). Our results showed a significantly lower average mitochondrial length (a typical indicator of mitochondrial fission) in HCC tissues than in adjacent nontumor tissues (Fig. 1A). Furthermore, western blot analysis for mitochondrial dymamic mediators DNM1L, FIS1, MFN1, MFN2 and OPA1 (Fig. 1B and S1A) demonstrated that the mitochondrial fission protein DNM1L was remarkably upregulated and the mitochondrial fusion protein MFN1 was downregulated in HCC tissues when compared with nontumor tissues. In addition, a consistent pattern of mRNA expression was also observed by qRT-PCR (Fig. 1C and S1B). Immunohistochemical staining analysis in a large cohort of 128 HCC patients further confirmed the above-mentioned results (Fig. 1D). Finally, we found that HCC patients with high DNM1L expression, low MFN1 expression or high expression ratio of DNM1L to MFN1 had a significantly poorer overall survival than those with low DNM1L expression, high MFN1 expression or low expression ratio of DNM1L to MFN1, respectively (Log rank P = 0.024, 0.017 and 0.007, respectively, Fig. 1E to G).
Figure 1.

Mitochondrial dynamics in HCC tissues and their effects on prognosis of HCC patients. (A) Representative transmission electron microscopy images of mitochondrial network in paired tissues from HCC patients (n=15). Asterisks, arrows and triangles indicate elongated, intermediate (mid) and fragmented mitochondria, respectively. N, nucleus. Scale bar: 2 µm. (B and C) Western blot and qRT–PCR analyses for expression levels of DNM1L, FIS1, MFN1, MFN2 and OPA1 in 39 paired tissues from HCC patients. T, tumor; P, peritumor. The relative expression ratio of tumor to peritumor was log2-transformed. The serial number of patient was rearranged for western blot according to expression level, while qRT-PCR data were displayed according to serial patient ID number. (D) Representative immunohistochemical (IHC) staining images of DNM1L, MFN1 and MFN2 in paired HCC tissues (n = 128). *, P<0.05; **, P<0.01. Scale bar: 50μm. (E to G) Kaplan-Meier curve analysis of overall survival in HCC patients by the expression of DNM1L and MFN1. Death/total and recurrence/total number of patients in each subgroup were presented.
Mitochondrial fission promotes the mitochondrial function and survival of HCC cells both in vitro and in vivo
To explore the effect of mitochondrial dynamics on cell survival, in vitro viability and in vivo growth of HCC cell lines with different mitochondrial networking status was assessed. Considering that TP53/TRP53/p53 (note that the mouse gene nomenclature is TRP53, but we use TP53 to refer to both the human and mouse genes or proteins (TP53) for simplicity) is frequently mutated and plays important role in cell survival, HCC cells with both wild-type TP53 (Bel7402 and SMMC7721) and TP53 point mutations (Huh-7:Y220C and MHCC97L: R249S) were selected for the establishment of mitochondrial fission cell models (Fig. S2A to E). MitoTracker Green staining analysis indicated that mitochondrial elements became significantly elongated and interconnected in both Bel7402 and Huh-7 cells with DNM1L knockdown or MFN1 overexpression when compared with those in control cells (Fig. 2A and S3A). In contrast, the percentage of fragmented mitochondria was remarkably increased in both SMMC7721 and MHCC97L cells with DNM1L overexpression or MFN1 knockdown (Fig. 2B and S3B). To assess whether mitochondrial fission is required for the maintenance of mitochondrial homeostasis, mitochondrial functional parameters were measured in HCC cells with DNM1L knockdown or DNM1L overexpression. As shown in Fig. 2C, our data indicated that DNM1L knockdown significantly induced the depolarization of mitochondrial membrane potential when compared with the control group. In contrast, DNM1L overexpression exhibited an opposite results in HCC cells upon treatment with CCCP (an uncoupler of oxidative phosphorylation). Moreover, oxidation consumption rate was significantly inhibited by DNM1L knockdown while DNM1L overexpression exhibited an opposite effect (Fig. 2D). All these results indicate that mitochondrial fission notably promotes mitochondrial function in HCC cells.
Figure 2.
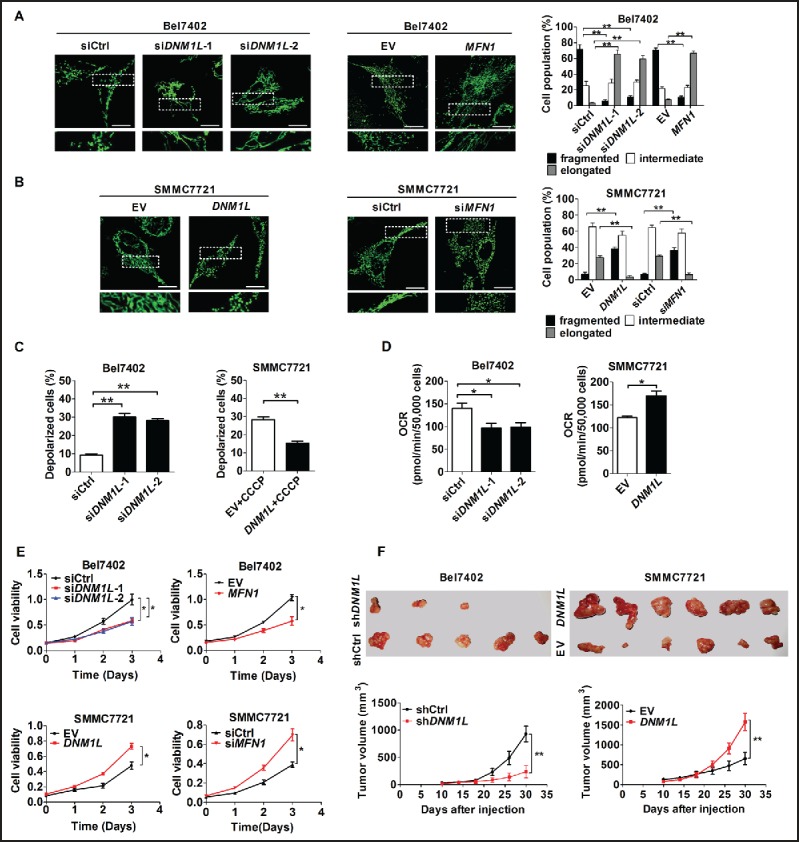
The effects of mitochondrial fission on mitochondrial function and survival of HCC cells in vitro and in vivo. (A and B) Confocal microscopy analysis of mitochondrial network in different HCC cells as indicated. Scale bars: 5 µm. siDNM1L-1 and siDNM1L-2, siRNAs against DNM1L; siMFN1, siRNA against MFN1; siCtrl, control siRNA; DNM1L, expression vector encoding DNM1L; MFN1, expression vector encoding MFN1; EV, empty vector. (C) Depolarization of mitochondrial membrane potential was analyzed by JC-1 staining in HCC cells with treatment as indicated. (D) Oxygen consumption rate (OCR) was measured with a liquid-phase oxygen electrode in HCC cells with treatment as indicated. (E) HCC cells were transiently transfected with siRNA or expression vector as indicated. Cells were reseeded for MTS cell viability assay 24 h after transfection. (F) Tumor growth curves of subcutaneous xenograft tumor model developed from HCC cells which were stably transfected with shRNA (n = 5) or forced-expression vector of DNM1L (n = 6) as indicated (lower panel). Tumor size including tumor length (L) and width (W) was measured using vernier calipers every 4 d from d 10 after transplantation. The tumor volumes were calculated according to the formula (L x W2)/2 and presented as mean ± SEM. Tumors from sacrificed mice were dissected 30 d after transplantation and were also shown in upper panel. shDNM1L, shRNA expression vector against DNM1L; shCtrl, control shRNA.
Furthermore, cell viability was significantly decreased in HCC cells with DNM1L knockdown or MFN1 overexpression, while it was remarkably increased in those with DNM1L overexpression or MFN1 knockdown, no matter what the TP53 mutation status is (Fig. 2E and S3C). We next examined the effect of altered mitochondrial fission on tumor growth in vivo by constructing xenograft nude mice model using HCC cell lines with stable DNM1L knockdown or overexpression (Fig. S3D). As shown in Figure S3E, TEM analysis for Bel7402 and SMMC7721 xenograft tumors demonstrated that DNM1L knockdown significantly inhibited mitochondrial fission and mitochondrial number while DNM1L overexpression exhibited an opposite effect, which is highly consistent with those obtained from cell models and provided further evidence for mitochondrial fission. Moreover xenograft tumors developed from Bel7402 cells with stable DNM1L knockdown exhibited a significant decrease in growth capacity when compared with control tumors (P = 0.005), whereas the growth capacity of xenografts developed from SMMC7721 cells with stable DNM1L overexpression were much higher than controls (P = 0.007, Fig. 2F).
Increased mitochondrial fission inhibits mitochondria-dependent apoptosis
To elucidate the detailed mechanism underlying the effect of mitochondrial fission on cell survival, we first assessed the functional role of DNM1L and MFN1 in apoptosis of HCC cells with different TP53 mutation statuses. The percentages of total (both early and late) apoptotic cells were significantly higher in both Bel7402 and Huh-7 cells with DNM1L knockdown or MFN1 overexpression than those in control cells (Fig. 3A and S4A). Furthermore, the increased mitochondrial fission by DNM1L overexpression or MFN1 knockdown remarkably inhibited CCCP-induced apoptosis in both SMMC7721 and MHCC97L cells (Fig. 3B and S4B). Moreover, CYCS (cytochrome c, somatic) release and the cleavage of CASP9 (caspase 9) and CASP3 were significantly induced by DNM1L knockdown while all of them were remarkably inhibited by DNM1L overexpression upon CCCP treatment (Fig. 3C and D, as well as S4C and D). Additionally, cell apoptosis induced by DNM1L knockdown was significantly attenuated by the treatment with an apoptosis inhibitor (Z-VAD-FMK), suggesting that apoptosis is the main mechanism of cell death mediated by DNM1L inhibition (Fig. 3E and S4E). We further confirmed the protective role of mitochondrial fission against apoptosis in xenograft tumor models. When compared with control, those xenografts developed from Bel7402 cells with DNM1L stable knockdown exhibited a marked increase of positive TUNEL staining (Fig. 3F). In contrast, forced expression of DNM1L significantly reduced the positive TUNEL staining in xenografts developed from SMMC7721 cells (Fig. 3G).
Figure 3.
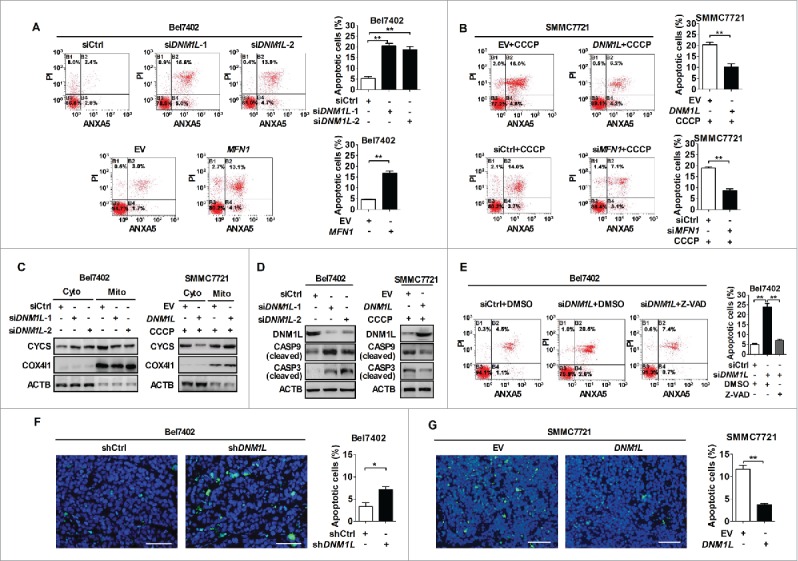
Increased mitochondrial fission inhibits mitochondria-dependent apoptosis. (A and B) Flow cytometry analysis of apoptosis by ANXA5/Annexin V (an indicator of apoptosis) and PI staining in both Bel7402 and SMMC7721 cells 48 h after transfection with siRNA or expression vector as indicated. SMMC7721 cells were also treated with CCCP (150 μM) for 4 h before apoptosis analysis. (C) Western blot analyses for protein levels of CYCS (cytochrome c) in cytoplasm and mitochondria of HCC cells with treatment as indicated. ACTB and COX4I1/COX IV were used as loading controls for cytoplasm and mitochondria, respectively. Cyto, cytoplasm; Mito, mitochondria. (D) Western blot analyses for protein levels of DNM1L, cleaved CASP9 and cleaved CASP3 in HCC cells with treatment as indicated. (E) Apoptosis analysis by flow cytometry in Bel7402 cells 48 h after treatment with siRNA and caspase inhibitor Z-VAD as indicated. Z-VAD (20 μM) treatment for 24 h were applied before cell harvest. Z-VAD, Z-VAD-FMK. (F and G) TUNEL staining in tumor tissues of nude mice xenograft model developed from different HCC cells stably transfected with different expression vector as indicated. Blue, Hochest 33342; Green, TUNEL-positive nucleus. *, P < 0.05; **, P < 0.01. Scale bar: 50 μm.
Increased mitochondrial fission affects apoptosis of HCC cells through coordinately regulating the NFKB and TP53 pathways
To investigate the molecular mechanism by which increased mitochondrial fission regulates apoptosis of HCC cells, we examined the expression levels of several key proteins involved in the regulation of apoptosis. Western blot analysis showed that the antiapoptotic molecules BCL2 and BCL2L1/BCL-XL were significantly decreased in Bel7402 cells with DNM1L knockdown, when compared with those in control cells. In contrast, overexpression of DNM1L exhibited an opposite effect (Fig. 4A). In addition, our data further indicated that phosphorylation of BCL2 was similarly affected by DNM1L (Fig. S5A). Considering the critical roles of the TP53 and NFKB pathways in the regulation of cell apoptosis, we further investigated whether mitochondrial fission regulates both pathways to affect the expression of above-mentioned molecules in HCC cells. Western blot analysis showed that DNM1L knockdown significantly inhibited the transport of RELA (a key subunit of NFKB) from cytoplasm to nucleus and promoted TP53 expression in Bel7402 cells. As expected, DNM1L overexpression clearly exhibited an opposite effect in SMMC7721 cells (Fig. 4A).
Figure 4.
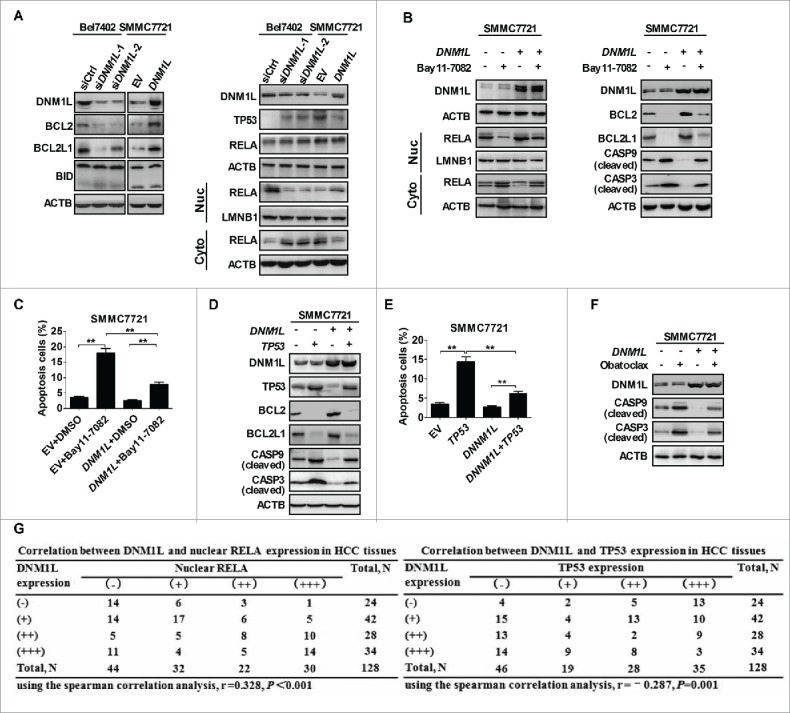
Increased mitochondrial fission affects apoptosis of HCC cells through coordinately regulating the NFKB and TP53 pathways. (A) Western blot analyses for protein levels of DNM1L, apoptosis-related molecules BCL2, BCL2L1, BID, TP53 and RELA in HCC cells with treatment as indicated. LMNB1 (a nuclear envelope marker) and ACTB were used as loading controls in the nuclear and cytoplasmic fractions, respectively. (B) Western blot analyses for protein levels of DNM1L and apoptosis-related molecules in whole-cells or RELA in cytoplasm and nucleus of SMMC7721 cells transiently transfected with the DNM1L expression vector and then followed by treatment with the NFKB inhibitor Bay11-7082 (12.5 μM) for 12 h. (C) Cell apoptosis was respectively evaluated in SMMC7721 cells with treatment as indicated. (D) Western blot analyses for protein levels of DNM1L, TP53 and apoptosis-related molecules in SMMC7721 cells transiently tansfected with DNM1L and/or TP53 expression vectors as indicated. (E) Cell apoptosis was evaluated in SMMC7721 cells tansfected with different expression vectors as indicated. (F) Western blot analyses for protein levels of cleaved CASP9 and cleaved CASP3 in HCC cells treated with obatoclax (5 μM) for 12 h before cell harvest. (G) The correlation was evaluated between the protein expression levels of DNM1L and TP53 or nuclear RELA in 128 HCC tissues based on IHC staining.
We next investigated the functional role of NFKB and TP53 pathways in HCC cell survival regulated by mitochondrial fission. Nuclear transport of active RELA was inhibited by a specific NFKB inhibitor, Bay11-7082, in SMMC7721 cells (Fig. 4B). Our data also showed that Bay11-7082 treatment significantly decreased the expression of BCL2, BCL2L1, and increased the expression of cleaved CASP9 and CASP3 in SMMC7721 cells (Fig. 4B). Moreover, SMMC7721 cells treated with Bay11-7082 exhibited a significantly increased apoptosis. (Fig. 4C and S5B). In addition, overexpression of TP53 exhibited a very similar effect with Bay11-7082 treatment on the expression of apoptosis-related key molecules as well as on DNM1L-mediated cell apoptosis (Fig. 4D, 4E and S5C). In addition, the treatment with obatoclax, an inhibitor of BCL2 family members, significantly increased levels of cleaved CASP3 and cleaved CASP9 when compared with control groups (Fig. 4F), further demonstrating that BCL2 and BCL2L1 play an important role in activation of caspases induced by DNM1L-mediated mitochondrial fission. To provide further supporting evidence, we detected the expression levels of TP53 and nuclear RELA in the same panel of HCC tissue samples (Fig. S5D). Spearman rank correlation analysis indicated a negative significant correlation between immunohistochemistry (IHC) scores of DNM1L and TP53 (r = −0.287, P = 0.001) and a significant positive correlation between DNM1L and nuclear RELA (r = 0.328, P<0.001) (Fig. 4G). Collectively, our data demonstrate that increased mitochondrial fission inhibited apoptosis through coordinately regulating NFKB and TP53 pathways.
Inhibition of apoptosis by increased mitochondrial fission is alternatively regulated by activation of autophagy
A series of studies have demonstrated that autophagy can protect cells by preventing them from apoptosis.17,18 We thus clarified whether autophagy plays a critical role in mitochondrial fission-mediated inhibition of HCC cell apoptosis. Western blot analysis showed that DNM1L knockdown or MFN1 overexpression significantly reduced the expression level of BECN1/Beclin1 and LC3B-II, markers of autophagy, and increased the expression of SQSTM1/p62, a receptor and substrate protein degraded by autophagy, in HCC cells (Fig. 5A). In contrast, DNM1L overexpression or MFN1 knockdown remarkably increased the expression level of BECN1 and production of LC3B-II, and reduced the expression level of SQSTM1 (Fig. 5B). Moreover, GFP-LC3B fluorescence analysis was used to further evaluate the effect of DNM1L on autophagosome formation. Our data indicated that Bel7402 cells with DNM1L knockdown had significantly less GFP-LC3B dots than control cells, whereas SMMC7721 cells with DNM1L overexpression had more accumulation of GFP-LC3B dots (Fig. 5C and 5D). Most of GFP-LC3B dots did not overlap with mitochondria indicated by MitoTracker Red, suggesting a global autophagy, but not specific mitophagy. We further evaluated the mitophagic flux by detecting the expression of mitophagy-specific markers PINK1 and PARK2 (Parkin) in HCC cells. As shown in Fig. 5A and 5B, our results showed that both DNM1L and MFN1 have no notable effect on the expression level of PINK1 and PARK2 as well as mitochondrial translocation of PARK2 in HCC cells, which provided further evidence supporting the absence of mitophagic flux in our cell models. All these results support the idea that mitochondrial fission promotes global autophagy in HCC cells.
Figure 5.

Inhibition of apoptosis by increased mitochondrial fission is alternatively regulated by activation of autophagy. (A and B) Western blot analyses for total protein levels of DNM1L, MFN1, BECN1, LC3B-I/-II, SQSTM1, PINK1 and PARK2 in HCC cells with different treatments as indicated. Protein levels of PARK2 in mitochondria were examined by purifying mitochondria from HCC cells. COX4I1/COX IV were used as loading controls for mitochondria. (C) Representative images of fluorescent LC3B puncta (green) and mitochondria (red) in HCC cells with different treatment as indicated. Scale bar: 5 µm. (D) Numbers of GFP-LC3B puncta per cell were analyzed in HCC cells with treatment as indicated. (E) Western blot analyses for DNM1L, BECN1, LC3B-I/-II, SQSTM1, PINK1 and PARK2 in SMMC7721 cells transiently transfected with DNM1L expression vector and then followed by treatment with the NFKB inhibitor Bay11-7082 (left panel) and for DNM1L, TP53, BECN1, LC3B-I/-II, SQSTM1, PINK1 and PARK2 in those cells transiently tansfected with DNM1L and/or TP53 expression vectors (middle panel) as well as for DNM1L, BECN1, LC3B-I/-II and SQSTM1 in cells with DNM1L knockdown or overexpression, which were transiently tansfected with BECN1 siRNA or BECN1 expression vector (right panel) as indicated. (F) Cell viability was evaluated by MTS assay in HCC cells with different treatment as indicated. (G) Apoptosis analysis by flow cytometry in HCC cells with treatment as indicated.
Mounting evidence has shown that the NFKB and TP53 pathways play central roles in regulation of autophagy. Therefore, it is reasonable to hypothesize that the activation of autophagy by mitochondrial fission may be mainly mediated by both pathways in HCC cells. Western blot analysis showed that both treatment with NFKB inhibitor Bay11-7082 and TP53 overexpression considerably suppressed the DNM1L-induced autophagy in SMMC7721 cells (Fig. 5E). Moreover, cell viability in both Bel7402 with DNM1L knockdown and SMMC7721 with DNM1L overexpression was significantly decreased upon treatment with the autophagy inhibitor 3-MA (Fig. 5F). Additionally, BECN1 overexpression in Bel7402 cells with DNM1L siRNA transfection remarkably affected the expression level of autophagy-related proteins and increased cell viability when compared with the control group. In contrast, BECN1 knockdown in SMMC7721 cells with DNM1L overexpression exhibited an opposite effect (Fig. 5E and F, as well as S6A). Furthermore, the treatment with 3-MA remarkably facilitated the induction of apoptosis in HCC cells with different DNM1L expression status (Fig. 5G and S6B and C). Taken together, our data suggest an alternative regulation mechanism by which mitochondrial fission promotes the activation of autophagy and thus inhibits the apoptosis of HCC cells.
Crosstalk between the TP53 and NFKB pathways is involved in the regulation of cell survival by mitochondrial fission
Comprehensive investigations have demonstrated that crosstalk between the TP53 and NFKB pathways occurs at multiple levels and is considered as a highly context-specific event.19,20 Therefore, we explored whether mitochondrial fission induced the cross-regulation of the NFKB and TP53 pathways and thus promoted HCC cell survival. As shown in Fig. S7A, the inhibition of NFKB activity by Bay11-7082 significantly increased the expression of TP53 and thus reversed the DNM1L-mediated TP53 downregulation. Equally, the forced expression of TP53 significantly inhibited the nuclear translocation of RELA and thus remarkably reduced the DNM1L-mediated activation of NFKB (Fig. S7B). Furthermore, the reciprocal regulation of TP53 and NFKB was further confirmed in Hep3B cells with deletion of the TP53 gene, by exogenously re-expressing TP53. As shown in Fig. S7D, treatment with Bay11-7082 significantly inhibited the nuclear translocation of RELA and thus considerably upregulated the expression level of TP53 in Hep3B cells with or without DNM1L overexpression (Fig. S7C) when the TP53 expression vector was transfected. Similarly, the inhibitory effect of exogenous TP53 expression on DNM1L-mediated activation of NFKB was also clearly observed in Hep3B cells (Fig. S7D). Furthermore, our data showed that simultaneous inhibition of NFKB and exogenous TP53 expression significantly promoted cell apoptosis, and reduced autophagy. Importantly, the effect of DNM1L-mediated mitochondrial fission on autophagy and apoptosis can be remarkably reversed by both treatments in Hep3B cells (Fig. S7E and Fig. S7F).
Increased mitochondrial fission regulates the activity of NFKB and TP53 through the ROS-modulated AKT-IKK-NFKBIA and AKT-MDM2 pathways
Mitochondria are a major source of intracellular reactive oxygen species (ROS), which functions as second messengers to regulate many key signaling pathways, such as NFKB and TP53. Therefore, we hypothesized that ROS-mediated specific pathways may be involved in the regulation of NFKB and TP53 activity by mitochondrial fission in HCC cells. Flow cytometry analysis indicated that DNM1L knockdown or MFN1 overexpression significantly reduced the production of ROS, whereas DNM1L overexpression or MFN1 knockdown significantly elevated the ROS levels (Fig. 6A and S8A). Cell survival in the context of oxidative stress is attributed in part to the activation of the AKT signaling pathway, which is important for MDM2-mediated TP53 degradation and NFKB activition.21,22,23 Thus, we explored whether mitochodrial fission regulates both TP53 and NFKB pathways by ROS-mediated activation of AKT and its target molecules. As expected, DNM1L knockdown or MFN1 overexpression significantly decreased the level of p-AKT and its target molecules p-MDM2 and p-IKK and upregulated the expression level of NFKBIA in Bel7402 cells, whereas DNM1L overexpression or MFN1 knockdown exhibited an opposite effect in SMMC7721 cells (Fig. 6B).
Figure 6.

Increased mitochondrial fission regulates the activity of NFKB and TP53 through the ROS-modulated AKT-IKK-NFKBIA and AKT-MDM2 pathways. (A) Intracellular ROS levels were analyzed by flow cytometry in HCC cells treated as indicated. (B) Western blot analyses for protein levels of AKT, phosphorylated AKT (p-AKT), phosphorylated MDM2 (p-MDM2), phosphorylated IKK (p-IKK) and NFKBIA in HCC cells treated as indicated. (C and D) Western blot analyses for protein levels of DNM1L, AKT, p-AKT, p-MDM2 and TP53 in HCC cells transiently transfected with DNM1L siRNA or expression vector and then followed by treatment with 100 µM H2O2 or 20 mM NAC for 12 h as indicated. (E and F) Western blot analyses for protein levels of DNM1L, AKT, p-AKT, p-IKK and NFKBIA or nuclear and cytosolic RELA in HCC cells with treatment as indicated.
To further demonstrate the critical role of ROS in mitochondrial fission-mediated AKT activation and subsequent regulation of the TP53 and NFKB pathways, we changed the ROS levels by treatment with H2O2 and NAC (a ROS scavenger) (Fig. S8B). Western blot analysis showed that H2O2 treatment significantly increased the protein levels of p-AKT and p-MDM2, and decreased the expression of TP53. In contrast, ROS elimination by NAC exhibited an opposite effect. In addition, we clearly observed that the effect of DNM1L knockdown and overexpression on TP53 expression was significantly reversed by H2O2 and NAC treatment, respectively (Fig. 6C and D). Similarly, the influence of ROS on the DNM1L-mediated activation of NFKB pathway was also observed. As shown in Fig. 6E and F, our data showed that the p-IKK were increased and followed by the degradation of NFKBIA and nuclear translocation of RELA upon exposure to H2O2. In contrast, the opposite effect was obtained upon treatment with NAC. Collectively, all our findings indicate that increased mitochondrial fission coordinately increased the activity of NFKB and degradation of TP53 through the ROS-modulated AKT-IKK-NFKBIA and AKT-MDM2 pathways.
The DNM1L inhibitor Mdivi-1 exhibits a therapeutic effect on HCC in vitro and in vivo
To explore the therapeutic role of Mdivi-1 (a DNM1L selective inhibitor) on HCC, we first investigated the effect of Mdivi-1 on mitochondrial fission and cell survival. Our results showed that treatment with Mdivi-1 significantly increased the percentage of Bel7402 and Huh-7 cells with elongated mitochondria and considerably reduced the viability of both Bel7402 and Huh-7 cells (Fig. 7A and 7B). Flow cytometry analysis demonstrated that Mdivi-1 treatment significantly promoted apoptosis in both Bel7402 and Huh-7 cells (Fig. 7C and Fig. S9). We then tested the in vivo effect of Mdivi-1 treatment on tumor growth. Our results indicated that Mdivi-1 injection significantly inhibited the growth of xenograft tumors developed from Bel7402 cells (Fig. 7D). In addition, we observed that there was a significant increase of positive TUNEL staining cells in xenograft tumors treated with Mdivi-1 (Fig. 7E). Taken together, our data suggest that the inhibition of mitochondrial fission by the DNM1L selective inhibitor Mdivi-1 may become a promising novel therapeutic strategy for HCC.
Figure 7.

The DNM1L inhibitor Mdivi-1 exhibits a therapeutic effect on HCC in vitro and in vivo. (A) Confocal microscopy analysis of the mitochondrial network in HCC cells treated with 50 µM Mdivi-1 or DMSO for 12 h as indicated. (B) Cell viability for HCC cells treated with Mdivi-1 or DMSO was evaluated using the MTS assay. (C) Cell apoptosis was measured in HCC cells treated with Mdivi-1 or DMSO as indicated. (D) Bel7402 tumor-bearing mice were treated with Mdivi-1 (0.75 mg/mice) or DMSO by intratumor injection. Dissected tumors from sacrificed mice are shown in upper panel. Tumor growth curves of the subcutaneous xenograft tumor model are shown in lower panel. (E) TUNEL staining in tumor tissues of nude mice treated as indicated. Scale bar: 50 µm. (F) Schematic depicting the effect of increased mitochondrial fission on the HCC cell survival and underlying mechanism.
Discussion
Mitochondrial morphology is regulated by continuous fusion and fission events that are essential for maintaining a normal mitochondrial function. Deregulated mitochondrial dynamics are implicated in many disorders.24 In the present study, we discovered a clear shift of mitochondrial dynamics from fusion to fission in HCC cells, which can be explained by upregulated DNM1L and/or downregulated MFN1. In contrast, other regulators of mitochondrial dynamics such as FIS1, MFN2 and OPA1 had no significant differences in expression. These findings may be explained by the fact that mitochondrial dynamic regulation is very complicated and diverse regulatory mechanisms exist in different diseases.24 For example, Rehman et al. have reported that DNM1L is upregulated and MFN2 is downregulated in lung cancer cells, while OPA1 is not changed.3 Moreover, the identification of poor clinical outcomes in HCC patients with pro-fission signature highlights the translational relevance of our findings. While a similar increase in mitochondrial fission due to increased DNM1L and decreased MFN2 has been reported in lung cancer cells.3 The upregulated expression of DNM1L and subsequent increased mitochondrial fission has also been reported in breast cancer cells with increased metastatic abilities. These observations support the interpretation that dysregulated mitochondrial dynamics in cancer cells can contribute to tumor progression.
During recent years, the expanding field of mitochondrial network biology has drawn much interest, as it is becoming clear that the network state has a significant effect on cell survival. In the present study, based on both cell models with altered mitochondrial fission by up- or downregulating MFN1 or DNM1L, we found that increased mitochondrial fission significantly promoted HCC cell survival by increasing autophagy and resistance to apoptosis. Consistent with our findings, previous studies have reported that mitochondrial fission induced by DNM1L overexpression or MFN1 knockdown is involved in the apoptosis resistance of both cancer and nonmalignant cells. For example, the apoptotic efficacy of ceramide, which causes a Ca2+-dependent perturbation of mitochondrial structure and function, is drastically reduced in DNM1L-overexpressing HeLa cells.25 Consistently, DNM1L knockdown and/or MFN2 overexpression significantly enhances spontaneous apoptosis in vitro and in vivo in several cancer types, including colon, breast and lung cancers.3,7,26 In addition, DNM1L downregulation or MFN2 overexpression induces mitochondrial elongation, accumulation of damaged mitochondria, and increased apoptosis in cardiomyocytes.11,27 Mitochondrial fission is also believed to be an early event during apoptosis, since collapse of mitochondrial membrane potential during apoptosis induces mitochondrial fission.28 However, it is still unclear whether mitochondrial fission induced by a mitochondrial membrane potential defect plays an active role in the intrinsic pathway of apoptosis or whether it is simply an epiphenomenon.29 Besides apoptosis, mitochondrial fission is also a quick direct consequence of inner membrane depolarization induced by CCCP.28 In our study, mitochondrial fission was induced by DNM1L overexpression or MFN1 knockdown, which maintains normal mitochondrial membrane potential and function and thus is totally different from those induced by mitochondrial membrane depolarization during CCCP-induced apoptosis.
A body of evidence has indicated that mitochondrial fission appears to be a prerequisite for mitophagy both in mammal and yeast cells.12,30 Moreover, mitochondrial fission is important in mediating global autophagy in cardiomyocytes under glucose deprivation,11 which protects heart cells against energy stress. Similar results have also been demonstrated in cancer-associated myofibroblasts and a neuroblastoma cell line as a model for Parkinson disease.13,31 Consistently, our results indicated that abnormally elevated mitochondrial fission induced a robust ROS production to activate the global autophagy, rather than only specific mitophagy, to function as an alternative mechanism underlying the inhibition of apoptosis in HCC cells. Our data showed that both DNM1L and MFN1 have no notable effect on the expression level of PINK1 and PARK2 in HCC cells, which provided further evidence supporting the absence of mitophagic flux in our cell models. However, several studies in mouse embryonic fibroblasts and insulinoma β-cells also indicate that inhibition of mitochondrial fission only results in reduction of mitophagy but not nonspecific global autophagy.12,32 We speculated that, under cellular stresses, such as glucose deprivation or tumorigenesis, mitochondrial fission is more likely to induce a consistent constitutive global autophagy to promoted cell survival by degrading redundant or damaged proteins and organelles.
It has been reported that the binding of BCL2 homologs to the BH3 domain of BECN1 prevents the assembly of an autophagy-competent complex and thus inhibits BECN1-induced autophagy. However, in some conditions, the BECN1–BCL2 homolog interaction can be disrupted by phosphorylation of BCL2, which results in the assembly of an autophagy-competent complex and apoptosis resistance.33 Moreover, previous studies have shown that many kinases such as the IKK complex, MAPK1/ERK2-MAPK3/ERK1, and PRKC can induce phosphorylation of BCL2 and thus potentially regulate the dissociation of the BCL2-BECN1 complex.34-37 In the present study, we demonstrated that increased mitochondrial fission not only induced BCL2 expression to play an antiapoptosis role but also promoted phosphorylation of BCL2 and thus lost its inhibitory function on BECN1-induced autophagy, which is compatible with another finding that mitochondrial fission promotes global autophagy by upregulating BECN1 in HCC cells.
Mitochondria are an important source of ROS within most mammalian cells. In general, moderate levels of ROS may function as signals to promote cell proliferation and survival, whereas an abnormal increase of ROS could induce cell death.38 ROS regulate the level of cell autophagy.39 Yu et al. have demonstrated that mitochondrial fission plays an important role in ROS production of cardiovascular cells.40 Consistently, we also observed a promoting effect of increased mitochondrial fission on ROS production in HCC cells. Moreover, we found that elevated ROS inhibit cell apoptosis by activating AKT pathway and promotes global autophagy through upregulating BECN1. Previously, elevated oxidative status has been found in many types of cancer cells. A body of evidence indicates that ROS play central roles in the key intracellular signal transduction pathways, which contributes to tumorigenesis.14 For example, it has been demonstrated that hydrogen peroxide can reversibly inhibit the tumor suppressor PTEN, thus resulting in the activation of the PI3K-AKT pathway, which has been widely reported in many kinds of cancers, including HCC.14 Our previous study has demonstrated that AKT-mediated phosphorylation of MDM2 promotes the ubiquitination and degradation of TP53 in HCC cells. Therefore, the current study further demonstrated that the AKT-MDM2-TP53 pathway could also be induced by increased mitochondrial fission and subsequent ROS production in HCC cells. In addition, the NFKB pathway, which is typically activated by NFKBIA release from the inactive complex via IKK-mediated NFKBIA phosphorylation, transcriptionally controls a large set of target genes and is frequently activated in many kinds of cancers, including HCC.41 Induction of IKK phosphorylation and subsequent NFKB activation by AKT has been observed in HCC cells both in vivo and in vitro.42,43 Consistently, our study provided first evidence that AKT-mediated activation of NFKB can be induced by mitochondrial fission. We further demonstrated that the crosstalk between NFKB and TP53 induced by increased mitochondrial fission is critical for survival of HCC cells. This crosstalk is also evidenced in tumor cells treated with TNF.44 In addition, studies have documented that mutation of the TP53 gene is a common genetic change in HCC, present in about 30% of cases.45 Moreover, our study also confirmed the similar effect of mitochondrial fission on cell survival in 2 HCC cells (Huh-7 and MHCC97L) with common TP53 point mutations, although only the activation of the NFKB pathway is possibly involved in MHCC97L cells, with mutation at TP53 codon 249 leading to loss of transcription activity.46
In the present study, we established HCC cell models with altered mitochondrial fission by up- or downregulating DNM1L or MFN1 to demonstrate the functional roles of mitochondrial fission in HCC. However, we cannot totally exclude the possibility that DNM1L or MFN1 alone, more than mitochondrial fragmentation, could lead to the tumor-resistance effects. Moreover, we recognize that a biochemical way of inducing fission without affecting any other aspect of cell function still needs direct evidence, which is not currently available. The mitochondrial division inhibitor Mdivi-1 is a derivative of quinazolinone and a selective inhibitor of mitochondrial fission. It blocks the self-assembly of DNM1L and causes the rapid and reversible formation of netlike mitochondria in wild-type cells.47 Due to its safety and protective benefits shown in vitro and in vivo, Mdivi-1 has been supposed to represent a novel class of therapeutics for stroke, myocardial infarction, neurodegenerative diseases and cancers.3,48–50 In the present study, our data clearly showed that Mdivi-1 treatment robustly increased the apoptosis of HCC cells both in vitro and in vivo. In agreement with this result, cell apoptosis induced by Mdivi-1 has been reported in human ovarian, breast cancer cell lines and xenograft models of lung cancer.3,51,52 Therefore, these findings raise the interesting possibility that Mdivi-1 may represent therapeutics for human cancers.
In summary, our findings demonstrate that mitochondrial fission is frequently upregulated in HCC tissues, which contributed to poor prognosis of patients. Increased mitochondrial fission plays a critical role in regulation of HCC cell survival by promoting autophagy and resistance to apoptosis, which was mediated through ROS-mediated AKT activation and subsequent coordinated regulation of the TP53 and NFKB pathways (Fig. 7F). Moreover, treatment with the mitochondrial division inhibitor midiv-1 significantly suppressed in vivo tumor growth, suggesting a potential novel treatment strategy for HCC.
Materials and methods
Cell culture and tissue collection
Human HCC cell lines Bel7402 and SMMC7721 were routinely cultured in RPMI-1640 medium supplemented with 10% FBS. Huh-7, MHCC97L and Hep3B were maintained in DMEM supplemented with 10% FBS. In addition, 128 tissue samples from HCC patients were collected at Xijing Hospital affiliated with the Fourth Military Medical University in Xi'an, China. The eligibility criteria for HCC patient recruitment were set as follows: (1) histologically-confirmed hepatocellular carcinoma (HCC); (2) receiving surgical resection; (3) availability of complete clinical and follow-up data; (4) no preoperative anticancer treatment; (5) no history of other malignancy; and (6) alive at least 1 mo after surgery. The demographic information, clinical and follow-up data of each patient was collected by well-trained staff interviewers or clinical specialists and summarized in Table 1. The latest follow-up date was July 2013 and the median follow-up duration was 22.9 mo (ranging from 2.3 to 45 mo). Overall survival was defined as the time from surgery to HCC-specific death. The study was approved by the Ethics Committee of the Fourth Military Medical University and written informed consent was obtained from all participants.
Table 1.
Distribution of HCC patients' characteristics.
| Variable | All patients, n(%) n = 128 |
|---|---|
| Gender, n(%) | |
| Female | 17 |
| Male | 111 |
| Age, years | |
| <53 | 66 |
| ≥53 | 62 |
| HBsAg, n(%) | |
| Negetive | 12 |
| Positve | 116 |
| Serum AFP, n(%), ng/ml | |
| <200 | 69 |
| ≥200 | 59 |
| Differentiation, n(%) | |
| I+II | 38 |
| III+IV | 90 |
| TNM stage, n(%) | |
| I+II | 103 |
| III+IV | 25 |
| Treatment, n(%) | |
| Surgery | 80 |
| Surgery+TACE | 48 |
| Survival | |
| Dead | 53 |
| Alive | 75 |
Knockdown, forced expression of target genes
For the generation of shRNA expression vectors, a small hairpin RNA (shRNA) containing specific sequences targeting the human DNM1L mRNA sequence (5'-CUACUUCCUGAAAACAAC-3') was cloned into the pSilenc- er™ 3.1-H1 puro vector (Ambion, AM5768). A control shRNA was also cloned into the pSilenc- er™ 3.1-H1 puro vector, which was used as a silencing negative control. For overexpression, the coding sequences of DNM1L, MFN1 and BECN1 were amplified from cDNA derived from SMMC7721 cells using primers listed in Table 2 and cloned into the pcDNA™3.1(+) vector (Invitrogen, V790-20). The expression plasmids for TP53 and the corresponding empty vector were kindly provided by Dr. Lingqiang Zhang (State Key Laboratory of Proteomics, Beijing Proteome Research Center, Beijing Institute of Radiation Medicine, Beijing, China). The expression plasmids for LC3B were kindly provided by Dr. Jian Zhang (Department of Biochemistry and Molecular Biology, The Fourth Military Medical University, Xi'an, China). For transfection, Bel7402, Huh-7, SMMC7721, MHCC97L and Hep3B cells were seeded in 6-well plates to 60% to 80% confluence. Then the vectors were respectively transfected into HCC cells using the Lipofectamine 2000 reagent (Invitrogen, 11668019) according to the manufacturer's instructions. Stable transfectants were generated after selection with G418 (Sigma-Aldrich, A1720) for 3 wk. All siRNAs were synthesized by GenePharma (Shanghai, China). The sequences of siRNA for DNM1L, MFN1 and BECN1 are provided in Table 2. The siRNAs were transfected with Lipofectamine 2000 (Invitrogen, 11668019) reagent according to the manufacturer's protocol.
Table 2.
Sequence of primers and siRNA.
| 1. Primers used in q-PCR analysis | ||
|---|---|---|
| DNM1L | forward primer | GGAGACTCATCTTTGGTGAAGAG |
| reverse primer | AAGGAGCCAGTCAAATTATTGC | |
| FIS1 | forward primer | GTCCAAGAGCACGCAGTTTG |
| reverse primer | ATGCCTTTACGGATGTCATCATT | |
| MFN1 | forward primer | TGGCTAAGAAGGCGATTACTGC |
| reverse primer | TCTCCGAGATAGCACCTCACC | |
| MFN2 | forward primer | CTCTCGATGCAACTCTATCGTC |
| reverse primer | TCCTGTACGTGTCTTCAAGGAA | |
| OPA1 | forward primer | TGTGAGGTCTGCCAGTCTTTA |
| reverse primer | TGTCCTTAATTGGGGTCGTTG | |
| GAPDH | forward primer | GGAGCGAGATCCCTCCAAAAT |
| reverse primer | GGCTGTTGTCATACTTCTCATGG | |
| 2. Primers used in gene cloning | ||
| DNM1L | forward primer | CCGGAATTCTAGCCAGTCTCCACATGAGC |
| reverse primer | CGCGGATCCGGCCCCGTGTTTTCAGAGT | |
| MFN1 | forward primer | ACGAATTCCTTGCCACCATGGCAGAACCT |
| reverse primer | ACCTCGAGTGTTAGGATTCTTCATTGCTTG | |
| BECN1 | forward primer | GGGGATCCGGAAGTTTTCGGCGGCTA |
| reverse primer | GGGGGAATTCGAAGAAAGGGAAAGGAGT | |
| 3. siRNA | ||
| DNM1L siRNA1 | sense | CCUGCUUUAUUUGUGCCUGAGG |
| antisense | CCUCAGGCACAAAUAAAGCAGG | |
| DNM1L siRNA2 | sense | ACUAUUGAAGGAACUGCAAAAUAUATT |
| antisense | UAUAUUUUGCAGUUCCUUCAAUAGUTT | |
| MFN1 siRNA | sense | GGAUCACAUUUUGUUGAAGTT |
| antisense | CUUCAACAAAAUGUGAUCCTT | |
| BECN1 siRNA | sense | CUCAGGAGAGGAGCCAUUU |
| antisense | AAAUGGCUCCUCUCCUGAG | |
| Control siRNA | sense | UUCUCCGAACGUGUCACGUTT |
| antisense | ACGUGACACGUUCGGAGAATT | |
Antibodies and reagents
The primary antibodies used in this study and their working concentration are listed in Table 3. The DNM1L inhibitor Mdivi-1 and NFKB inhibitor Bay11-7082 were purchased from Sigma-Aldrich (M0199 and B5556). The cell-permeating Caspase inhibitor Z-VAD-FMK was purchased from Beyotime Biotechnology (C1202). The BCL2 family inhibitor obatoclax was purchased from Selleck Chemicals (GX15-070).
Table 3.
Primary antibodies used for western blot and immunohistochemistry.
| Antibody | Company (Cat. No.) | Working dilutions |
|---|---|---|
| DNM1L | abcam (ab56788) | WB: 1/800 IHC: 1/250 |
| p-AKT | Cell Signaling Technology (4051) | WB: 1/600 |
| MFN2 | abcam (ab101055) | WB: 1/100 IHC:1/150 |
| OPA1 | abcam (ab90857) | WB: 1/500 |
| FIS1 | abcam (ab156865) | WB: 1/1000 |
| ACTB | Beijing TDY BIOTEC CO., Ltd. (TDY051C) | WB: 1/3000 |
| MFN1 | abcam (ab104585) | WB: 1/500 |
| CYCS | PROTEINTECH GROUP, INC. (10993-1-AP) | WB: 1/750 |
| COX4I1 | ABGENT(AP9153a) | WB: 1/750 |
| CASP9 | PROTEINTECH GROUP, INC. (66169-1-Ig) | WB: 1/1000 |
| CASP3 | PROTEINTECH GROUP, INC. (25546-1-AP) | WB: 1/750 |
| BCL2 | PROTEINTECH GROUP, INC. (12789-1-AP) | WB: 1/1000 |
| BCL2L1 | PROTEINTECH GROUP, INC. (66020-1-Ig) | WB: 1/1000 |
| BID | Cell Signaling Technology (2002) | WB: 1/1000 |
| RELA | PROTEINTECH GROUP, INC. (10745-1-AP) | WB: 1/1000 |
| RELA | abcam(ab7970) | IHC:1/1000 |
| LMNB1 | Beijing TDY BIOTEC CO., Ltd. (TDY049) | WB: 1/2000 |
| TP53 | Cell Signaling Technology (9282) | WB: 1/500 IHC:1/100 |
| BECN1 | PROTEINTECH GROUP, INC. (1306-1-AP) | WB: 1/1000 |
| LC3B-I/-II | abcam (ab51520) | WB: 1/3000 |
| SQSTM1 | PROTEINTECH GROUP, INC. (18420-1-AP) | WB: 1/1000 |
| PINK1 | abcam (ab75487) | WB: 1/200 |
| PARK2 | PROTEINTECH GROUP, INC. (14060-1-AP) | WB: 1/500 |
| p-MDM2 | Affinty (AF3376) | WB: 1/1000 |
| p-IKK | Santa Cruz Biotechnology (C-23470-R) | WB: 1/200 |
| NFKBIA | PROTEINTECH GROUP, INC. (10268-1-AP) | WB: 1/1000 |
| p-BCL2 (S70) | Cell Signaling Technology (2827) | WB: 1/1000 |
Quantitative real-time reverse transcription PCR (qRT-PCR)
Total RNA was extracted from cultured HCC cells or human HCC tissue samples using the TRIzol Reagent (Invitrogen. 15596018). Genomic DNA digestion and reverse transcription were performed using the PrimeScript RT Reagent kit with gDNA Eraser (Takara, RR047A) according to the manufacturer's instructions. For the qRT-PCR analysis, cDNA were amplified using a SYBR Green PCR Kit (Takara, 639676). The cycling parameters were 95°C for 15 s, 55°C for 15 s and 72°C for 15 s for 40 cycles. A melting-curve analysis was then performed to check the specificity of PCR. The Ct value was measured during the exponential amplification phase. The relative expression level (defined as fold change) of the target gene was determined using a 2−△△CT method. GAPDH was used as an internal control. The expression level was normalized to the fold change detected in the corresponding control cells, which was defined as 1.0. For mRNA expression level of target genes in HCC tissues, the fold change between tumor and adjacent nontumor tissues was log2-transformed for further analysis.
Western blot and immunohistochemistry
HCC tissues and cell lines were processed for western blot and IHC as previously described.22 The band intensity on the western blots was quantified using Quantity One software (Bio-Rad, Hercules, CA). The fold change between tumor and adjacent nontumor tissues were log2-transformed for further analysis. For IHC, the expression level of target proteins was independently evaluated by 2 pathologists who were blind to the clinical data, according to the proportion and intensity of positive cells that were determined within 5 microscopic visual fields per slide (200-fold magnification). A proportion score, which represents the estimated proportion of positively stained tumor cells, was assigned as follows: < 10%, 0; 10 to 25%, 1; 26 to 50%, 2; 51 to 75%, 3; and > 75%, 4. An intensity score, which represents the average intensity of the positive tumor cells, was assigned as follows: 0 (no staining), 1 (intensity lower than positive control), 2 (intensity equal to positive control), 3 (intensity higher than positive control), or 4 (significantly strong). The proportion and intensity scores were then multiplied to obtain a total score, which ranged from 0 to 16. A total score of < 2, ≥ 2 to < 7, ≥ 7 to < 12, and ≥ 12 was defined as being negative (−), weak positive (+), moderate positive (++), and strong positive (+++), respectively. PBS buffer: 2 mM KH2PO4, 10 mM Na2HPO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4. PBS-Tween buffer: PBS buffer with 0.1 % v/v Tween 20.
Mitochondrial network imaging by electron microscopy and confocal microscopy
Conventional transmission electron microscopy analysis was performed as described previously.22 In brief, human HCC and xenograft tumor tissues were fixed by glutaraldehyde. Then the specimens were OsO4 postfixed, alcohol dehydrated, and embedded in araldite. Thin sections were stained with uranyl acetate and lead citrate and analyzed with a Tecnai G2 electron microscope (FEI, cHillsboro, Oregon), at 11500 magnifications. The fluorescent dye MitoTracker green FM (Molecular Probes, M7514) and MitoTracker Red FM (Molecular Probes, M22425) were used to monitor mitochondrial morphology in living cells according to the manufacturer's instructions. Then cells were viewed with an Olympus FV 1000 laser-scanning confocal microscope (Olympus Corporation, Tokyo, Japan). For morphometric analysis, the length of mitochondria was measured using the ImageJ software (NIH, Bethesda, MD). In addition, the number of mitochondria was counted and averaged in 20 cells per sample.
Nude mice xenograft model
Six-wk-old BALB/c nude mice with the average body weight of 18 to 22 g were randomly divided into groups. Xenografts were initiated by subcutaneous injection of 107 Bel7402-shCtrl and Bel7402-shDNM1L cells (on the right and left sides, respectively) into the back of nude mice (n = 6) or by SMMC7721-EV and SMMC7721-DNM1L cells (on the right and left sides, respectively) into nude mice (n = 6). Thirty d later, the mice were sacrificed and the tumor nodules were harvested and photograghed. For Mdivi-1 treatment, Mdivi-1 at the dose of 0.75 mg/mice was injected into each tumor twice a week when tumors reached 2 to 3 mm diameter. Equivalent volumes of DMSO (Sigma-Aldrich, D2650) were used as a control therapy. One mo later, the mice were sacrificed. The study was approved by the ethics committee of the Fourth Military Medical University for animal research.
Cell viability and apoptosis assays
Cell viability was determined by the MTS assay (Promega Corporation, G3581) according to the manufacturer's instructions. Briefly, 1 × 103 HCC cells were plated in each well of a 96-well culture plate. After 12 h, cell viability was measured by addition of 20 μl of MTS (0.2%)-PMS (0.092%; phenazine methosulfate, 20:1) solution and incubation for 2 h. The microplates were read in a spectrophotometer at a wavelength of 490 nm. Each sample was analyzed in triplicate. Cell apoptosis was determined with an ANXA5/annexin V-FITC Apoptosis Detection Kit (BestBio, BB-4101-2) following the manufacturers' instructions. Briefly, HCC cells seeded in 6-well plates were collected and resuspended with 500 μL binding buffer at a concentration of 106 cells/mL. After adding 5 μL ANXA5-FITC and 5 μL PI, cells were mixed and incubated at room temperature in the dark for 15 min. The samples were analyzed with a flow cytometer (Beckman, Fullerton, CA). For analysis of apoptosis in xenograft tissues, terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL) assay (Roche Applied Science, 11684795910) was performed according to the manufacturer's protocol. Images of TUNEL and DAPI-stained sections were obtained by a fluorescence microscope (DM5000B; Leica, Heerbrugg, Switzerland). Only TUNEL- and DAPI-positive nuclei that were located within tumor tissues were counted as apoptotic nuclei. The apoptosis index was calculated as the percentage of TUNEL-positive nuclei after at least 500 cells were counted. Results were expressed as the mean number of TUNEL-positive apoptotic HCC cells in each group.
Detection of reactive oxygen species, mitochondrial membrane potential and oxygen consumption rate
Cellular reactive oxygen species (ROS) were detected by the fluorescent probe DCFH-DA (Beyotime Biotechnology, S0033) according to the manufacturer's protocols. Briefly, DCFH-DA was diluted to a final concentration of 10 μM with serum free medium. Then cell suspension was incubated with DCFH-DA at 37°C for 20 min. The fluorescence in each group was assessed by flow cytometry. For the detection of mitochondrial membrane potential, JC-1 dye was purchased from Beyotime Biotechnology (C2006)and stored in DMSO. Cells were adjusted to a density of 0.5 ×106/mL and stained with 2 μg/mL JC-1 for 30 min at 37°C. Cells were then resuspended and analyzed by flow cytometry. We evaluated cellular OCR using a liquid-phase oxygen electrode system and software according to the manufacturer's protocol (Hansatech Instruments, Pentney, Norfolk, UK). The amplified signal from the O2 sensor was recorded at sampling intervals of 0.5 min.
Evaluation of fluorescent LC3B puncta
HCC cells were transiently transduced with pcDNA3.1-GFP-LC3B followed by staining with MitoTracker Red FM (Molecular Probes, M22425). Autophagy was quantified by calculating the average number of GFP-LC3B puncta per cell in 5 high-power fields (HPF, 400×).
Statistical analysis
Experiments were repeated 3 times, where appropriate. Data representing mean ± SEM. SPSS 17.0 software (SPSS, Chicago, IL) was used for all statistical analyses and P < 0.05 was considered significant. Unpaired Student t tests were used for comparisons between 2 groups where appropriate. Correlations between measured variables were tested by Pearson or Spearman rank correlation analyses. For prognosis analysis, variables (the IHC score of DNM1L, MFN1 and the expression ratio of DNM1L/MFN1) were divided into high or low level by the median value for further analysis. The Kaplan-Meier survival curve and log-rank test were used to distinguish subgroup patients who had different overall survival.
Supplementary Material
Abbreviations
- ACTB
actin, β
- BCL2
B-cell CLL/lymphoma 2
- BCL2L1/BCL-XL
BCL2 like 1
- BECN1/Beclin1
Beclin 1, autophagy related
- CASP3
caspase 3
- CASP9
caspase 9
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
- COX4I1
cytochrome c oxidase subunit 4I1
- CYCS
cytochrome c, somatic
- DNM1L/DRP1
dynamin 1-like
- FIS1
fission, mitochondrial 1
- HCC
hepatocellular carcinoma
- IHC
immunohistochemistry
- LMNB1
lamin B1
- MAP1LC3B/LC3B
microtubule associated protein 1 light chain 3 β
- Mdivi-1
mitochondrial division inhibitor 1
- MDM2
MDM2 proto-oncogene
- E3
ubiquitin protein ligase
- MFN1
mitofusin 1
- MFN2
mitofusin 2
- NFKB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NFKBIA
nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, α
- OPA1
optic atrophy 1 (autosomal dominant)
- PARK2
Parkin RBR E3 ubiquitin protein ligase
- PINK1
PTEN- induced putative kinase 1
- RELA
v-rel avian reticuloendotheliosis viral oncogene homolog A
- qRT-PCR
quantitative real-time reverse transcription PCR
- ROS
reactive oxygen species
- SQSTM1/p62
sequestosome 1
- TEM
transmission electron microscopy
- TP53
tumor protein p53
- TUNEL
TdT-mediated dUTP nick-end labeling
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Natural Science Foundation of China (grants 81572410, 81320108021 and 81171966) and National Basic Research Program (grant 2015CB553703).
References
- [1].Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 2010; 11:872-84; PMID:21102612; http://dx.doi.org/ 10.1038/nrm3013 [DOI] [PubMed] [Google Scholar]
- [2].Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell 2006; 125:1241-52; PMID:16814712; http://dx.doi.org/ 10.1016/j.cell.2006.06.010 [DOI] [PubMed] [Google Scholar]
- [3].Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J 2012; 26:2175-86; PMID:22321727; http://dx.doi.org/ 10.1096/fj.11-196543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jin B, Fu G, Pan H, Cheng X, Zhou L, Lv J, Chen G, Zheng S. Anti-tumour efficacy of mitofusin-2 in urinary bladder carcinoma. Medical oncology 2011; 28 Suppl 1:S373-80; PMID:20803103; http://dx.doi.org/ 10.1007/s12032-010-9662-5 [DOI] [PubMed] [Google Scholar]
- [5].Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, Abel PW, Tu Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013; 32:4814-24; PMID:23128392; http://dx.doi.org/ 10.1038/onc.2012.494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chiang YY, Chen SL, Hsiao YT, Huang CH, Lin TY, Chiang IP, Hsu WH, Chow KC. Nuclear expression of dynamin-related protein 1 in lung adenocarcinomas. Mod Pathol 2009; 22:1139-50; PMID:19525928; http://dx.doi.org/ 10.1038/modpathol.2009.83 [DOI] [PubMed] [Google Scholar]
- [7].Inoue-Yamauchi A, Oda H. Depletion of mitochondrial fission factor DRP1 causes increased apoptosis in human colon cancer cells. Biochem Biophys Res Commun 2012; 421:81-5; PMID:22487795; http://dx.doi.org/ 10.1016/j.bbrc.2012.03.118 [DOI] [PubMed] [Google Scholar]
- [8].Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, Ponpuak M, Master S, Pilli M, White E, et al.. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol 2010; 12:1154-65; PMID:21102437; http://dx.doi.org/ 10.1038/ncb2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chang Y, Yan W, He X, Zhang L, Li C, Huang H, Nace G, Geller DA, Lin J, Tsung A. miR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology 2012; 143:177-87e8; PMID:22504094; http://dx.doi.org/ 10.1053/j.gastro.2012.04.009 [DOI] [PubMed] [Google Scholar]
- [10].Hu F, Han J, Zhai B, Ming X, Zhuang L, Liu Y, Pan S, Liu T. Blocking autophagy enhances the apoptosis effect of bufalin on human hepatocellular carcinoma cells through endoplasmic reticulum stress and JNK activation. Apoptosis 2014; 19:210-23; PMID:24114361; http://dx.doi.org/ 10.1007/s10495-013-0914-7 [DOI] [PubMed] [Google Scholar]
- [11].Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, et al.. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 2015; 116:264-78; PMID:25332205; http://dx.doi.org/ 10.1161/CIRCRESAHA.116.303356 [DOI] [PubMed] [Google Scholar]
- [12].Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al.. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008; 27:433-46; PMID:18200046; http://dx.doi.org/ 10.1038/sj.emboj.7601963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Guido C, Whitaker-Menezes D, Lin Z, Pestell RG, Howell A, Zimmers TA, Casimiro MC, Aquila S, Ando' S, Martinez-Outschoorn UE, et al.. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 2012; 3:798-810; PMID:22878233; http://dx.doi.org/ 10.18632/oncotarget.574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Clerkin JS, Naughton R, Quiney C, Cotter TG. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett 2008; 266:30-6; PMID:18372105; http://dx.doi.org/ 10.1016/j.canlet.2008.02.029 [DOI] [PubMed] [Google Scholar]
- [15].Garg AD, Dudek AM, Ferreira GB, Verfaillie T, Vandenabeele P, Krysko DV, Mathieu C, Agostinis P. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy 2013; 9:1292-307; PMID:23800749; http://dx.doi.org/ 10.4161/auto.25399 [DOI] [PubMed] [Google Scholar]
- [16].Zhang B, Davidson MM, Zhou H, Wang C, Walker WF, Hei TK. Cytoplasmic irradiation results in mitochondrial dysfunction and DRP1-dependent mitochondrial fission. Cancer research 2013; 73:6700-10; PMID:24080278; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol 2010; 22:177-80; PMID:20022734; http://dx.doi.org/ 10.1016/j.ceb.2009.11.015 [DOI] [PubMed] [Google Scholar]
- [18].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/ 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schneider G, Henrich A, Greiner G, Wolf V, Lovas A, Wieczorek M, Wagner T, Reichardt S, von Werder A, Schmid RM, et al.. Cross talk between stimulated NF-kappaB and the tumor suppressor p53. Oncogene 2010; 29:2795-806; PMID:20190799; http://dx.doi.org/ 10.1038/onc.2010.46 [DOI] [PubMed] [Google Scholar]
- [20].Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol 1999; 19:3485-95; PMID:10207072; http://dx.doi.org/ 10.1128/MCB.19.5.3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dolado I, Nebreda AR. AKT and oxidative stress team up to kill cancer cells. Cancer Cell 2008; 14:427-9; PMID:19061832; http://dx.doi.org/ 10.1016/j.ccr.2008.11.006 [DOI] [PubMed] [Google Scholar]
- [22].Huang Q, Li J, Xing J, Li W, Li H, Ke X, et al.. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J Hepatol 2014; 61:859-66; PMID:24801417; http://dx.doi.org/ 10.1016/j.jhep.2014.04.035 [DOI] [PubMed] [Google Scholar]
- [23].Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer 2009; 125:2863-70; PMID:19609947; http://dx.doi.org/ 10.1002/ijc.24748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 2009; 89:799-845; PMID:19584314; http://dx.doi.org/ 10.1152/physrev.00030.2008 [DOI] [PubMed] [Google Scholar]
- [25].Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell 2004; 16:59-68; PMID:15469822; http://dx.doi.org/ 10.1016/j.molcel.2004.09.026 [DOI] [PubMed] [Google Scholar]
- [26].Qian W, Choi S, Gibson GA, Watkins SC, Bakkenist CJ, Van Houten B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J Cell Sci 2012; 125:5745-57; PMID:23015593; http://dx.doi.org/ 10.1242/jcs.109769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, Cheng H, Chen KH, Xiao RP. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem 2007; 282:23354-61; PMID:17562700; http://dx.doi.org/ 10.1074/jbc.M702657200 [DOI] [PubMed] [Google Scholar]
- [28].Legros F, Lombes A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell 2002; 13:4343-54; PMID:12475957; http://dx.doi.org/ 10.1091/mbc.E02-06-0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Landes T, Martinou JC. Mitochondrial outer membrane permeabilization during apoptosis: the role of mitochondrial fission. Biochim Biophys Acta 2011; 1813:540-5; PMID:21277336; http://dx.doi.org/ 10.1016/j.bbamcr.2011.01.021 [DOI] [PubMed] [Google Scholar]
- [30].Mao K, Klionsky DJ. Mitochondrial fission facilitates mitophagy in Saccharomyces cerevisiae. Autophagy 2013; 9:1900-1; PMID:24025250; http://dx.doi.org/ 10.4161/auto.25804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 2009; 284:13843-55; PMID:19279012; http://dx.doi.org/ 10.1074/jbc.M808515200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal 2011; 14:1939-51; PMID:21128700; http://dx.doi.org/ 10.1089/ars.2010.3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88; PMID:18570871; http://dx.doi.org/ 10.1016/j.molcel.2008.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bodur C, Kutuk O, Tezil T, Basaga H. Inactivation of Bcl-2 through IkappaB kinase (IKK)-dependent phosphorylation mediates apoptosis upon exposure to 4-hydroxynonenal (HNE). J Cell Physiol 2012; 227:3556-65; PMID:22262057; http://dx.doi.org/ 10.1002/jcp.24057 [DOI] [PubMed] [Google Scholar]
- [35].Blagosklonny MV. Unwinding the loop of Bcl-2 phosphorylation. Leukemia 2001; 15:869-74; PMID:11417471; http://dx.doi.org/ 10.1038/sj.leu.2402134 [DOI] [PubMed] [Google Scholar]
- [36].Ruvolo PP, Deng X, Carr BK, May WS. A functional role for mitochondrial protein kinase Calpha in Bcl2 phosphorylation and suppression of apoptosis. J Biol Chem 1998; 273:25436-42; PMID:9738012; http://dx.doi.org/ 10.1074/jbc.273.39.25436 [DOI] [PubMed] [Google Scholar]
- [37].Tamura Y, Simizu S, Osada H. The phosphorylation status and anti-apoptotic activity of Bcl-2 are regulated by ERK and protein phosphatase 2A on the mitochondria. FEBS Lett 2004; 569:249-55; PMID:15225643; http://dx.doi.org/ 10.1016/j.febslet.2004.06.003 [DOI] [PubMed] [Google Scholar]
- [38].Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal 2008; 10:1343-74; PMID:18522489; http://dx.doi.org/ 10.1089/ars.2007.1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 2011; 36:30-8; PMID:20728362; http://dx.doi.org/ 10.1016/j.tibs.2010.07.007 [DOI] [PubMed] [Google Scholar]
- [40].Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res 2008; 79:341-51; PMID:18440987; http://dx.doi.org/ 10.1093/cvr/cvn104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol 2007; 8:49-62; PMID:17183360; http://dx.doi.org/ 10.1038/nrm2083 [DOI] [PubMed] [Google Scholar]
- [42].An J, Wang X, Guo P, Zhong Y, Zhang X, Yu Z. Hexabromocyclododecane and polychlorinated biphenyls increase resistance of hepatocellular carcinoma cells to cisplatin through the phosphatidylinositol 3-kinase/protein kinase B pathway. Toxicol Lett 2014; 229:265-72; PMID:24960055; http://dx.doi.org/ 10.1016/j.toxlet.2014.06.025 [DOI] [PubMed] [Google Scholar]
- [43].Factor V, Oliver AL, Panta GR, Thorgeirsson SS, Sonenshein GE, Arsura M. Roles of Akt/PKB and IKK complex in constitutive induction of NF-kappaB in hepatocellular carcinomas of transforming growth factor α/c-myc transgenic mice. Hepatology 2001; 34:32-41; PMID:11431731; http://dx.doi.org/ 10.1053/jhep.2001.25270 [DOI] [PubMed] [Google Scholar]
- [44].Weisz L, Damalas A, Liontos M, Karakaidos P, Fontemaggi G, Maor-Aloni R, Kalis M, Levrero M, Strano S, Gorgoulis VG, et al.. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor α in cancer cells. Cancer Res 2007; 67:2396-401; PMID:17363555; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2425 [DOI] [PubMed] [Google Scholar]
- [45].Liu J, Ma Q, Zhang M, Wang X, Zhang D, Li W, Wang F, Wu E. Alterations of TP53 are associated with a poor outcome for patients with hepatocellular carcinoma: evidence from a systematic review and meta-analysis. Eur J Cancer 2012; 48:2328-38; PMID:22459764; http://dx.doi.org/ 10.1016/j.ejca.2012.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Frebourg T, Barbier N, Kassel J, Ng YS, Romero P, Friend SH. A functional screen for germ line p53 mutations based on transcriptional activation. Cancer Res 1992; 52:6976-8; PMID:1458490 [PubMed] [Google Scholar]
- [47].Tanaka A, Youle RJ. A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Mol Cell 2008; 29:409-10; PMID:18313377; http://dx.doi.org/ 10.1016/j.molcel.2008.02.005 [DOI] [PubMed] [Google Scholar]
- [48].Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al.. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 2008; 14:193-204; PMID:18267088; http://dx.doi.org/ 10.1016/j.devcel.2007.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ferrari LF, Chum A, Bogen O, Reichling DB, Levine JD. Role of Drp1, a key mitochondrial fission protein, in neuropathic pain. J Neurosci 2011; 31:11404-10; PMID:21813700; http://dx.doi.org/ 10.1523/JNEUROSCI.2223-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 2009; 119:1275-85; PMID:19349686; http://dx.doi.org/ 10.1172/JCI37829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang J, Hansen K, Edwards R, Van Houten B, Qian W. Mitochondrial division inhibitor 1 (mdivi-1) enhances death receptor-mediated apoptosis in human ovarian cancer cells. Biochem Biophys Res Commun 2015; 456:7-12; PMID:25446129; http://dx.doi.org/ 10.1016/j.bbrc.2014.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Qian W, Wang J, Roginskaya V, McDermott LA, Edwards RP, Stolz DB, Llambi F, Green DR, Van Houten B. Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget 2014; 5:4180-94; PMID:24952704; http://dx.doi.org/ 10.18632/oncotarget.1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.