

Abstract
The transcription factor Nrf2 (NF-E2-related-factor 2) regulates a battery of antioxidative stress-response genes and detoxication genes, and Nrf2 knockout lines of mice have been contributing critically to the clarification of roles that Nrf2 plays for cell protection. However, there are apparent limitations in use of the mouse models. For instance, rats exhibit more suitable features for toxicological or physiological examinations than mice. In this study, we generated 2 lines of Nrf2 knockout rats by using a genome editing technology; 1 line harbors a 7-bp deletion (Δ7) and the other line harbors a 1-bp insertion (+1) in the Nrf2 gene. In the livers of rats homozygously deleting the Nrf2 gene, an activator of Nrf2 signaling, CDDO-Im, could not induce expression of representative Nrf2 target genes. To examine altered toxicological response, we treated the Nrf2 knockout rats with aflatoxin B1 (AFB1), a carcinogenic mycotoxin that elicits gene mutations through binding of its metabolites to DNA and for which the rat has been proposed as a reasonable surrogate for human toxicity. Indeed, in the Nrf2 knockout rat livers the enzymes of the AFB1 detoxication pathway were significantly downregulated. Single dose administration of AFB1 increased hepatotoxicity and binding of AFB1-N7-guanine to hepatic DNA in Nrf2 knockout rats compared with wild-type. Nrf2 knockout rats repeatedly treated with AFB1 were prone to lethality and CDDO-Im was no longer protective. These results demonstrate that Nrf2 knockout rats are quite sensitive to AFB1 toxicities and this rat genotype emerges as a new model animal in toxicology.
Keywords: Nrf2, knockout rat, aflatoxin, hepatotoxicity.
INTRODUCTION
Nrf2 (NF-E2-related-factor 2) is a transcription factor and regulates genes responsible for xenobiotic detoxication, drug transport, and metabolism (Hirotsu et al., 2012). Keap1 (Kelch-like-ECH-associated-protein 1) controls this activity of Nrf2. Keap1 is a Cullin 3-based ubiquitin E3 ligase, residing in the cytoplasm (Kobayashi et al., 2004). Keap1 binds and ubiquitinates Nrf2, leading to degradation of Nrf2 through the 26S proteasome. Nrf2 activation is constitutively maintained at low levels under unstressed conditions. When cells encounter electrophilic or oxidative stresses, cysteine residues of Keap1 are modified and the ubiquitin ligase activity of Keap1 is inactivated. Therefore, Nrf2 is stabilized and accumulates in the nucleus where it binds to antioxidant responsive elements in gene promoters together with small Maf proteins. Intriguingly, cancer cells often manifest high levels of cytoprotection through acquisition of somatic mutations in either the NRF2 or KEAP1 gene, which localize in their mutually interacting domains (Padmanabhan et al., 2006; Ohta et al., 2008). These somatic mutations cause a disruption of the Keap1-Nrf2 binding, resulting in the stabilization and accumulation of Nrf2. Cancer cells with Nrf2 accumulation acquire chemo- and radio-resistance (Wang et al., 2008; Zhang et al., 2010) and metabolic reprogramming toward malignant proliferation (Mitsuishi et al., 2012).
To study the molecular basis of Nrf2 function and the contribution of Nrf2 to cytoprotection against various insults, we have generated Nrf2 (Itoh et al., 1997) and Keap1 (Okawa et al., 2006; Taguchi et al., 2010; Wakabayashi et al., 2003) knockout lines of mice. These lines of mice have been used widely and contributed substantially to our understanding of the Keap1-Nrf2 system (Taguchi and Yamamoto 2015). However, there are inherent limitations in the use of mice for toxicological and physiological studies. Rats are a common alternative species for toxicological studies; however, unlike the mouse model, methods to derive and propagate rat embryonic stem (ES) cells were only developed recently (Li et al., 2008), which has restricted severely the use of genetically engineered rats. Although there are many precious natural mutant rats, gene-targeted rats have been largely unavailable. Recent development of a series of genome editing technologies has changed this situation dramatically (Mashimo, 2014). These technologies enable us to generate many gene-knockout model animals simply by using the techniques widely used for the generation of transgenic mice.
With the advent of these gene editing technologies, rats are becoming alternative experimental animals that share many advantages previously associated with genetically engineered murine models (Jacob, 1999). In addition, compared with mice, rats are easier for the conduct of surgeries, provide larger size organs, and offer richer information in behavioral analyses than do mice. One of the most important advantages that rats offer to toxicological studies is reflected in observations that rats appear to mimic the detoxication metabolism of humans more closely than do mice (Wild et al., 1996). Rats are also more suitable for pharmacological studies. Activators of Nrf2 signaling have been developed as therapeutic drugs (Suzuki et al., 2013). Indeed, an Nrf2 activator, dimethyl fumarate (Tecfidera), has been approved for the treatment of multiple sclerosis and many more drug candidates are under development. To validate the efficacy and toxicity of such drugs, rat models are inherently more translational to humans than mouse models.
To initiate the validation of the Nrf2 knockout rat as a useful tool in understanding toxicological mechanisms and outcomes, we have challenged these animals with aflatoxin B1 (AFB1). AFB1 is a highly carcinogenic mycotoxin produced by Aspergillus species of molds. Upon eating foods contaminated with AFB1, AFB1 is metabolized in the liver to a reactive epoxide intermediate, AFB1-8,9-epoxide, by cytochrome P450s (Kensler et al., 2011) and AFB1-8,9-epoxide spontaneously forms adducts with guanine bases in DNA, resulting in AFB1-N7-guanine and AFB1-formamidopyrimidine. These modified bases elicit DNA mutations. Detoxication of AFB1 relies on glutathione S-transferases (GSTs) and aldo-keto reductases (AKRs). GSTA3 and/or GSTA5 catalyze conjugation of glutathione to AFB1-8,9-epoxide and produce AFB1-glutathione conjugate (AFB1-SG). AFB1-SG is sequentially converted to AFB1-N-acetyl cysteine (AFB1-NAC) endproduct. Alternatively, the AFB1-8,9-epoxide is converted to AFB1-dialdehyde phenolate through an AFB1-dihydrodiol intermediate. AFB1-dialdehyde phenolate reacts with proteins such as serum albumin. AFB1-dialdehyde phenolate is metabolized to AFB1-dialcohol by AKR7A.
It has been shown that CDDO-Im (2-cyano-3,12-dioxooleana-1,9-dien-28-imidazolide), an Nrf2 activator, protects rats against AFB1-induced hepatocellular carcinoma with striking potency and efficacy (Johnson et al., 2014). CDDO-Im treatment also decreases the burden of AFB1-N7-guanine in liver and increases the elimination of AFB1-NAC in urine, suggesting that Nrf2 enhances detoxication of AFB1 in vivo. However, further genetic analyses of the mechanisms of protection against AFB1 toxicity have been hampered because of the lack of Nrf2 knockout rats. Because mice possess much higher levels of hepatic GST activity toward the AFB1-epoxide than found in rats and humans (Wild and Turner, 2002), the mouse has not proven to be a suitable model to reproduce AFB1-induced hepatocellular carcinoma seen in exposed humans. Therefore, in this study, we have undertaken the generation of Nrf2 knockout rats by means of a genome editing technology and evaluated the impact of disruption of Nrf2 signaling AFB1 toxicity. We report here that the Nrf2 knockout rats are informative model animals to evaluate roles that Nrf2 plays in the regulation of AFB1 detoxication.
MATERIALS AND METHODS
Chemicals
CDDO-Im (2-cyano-3,12-dioxooleana-1,9-dien-28-imidazolide) was generously provided by Mochida Pharmaceutical (Tokyo). AFB1 (A6636) was purchased from Sigma-Aldrich (St Louis, Missouri). All other chemicals used were obtained from commercial sources and were of the highest grade available.
Animals
Male F344 rats or Nrf2 knockout C57BL/6J mice (Itoh et al., 1997) were provided water and MR diet (Nosan Co., Kanagawa) ad libitum. All rats and mice were maintained under semispecific-pathogen-free conditions and treated according to the regulations of The Standards for Human Care and Use of Laboratory Animals of Tohoku University and Guidelines for Proper Conduct of Animal Experiments of the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Generation of Nrf2 Mutant Rats
Generation of Nrf2 mutant rats followed the standard procedures as described previously (Mashimo et al., 2010). In brief, founder animals heterozygous for deletion of the Nrf2 were generated on F344 inbred background using Zinc finger nuclease (ZFN) technology (Sigma-Aldrich Co.). ZFN constructs were designed to target the upstream of exon 5 in the Nrf2 sequence, ACCACTGTCCCCAGCCCAgaggccACACTGACAGAG (Figure 1A). Off-target sites with the highest degree of similarity were identified by searching the rat genome (RGSCv3.4) for matches with the ZFN construct sequence with appropriate spacing of 5–6 bp. A list of these target sites is shown in Table 1. Approximately 2–3 pl of ZFN mRNA (10 ng/μl) was injected into the pronuclei of embryos collected from F344/Stm females. The cultured embryos were then transferred to the oviducts of pseudopregnant females (Crlj:WI, 8–10 weeks). A region of exon 5 genomic DNA was then amplified by PCR (94 °C for 30 s, 60 °C for 60 s, 72 °C for 45 s, 35 cycles) using forward primer (Nrf2 Small F: TGAAAATGGGAGTTATCGGG) and reverse primer (Nrf2 Small R: TGTGTTCAAGGTGGGATTTG). The PCR product of the wild-type sample was 334 bp. Amplified samples were then sequenced by ABI3100 (Applied Biosystems) using standard protocols. Each heterozygous pair was maintained to yield littermates of wild-type and homozygous Nrf2 mutant rats. Toes were clipped and genotyped as shown in Figures 1C and D. All animal care and experiments conformed to the Guidelines for Animal Experiments of Tohoku University and Kyoto University, and were approved by the Animal Research Committees of Tohoku University and Kyoto University. The resultant rats were deposited to the National BioResource Project for the Rat in Japan (NBRP-Rat) as F344-Nfe2l2em1Kyo and F344-Nfe2l2em2Kyo.
FIG. 1.
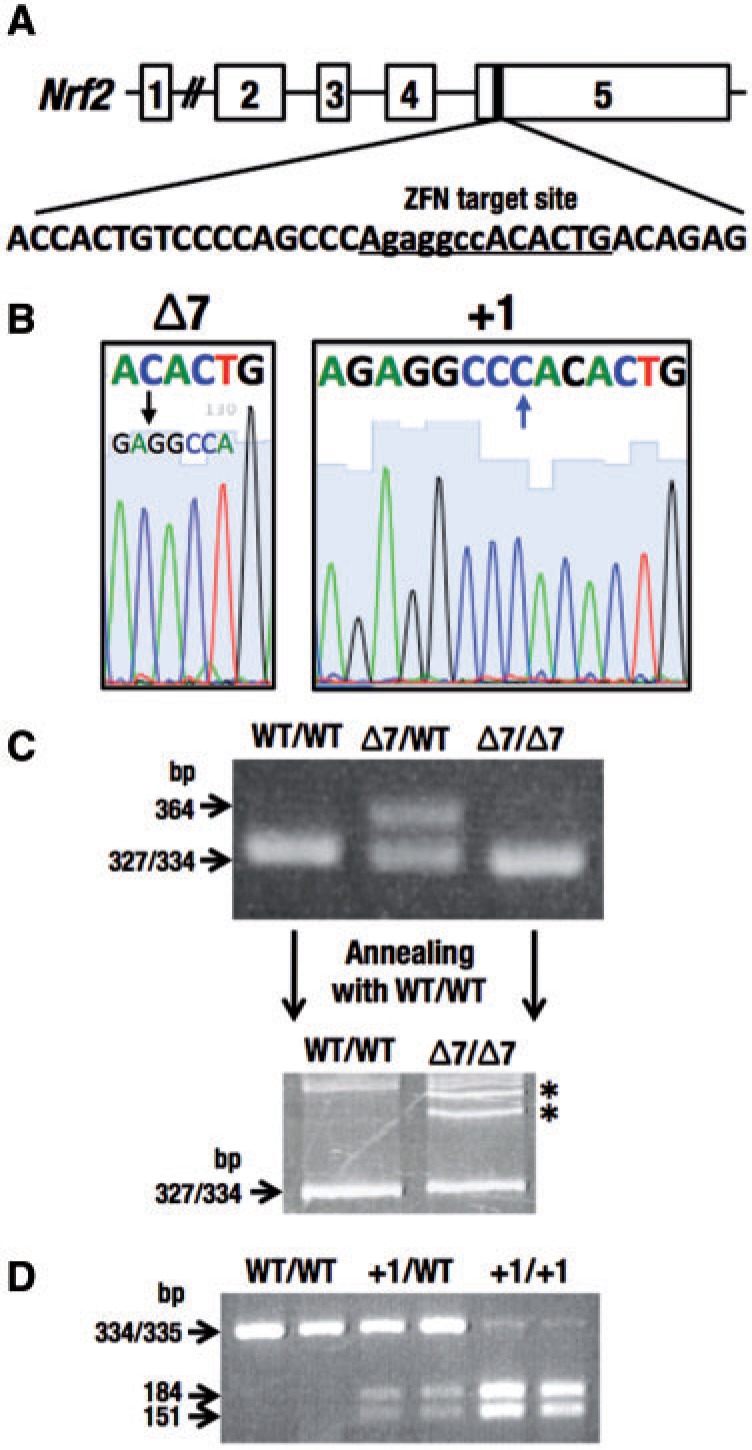
Generation of Nrf2 mutant rats. A, The designed target site of zinc finger nuclease (ZFN) in exon 5 of Nrf2 gene. B, Two lines with an Nrf2 mutation in the ZFN target site, Δ7 (left) and +1 (right). Sequences underlined in (A) were shown. C, Genotyping of Nrf2 Δ7 mutation by electrophoresis. Electrophoresis was performed using PCR products in 4% agarose gel (upper panel). The heterozygous sample of Δ7 mutation and wild-type had both 334 and 364 bp. The polyacrylamide gel electrophoresis-based genotyping analysis was analyzed to distinguish a difference between wild-type (334 bp) and Δ7 mutation (327 bp). Annealing and denaturation formed heteroduplex DNA (*), which migrated slower than homoduplex DNA in 9% acrylamide gel (lower panel). D, Genotyping of Nrf2 +1 mutation by a restriction enzyme, BmgT120I. The PCR product (335 bp) with +1 mutation was digested to 151 and 184 bp by BmgT120I.
TABLE 1.
Potential ZFN Off-Target Sites
| ID | Chr No. | Start Pos. | Sequencea | No. of Mismatch | Homodimer (+)/Heterodimer (−) | Gene |
|---|---|---|---|---|---|---|
| 1 | chr3 | 58368282 | AACAACTGGAAATAGCCCAAAGATACACTGAGGGAGATGGACA | 8 | — | — |
| 2 | chrX | 145527827 | AACCAATCTCCTCATCCCCGTTCTACATTGACAGTGATGGAGG | 8 | — | Hagh |
| 3 | chr10 | 14112775 | GGTTCATCTGGGTCTGTGTGTGGTTGGGGTGGGGTCAGAGGGC | 8 | — | — |
| 4 | chr10 | 109602919 | AACCACTGACTGCAGCCAAATGTCACACTGAAAGTTATGGTCT | 8 | — | — |
aBases differing from the consensus target sequence are shown in red. FokI catalytic sequences are shown in green.
Determination of an Intrahepatic Shunt
An intrahepatic shunt was determined by cannulation of the portal vein of 14- to 20-week-old rats, with slight modification to the previous method (Skoko et al., 2014). As a staining solution, bromophenol blue was used instead of trypan blue.
Single Dose of CDDO-Im
Nrf2 knockout rats with Δ7 or +1 mutations and wild-type rats at the age of 6–7 weeks, and wild-type mice and Nrf2 knockout mice were gavaged with a single dose of an Nrf2 activator, CDDO-Im (30 μmol/kg body weight) or vehicle of 10% cremophor-EL, 10% dimethyl sulfoxide (DMSO) and PBS. Rats and mice were sacrificed 6 h after dosing and the livers harvested.
Single Dose of AFB1 With or Without CDDO-Im
For detection of the AFB1-DNA adduct, rats were gavaged with CDDO-Im (30 μmol/kg body weight) 3 times every other day at 8 am. Twenty-four hours after the last treatment with CDDO-Im, rats were gavaged with 25 μg/100 g body weight of AFB1 dissolved in DMSO. Rats were then housed in metabolism cages and sacrificed 24 h after the administration of AFB1. The time schedule is presented in Figure 6A. The serum was analyzed using FUJI DRI-CHEM 7000 (FUJIFILM, Tokyo) to detect alanine transferase (ALT). Livers were immediately frozen in liquid nitrogen using a freeze clamp and stored at −80 °C. DNA was isolated by the method as described (Kensler et al., 1985) and analyzed for levels of AFB1-DNA adducts by liquid chromatography-mass spectrometry as described previously (Egner et al., 2003). Total DNA content was measured spectrophotometrically using diphenylamine. Immediately following the urine collection, samples were centrifuged at 150 × g and adjusted to an acidic pH using 0.5 mol/l ascorbic acid. Urines were analyzed for levels of AFB1-N7-guanine and AFB1-NAC by isotope dilution mass spectrometry (Egner et al., 2006). Levels were normalized to creatinine content as measured using a spectrophotometric creatinine kit (Eagle Diagnostic), as previously reported (Johnson et al., 2014). Serum AFB1-adducts were also measured by isotope dilution mass spectrometry as described (Scholl et al., 2006).
FIG. 6.

Effects of a single dose of AFB1 with CDDO-Im in wild-type rats and Nrf2 knockout rats with Δ7 mutation. A, Schedule of treatment of CDDO-Im and AFB1. Rats were gavaged with CDDO-Im (30 μmol/kg body weight) 3 times every other day at 8 am. Twenty-four hours after the last treatment with CDDO-Im, rats were gavaged with AFB1 (25 μg/100 g body weight). Rats in metabolic cages at days 1 and 7 were sacrificed 24 h after administration of AFB1. B, Groups of genotypes and treatments. Rats (200–265 g on the first day of administration of CDDO-Im) were used. C, Percentages of liver to body weight. D, Serum ALT. E, Urine volume. F, Metabolites of AFB1 in the liver, urine, and serum. The data represent mean ± SD (n = 6–7). *P < .05; **P < .01. Asterisks without brackets indicate the comparison with group 1.
Repeated Dose Toxicity of AFB1 With or Without CDDO-Im
The dose and schedule for administration of CDDO-Im and AFB1 to rats were identical to that of Yates (Yates et al., 2006). Rats were gavaged with CDDO-Im (30 μmol/kg body weight) for 3 successive weeks on Monday, Wednesday, and Friday at 8 am. Beginning on the 1 week, AFB1 (25 μg/rat) was gavaged at 12 am Monday through Friday for 2 weeks. Rats were sacrificed 5 weeks after the last doses of CDDO-Im and AFB1. The schedule is presented in Figure 7A.
FIG. 7.
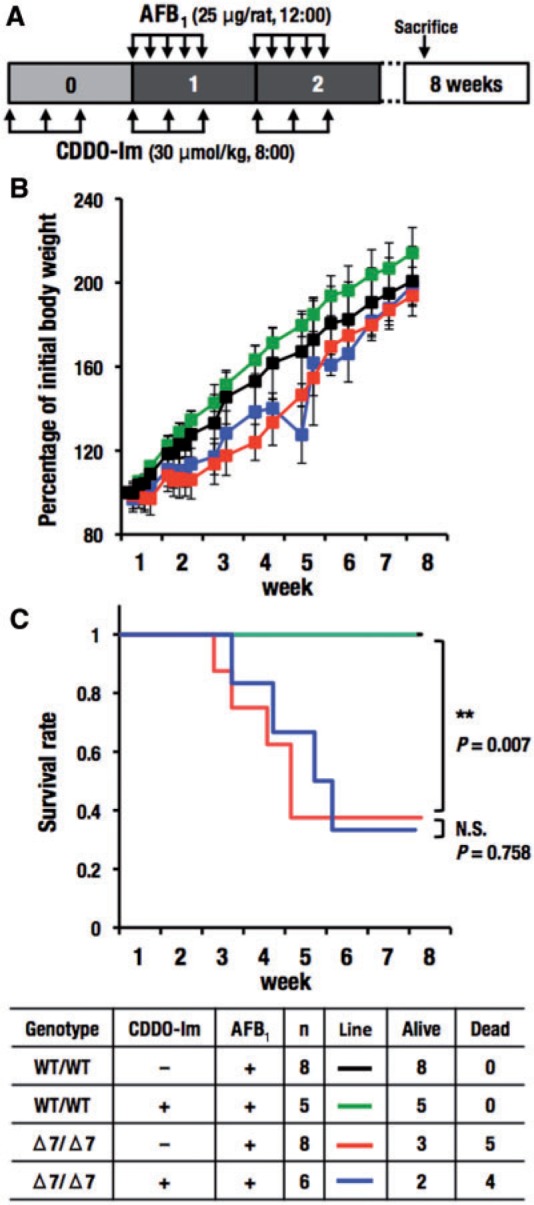
Effects of repeated doses of AFB1 with CDDO-Im in wild-type and Nrf2 knockout rats with Δ7 mutation. A, Schedule of treatment of CDDO-Im and AFB1. Rats were gavaged with CDDO-Im (30 μmol/kg body weight) for 3 successive weeks on Monday, Wednesday, and Friday at 8 am. Beginning on the first week, AFB1 (25 μg/rat) was gavaged at 12 am Monday through Friday for 2 weeks. Rats were sacrificed 5 weeks after the last doses of CDDO-Im and AFB1. B, Change of body weight. Rats (104–168 g on the first day of administration of AFB1) were used. Body weight on the first day of administration of AFB1 was set to 100%. The data represent mean ± SD (n = 5–8). C, Survival rate. Four groups were listed in the table. **P < .01. Asterisks with brackets indicate the comparison between indicated groups.
Quantitative Reverse Transcription Polymerase Chain Reaction Analysis
Total RNA was isolated from livers using Sepazol-RNA I Super G (Nacalai Tesque, Kyoto). RNA concentration was measured using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, Delaware). RNA was transcribed into cDNA using SuperScript III Reverse Transcriptase (Life Technologies, Carlsbad, California). Reverse transcription-quantitative PCR (RT-qPCR) was performed using the Applied Biosystems ABI7300 PCR system, and thunderbird SYBR qPCR Mix (TOYOBO, Osaka). The data were normalized to Gapdh expression. The primers used for RT-qPCR are listed in Table 2.
TABLE 2.
Primers Used in the Quantitative RT-qPCR
| Rat Gene | Oligonucleotide Sequence | Reference | |
|---|---|---|---|
| Akr7a2 | F | 5′-GAGCTTGGCTTGTCCAAC-3′ | Merrick et al. (2012a) |
| R | 5′-ATCCAGCCGTTGCTTTTA-3′ | ||
| Akr7a3 | F | 5′-CCGCTTCTTTGGGAATCCAT-3′ | Hayashi et al. (2012) |
| R | 5′-GGCGATGCCATTGAAGTGT-3′ | ||
| Gapdh | F | 5′-TTCAATGGCACAGTCAAGGC-3′ | Yeligar et al. (2010) |
| R | 5′-TCACCCCATTTGATGTTAGCG-3′ | ||
| Gclm | F | 5′-CTGCTAAACTGTTCATTGTAGG -3′ | Suh et al. (2004) |
| R | 5′-CTATTGGGTTTTACCTGTG -3′ | ||
| Gsta3 | F | 5′-AGTCCTTCACTACTTCGATGGCAG-3′ | Djordjevic et al. (2015) |
| R | 5′-CACTTGCTGGAACATCAAACTCC-3′ | ||
| Gsta5 | F | 5′-GTGCAGACCAAAGCCATT-3′ | Merrick et al. (2012b) |
| R | 5′-TGAGGGCTCTCTCCTTCA-3′ | ||
| Hmox1 | F | 5′-TTGTCTCTCTGGAATGGAAGG-3′ | Yeligar et al. (2010) |
| R | 5′-CTCTACCGACCACAGTTCTG-3′ | ||
| Keap1 | F | 5′-GGACGGCAACACTGATTC-3′ | Yamashita et al. (2014) |
| R | 5′-TCGTCTCGATCTGGCTCATA-3′ | ||
| Nqo1 | F | 5′-CATTCTGAAAGGCTGGTTTGA -3′ | Yeligar et al. (2010) |
| R | 5′-CTAGCTTTGATCTGGTTGTCA G -3′ | ||
Microarray Analysis
Total RNA from liver was labeled with Cy3. The samples were hybridized to Oligo DNA Microarray kit for Whole Rat or Mouse Genome (Agilent Technologies, Santa Clara, California) according to the manufacturer’s protocol. Arrays were scanned using the G2539A Microarray Scanner System (Agilent Technologies), and the resulting data were analyzed using GeneSpring GX software (Agilent Technologies). The microarray data obtained in this study have been submitted to the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/ last accessed April 15, 2016) and assigned the GEO accession number GSE77377. Mouse ChIP-seq data using Nrf2 and MafG antibodies were analyzed (Hirotsu et al., 2012). The samples were mouse Hepa1 cells treated with 100 μM diethylmaleate for 4 h.
Western Blot
Livers were homogenized in 9 volumes of 0.25 M sucrose containing 10 μM MG132, and 10 μM Na3VO3, 100 μM NaF and EDTA-free Complete (05056489001, Roche Diagnostics, Germany). Nucleic fraction was isolated as previously reported (Taguchi et al., 2010, 2014). Protein concentration was measured using a bicinchoninic acid protein assay kit (Pierce Biotechnology, Rockford, Illinois), with bovine serum albumin as the standard. The antibodies used for Western blot are listed in Table 3.
TABLE 3.
Antibodies Used in Western Blot
| Antibody | Catalog Number | Reference or Company |
|---|---|---|
| Anti-AKR7A2 | ab175295 | Abcam PLC, Cambridge, United Kingdom |
| Anti-AKR7A3 | 13209-1-AP | Proteintech Group, Inc., Chicago |
| Anti-GSTA3 | Mclellan et al. (1994) | |
| Anti-Keap1 | No. 111 | Watai et al. (2007) |
| Anti-Lamin B | sc-6217 | Santa Cruz Biotechnology Inc., Dallas, Texas |
| Anti-Nrf2 | No. 103 | Maruyama et al. (2008) |
| Anti-NQO1 | ab2346 | Abcam PLC, Cambridge, United Kingdom |
| Anti-αTubulin | T9026 | Sigma-Aldrich Co., LLC, St Louis |
Statistical Analysis
The average values were calculated, and the error bars indicate standard deviations. Differences were analyzed using the Student’s t test. The differences in a survival rate were analyzed using Longrank test. P < .05 was considered statistically significant.
RESULTS
Generation of Nrf2 Mutant Rats Using ZFN Technology
To generate Nrf2 knockout rats, we designed a ZFN construct that targeted exon 5 (Figure 1A), as we previously had generated an Nrf2 knockout line of mouse similarly by targeting exon 5 of the mouse Nrf2 gene (Itoh et al., 1997). To clarify whether the ZFN construct induces mutations only in the targeted region, we have searched for potential off-target sites in the rat genome that are most homologous to the target sequence. We could not find identical target sites in rat genome. Instead, we found 4 sites that carried 8-base mismatches to the on-target site (Table 1). This observation strongly argues that the ZFN construct that we have employed is specific to loci of interest. Although we could not exclude the formal possibility that the ZFN construct might cleave one (or more) of the off-target sites, we surmise that such off-target mutations could be easily segregated from the Nrf2 locus during the crossing, as the potential off-target sites were located in different chromosomes from the Nrf2 locus. Therefore, we have concluded that the ZFN construct we have employed is reliable to produce mutant alleles at the locus of interest.
Of the 11 pups obtained from ZFN-injected eggs, 2 pups appeared to be gene-edited in the Nrf2 locus through mono-allelic mutations. One mutation was a 7-bp deletion (Δ7) and the other was a 1-bp insertion (+1) within the ZFN target site (Figure 1B). Both mutations were expected to produce a truncated Nrf2 protein by introducing stop codons within exon 5. Genotyping analyses of these mutations were conducted by DNA sequencing. For initial genotyping of Nrf2 Δ7 mutation, we compared the PCR product of 334 bp in the wild-type lane with that of 327 bp in the Nrf2 Δ7 mutant lane. We found a band at 364 bp appearing in 4% agarose gel electrophoresis in the heterozygote lane (Figure 1C, upper column). The reason for the 364-bp band is a slower running hetero-duplex DNA in agarose gel. As it was technically difficult to identify the 7-bp difference between wild-type and Δ7 mutants by agarose gel electrophoresis (Figure 1C, upper column), we exploited a polyacrylamide gel electrophoresis-based genotyping analysis for the detection of minor differences based on the formation of hetero-duplexes (Zhu et al., 2014). The PCR products (5 μl) with a single band in the agarose gel annealed with the wild-type sample (5 μl) at 72 °C, 5 min and 96 °C, 5 min and maintained at room temperature. The annealing and denaturation processes formed hetero-duplex DNA, which migrated significantly slower than homo-duplex DNA in a 9% polyacrylamide gel (Figure 1C, lower column). The hetero-duplex DNA formed through annealing and denaturation was detected in 4% agarose gel as well as 9% acrylamide gel (data not shown). We reproducibly observed 2 hetero-duplex bands that probably correspond to the hybrids formed by plus and minus complementary strands of amplified wild-type and mutant allele (Espejo et al., 1998).
In contrast, the Nrf2 +1 mutation resulted in a restriction enzyme-recognition site. The mutation generated a sequence motif that could be cleaved by BmgT120I. Using the restriction fragment length polymorphism assay, the PCR product with +1 mutation (335 bp) was digested to 184- and 151-bp fragments by digestion with BmgT120I (Figure 1D). These multiple approaches enabled us to genotype precisely and reproducibly the 2 Nrf2 mutations in rats.
Apparent Phenotypes in Nrf2 Mutant Rats
We have identified 3 unexpected phenotypes in the Nrf2 knockout mouse; white incisors (Yanagawa et al., 2004), a congenital intrahepatic shunt (Skoko et al., 2014), and smaller liver size (Zhang et al., 2013). Penetration of the incisor and liver size phenotypes are very high, whereas the intrahepatic shunt phenotype is found about half of the mice in C57BL/6J background and much less frequently in ICR background mice.
Although iron deposition makes incisors of rodents red-yellowish, incisors of Nrf2 knockout mice are whitish, as Nrf2 regulates the genes responsible for iron transport and deposition in the teeth (Yanagawa et al., 2004). In this study, we found that Nrf2 knockout rats (both with Δ7 and +1 lines) also showed decolorized incisors compared with wild-type rats (Figure 2A), albeit the discoloration was much milder than that in Nrf2 knockout mice. We also examined the presence of an intrahepatic shunt in the Nrf2 knockout rats. Our Nrf2 knockout rats were in the F344 background. Neither the Δ7 nor the +1 mutant showed the presence of apparent shunt, as was the case for the C57BL/6J strain of mouse (Figure 2B). These results suggest that genetic background may play a strong influence on the elaboration of this phenotype. Liver sizes in Δ7 and the +1 mutant were significantly smaller than the littermate wild-type rats (Figure 2B).
FIG. 2.
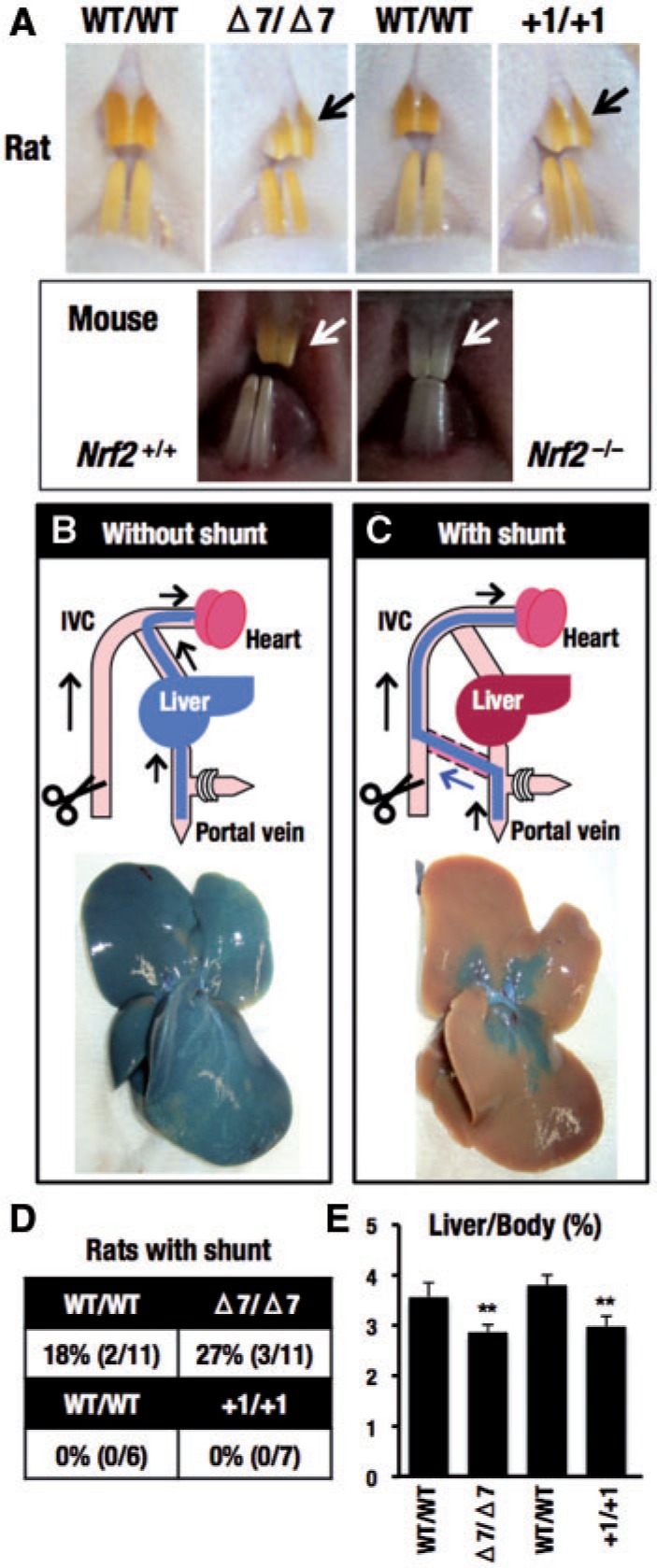
Comparison of obvious phenotypes between Nrf2 mutant mice and Nrf2 mutant rats. Nrf2 mutant rats with Δ7 or +1 mutations (14–20 weeks old, 6–11 rats) and Nrf2 knockout mice (11–15 weeks old) were used. A, Color of incisors. Absence (B) or presence (C) of a congenital intrahepatic shunt was determined by cannulation of the portal vein with a catheter, opening of the inferior vena cava and flushing the liver with saline. A solution of bromophenol blue was then perfused into the portal vein. The liver was then excised and photographed. Representative photographs are shown with illustrations. IVC, inferior vena cava. D, Percentages of rats with shunt were calculated. E, Percentages of liver weight to body weight were calculated after injection of bromophenol blue. **P < .01.
Confirmation of Nrf2 Deletion in Rats With Δ7 or +1 Mutations
Nrf2 is constantly degraded by the ubiquitin-proteasome system, and Nrf2 protein is maintained at quite low levels under normal conditions. To verify Nrf2 depletion within the Δ7 and +1 mutants, we administered CDDO-Im to rats in order to induced Nrf2 accumulation (Figure 3A, left panel). Showing very good agreement with our previous study (Yates et al., 2006), oral administration of CDDO-Im (30 μmol/kg body weight) for 6 h caused nuclear accumulation of Nrf2 in liver of wild-type rats. In contrast, CDDO-Im did not induce Nrf2 accumulation in rats with the Δ7 mutation. Similarly, under basal conditions Nrf2 was completely lost in rats with Δ7 mutations, which is significant when compared with the faint Nrf2 bands in wild-type rats. The loss of Nrf2 induction, as well as loss of basal expression of Nrf2, was quite reproducible in rats with +1 mutations (Figure 3A, right panel). On the other hand, Keap1 protein levels were constant in both wild-type and the Δ7 and +1 mutant rats.
FIG. 3.

Confirmation of Nrf2 deletion in Nrf2 Δ7 or +1 mutations using an Nrf2 activator, CDDO-Im. Single dose of CDDO-Im (30 μmol/kg body weight for 6 h) was orally gavaged to rats. A, Loss of Nrf2 induction by CDDO-Im in the liver of rats with Nrf2 Δ7 or +1 mutations. Both basal and CDDO-Im-inducible Nrf2 protein expressions in wild-type rats were lost in rats with Δ7 or +1 mutations. Keap1 was expressed constantly in all groups. Lamin B and αTubulin were an internal control in the nucleus and 1000 × g supernatant, respectively. B, Quantification of the mRNA levels in the liver of rats with Δ7 or +1 mutations using RT-qPCR. Nrf2 target genes, Ho-1 and Gclm, were upregulated in CDDO-Im-treated wild-type rats, but not in rats with Δ7 or +1 mutation. Keap1 was expressed constantly in all groups. Nrf2 were comparable with those the wild-type rats. Gapdh was used as an internal control. The data represent mean ± SD (n = 3–4). **P < .01. Asterisks without brackets indicate the comparison with vehicle-treated wild-type rats.
We targeted the last fifth exon of Nrf2 gene, and in this case nonsense-mediated decay was not expected (Maquat, 2004). The expression of Nrf2 mRNA levels was comparable with those of the wild-type rats (Figure 3B). Expression levels of Keap1 mRNA were also constant in all groups, showing very good correlation with the protein levels. In contrast, we found that in Δ7 mutant rats Ho-1 and Gclm mRNAs were not induced by CDDO-Im treatment (Figure 3B). The Ho-1 and Gclm are representative Nrf2 target genes that are usually induced significantly in wild-type rats by CDDO-Im treatment. Indeed, we verified the Ho-1 and Gclm mRNA induction as can be seen in Figure 3B. These changes in Ho-1 and Gclm mRNA expression were quite reproducible in the +1 mutant rats. Collectively, these data demonstrate that the 2 lines rats with Δ7 and +1 mutations in the Nrf2 gene are genuine Nrf2 knockout rats, with subsequent loss of the downstream transcriptional activity of Nrf2.
Expression of Nrf2-Target Genes in Microarray Analyses
In order to identify Nrf2-target genes in rats, we performed microarray analyses in which we compared gene expression in Nrf2 knockout rats with Δ7 or +1 mutations together with CDDO-Im-treated or vehicle-treated wild-type rats. In this analysis, we defined Nrf2-target genes with 2 criteria. First, we selected genes for which levels were induced more than 2-fold in wild-type rats treated with CDDO-Im compared with vehicle-treated wild-type rats. Second, we selected genes with more than 2-fold lower expression in CDDO-Im-treated Nrf2 knockout rats compared with CDDO-Im-treated wild-type rats.
As shown in Figure 4A, we found 291 probes and 367 pobes of Nrf2-target gene candidates satisfying these 2 criteria in Δ7 and +1 mutant rats, respectively. When we merged these 2 clusters of probes, we found 187 overlapping probes, which corresponded to 106 genes, were commonly induced by CDDO-Im, but the induction was canceled by the mutations in the rat Nrf2 gene. The genes that were commonly induced in wild-type by CDDO-Im and were canceled by the Δ7 or +1 mutation in the rat Nrf2 gene are listed in Figure 4B. This group of genes included ATP-binding cassette (ABC) transporters (Abcc3, Abcc4), aldo-keto reductase (AKR) family (Akr1b8, Akr1b10), aldehyde dehydrogenase (ALDH) family (Aldh1a7), transcription factors (Bach1, Myc), glutathione synthetase (Gclc, Gclm), GST family (Gsta3), antioxidative enzymes (Gpx2, Hmox1, Nqo1, Slc7a11, Srxn1, Txnrd1), small Maf (Maff, Mafg), and a metabolic enzyme (G6pdx). These genes have been shown to be Nrf2-target genes in various gene expression analyses using mouse and human cells. For instance, the inductions of Hmox1 (named as Ho-1) and Gclm by CDDO-Im were indeed suppressed in Nrf2 knockout rats with Δ7 and +1 mutations (see Figure 3B). To our surprise, however, only 23 genes were found commonly in mice that were induced by CDDO-Im and the induction was canceled by the Nrf2 gene knockout in the same experimental condition as that employed for rats. Showing very good agreement, only 53 genes of the 106 genes identified in rats were found in the positive gene data of mouse Nrf2 and MafG ChIP-seq analyses (Hirotsu et al., 2012). Even considering the differences in experimental conditions, these results imply the significant genetic variations in the regulation of detoxifying enzymes between these 2 species.
FIG. 4.

Nrf2-dependent gene induction by CDDO-Im. Microarray analyses were performed using wild-type rats and rats with Nrf2 Δ7 or +1 mutations in the absence or presence CDDO-Im. In this analysis, we defined Nrf2-target genes with 2 criteria. First, we selected genes whose levels were induced more than 2-fold in wild-type rats treated with CDDO-Im compared with that in vehicle-treated wild-type rats. Second, we selected genes downregulated more than 2-fold in CDDO-Im-treated Nrf2 knockout rats compared with CDDO-Im-treated wild-type rats. Number (A) and names (B) of the genes that were induced by CDDO-Im but for which induction was canceled by the Nrf2 Δ7 or +1 mutation. The 187 probes correspond to 106 genes. Fifty-three genes marked by shadowing were in common with the mouse genes to which both Nrf2 and MafG bound in ChIP-seq analyses (Hirotsu et al., 2012). Twenty-three genes with black dots were found to show Nrf2-dependent induction by CDDO-Im in a similar set of analyses in which wild-type and Nrf2 knockout mice were treated with CDDO-Im under the same condition as that used for rats.
Nrf2-Dependent Detoxication of AFB1
As shown in Figure 5A, AFB1 is metabolized to AFB1-8,9-epoxide by cytochrome P450s. AFB1-8,9-epoxide is then conjugated with glutathione by GSTA3 and GSTA5 or is converted to AFB1-dihydrodiol by hydrolysis. In the acidic condition, AFB1-dihydrodiol is converted to AFB1-dialdehyde phenolate, which is metabolized to AFB1-dialcohol by AKR7A2 and AKR7A3. Thus, GST and AKR isozymes are important for detoxication of AFB1.
FIG. 5.

Nrf2-dependent basal expression of enzymes responsible for AFB1 detoxication. A, The metabolic pathway of AFB1. B, Expression of enzymes responsible for AFB1 detoxication in Nrf2 knockout rats with Δ7 or +1 mutations. αTubulin was an internal control in the cytoplasmic fraction. *Nonspecific band. C, mRNA expression of genes related to AFB1 detoxication. The data represent mean ± SD (n = 3–4). *P < .05; **P < .01. Asterisks without brackets indicate the comparison with vehicle-treated, wild-type rats.
Using the Nrf2 knockout rats under the same conditions as for the data depicted in Figure 3, we examined whether enzymes responsible for detoxication of AFB1 were expressed in an Nrf2-dependent manner or not. The basal expression of GSTA3, AKR7A2, and AKR7A3 was decreased reproducibly in both lines of Nrf2 knockout rats compared with wild-type rats (Figure 5B), demonstrating the Nrf2-dependency in the induction of these gene transcripts. The expression of mRNAs related to detoxication of AFB1 (Gsta3, Gsta5, Akr7a2, and Akr7a3) was also examined (Figure 5C). CDDO-Im elevated expression of these mRNAs in wild-type rats, but not in Nrf2 knockout rats, indicating that the expression of these genes is Nrf2-dependent. Gsta3 was also identified as an Nrf2-depenent gene by microarray analyses as shown in Figure 4B.
Nrf2 Is Protective Against AFB1 Toxicity in Liver
In order to quantify potential modulation of levels of DNA or protein adducts following administration of AFB1 (see Figure 5A), single doses of AFB1 with or without prior CDDO-Im treatments were administered to the 4 groups of rats. The schedule for dosing with AFB1 and CDDO-Im is shown in Figure 6A and treatment assignments for the groups of rats are shown in Figure 6B. As shown in Figure 6C, simultaneous administration of CDDO-Im gave rise to the enlargement of livers in AFB1-treated wild-type rats (group 2) compared with vehicle-treated wild-type rats (group 1). In Nrf2 knockout rats with Δ7 mutation (groups 3 and 4), liver sizes were significantly smaller than wild-type rats, and the CDDO-Im treatment made no difference.
The AFB1-treatment increased serum ALT levels, a marker of liver injury, of wild-type rats, but ALT levels remained at normal levels with pretreatment with CDDO-Im (Figure 6D). These results are consistent with the previous observation that CDDO-Im is protective against AFB1 toxicity (Johnson et al., 2014). Importantly, the AFB1-treatment significantly increased ALT levels in Nrf2 knockout rats with Δ7 mutation compared with wild-type rats; however, pretreatment of CDDO-Im did not afford significant protection (Figure 6D). These results indicate that Nrf2 is an important determinant of the extent of hepatotoxicity of AFB1, and that CDDO-Im exerts protective effects through Nrf2 signaling.
To verify this notion, we quantified metabolites that are indicators of toxicity and detoxication of AFB1. As described earlier, the reactive metabolite AFB1-8,9-epoxide is detoxified either by glutathione conjugation or by hydration followed by further reduction through AKRs. In the latter case, resulting AFB1-dialdehyde reacts with proteins, such as serum albumin. Urine volumes were not affected in the 4-genotype groups (Figure 6E). We found that CDDO-Im decreased significantly the AFB1-N7-guanine levels isolated from liver DNA and excreted into urine of AFB1-treated wild-type rats (Figure 6F). CDDO-Im-treatment enhanced the urinary levels of AFB1-NAC, but did not significantly reduce levels of AFB1-Lys in serum. The former is an ultimate metabolite of AFB1-glutathione conjugate, whereas the latter is a product of AFB1-albumin interaction (see Figure 5A).
Levels of AFB1-N7-guanine were increased in both the liver and urine of Nrf2 knockout rats with Δ7 mutation. AFB1-NAC in urine and AFB1-Lys in serum of Nrf2 knockout rats with Δ7 mutation did not show significant differences from those of wild-type rats. The CDDO-Im pretreatment did not affect levels of these 3 metabolites, demonstrating that the protective alterations in the detoxication of AFB1 seen in wild-type rats do not occur in the Nrf2 knockout rats.
Nrf2 Knockout Rats Are Sensitive to AFB1 Toxicity
Our analyses of rats with a single AFB1 injection revealed that Nrf2 is critical for protection against the acute toxicity of AFB1. To ascertain this point further, we conducted subchronic injections of AFB1 to the Nrf2 knockout rats. The protocol of this experiment is shown in Figure 7A. Nrf2 knockout rats with Δ7 mutation did not gain body weight during the periods of administration of AFB1 (5 days in each week), but did gain weight during the 2-day dosing holiday and after AFB1 dosing stopped (Figure 7B). Co-treatment of CDDO-Im did not affect the ratios of body weight gain of both the AFB1-treated wild-type and Nrf2 knockout rats.
Strikingly, more than a half of the Nrf2 knockout rats (5/8 rats; 62.5%) died following AFB1 treatment (Figure 7C). Concomitant treatment of CDDO-Im did not protect the Nrf2 knockout rats against the toxicity of AFB1 (4/6 rats; 66.7%). The dose of AFB1 employed was not toxic for the wild-type rats. Body weights of the surviving Nrf2 knockout rats were almost similar to those of the wild-type rats. Based on the results shown in Figure 6 (metabolism) and Figure 7 (survival), we conclude that Nrf2 knockout rats are highly sensitive to AFB1 toxicity due to impaired capacity for AFB1 detoxication.
DISCUSSION
Nrf2 has been emerging as a key transcription factor that regulates expression of genes of cytoprotective enzymes. Especially in the field of toxicology, Nrf2 knockout mice have been serving as an excellent model system to examine physiological and pathological regulation of enzymes involved in detoxication pathways (Itoh et al., 1997; Taguchi et al., 2011). Genetic activation of Nrf2 by Keap1 knockout or knockdown gives rise to an elaborate counterpoint to the Nrf2 knockout mice (Okawa et al., 2006; Taguchi et al., 2010; Wakabayashi et al., 2003). Aberrant Nrf2 activation caused by somatic mutations of Keap1 or Nrf2 genes in a variety of cancers (Padmanabhan et al., 2006; Singh et al., 2006) appears to be linked to emergence of resistant foci toward insults of toxic chemicals (Solt et al., 1977). In this study, we established Nrf2 knockout rats as an additional, more facile model system for toxicological studies. We have generated and characterized 2 lines of Nrf2 knockout rats, and found that the Nrf2 knockout rats share many common phenotypes with the Nrf2 knockout mouse. Using the Nrf2 knockout rats, we examined a protective role that Nrf2 plays against toxic chemicals. These Nrf2 knockout rats broaden the opportunities for molecular toxicology in the species Rattus.
As an experimental model animal, rats have provided historically a number of important physiological, pathological, or toxicological insights. For instance, a number of naturally occurring mutant rats related to diseases are available; Dahl rats for susceptibility (S rat) or resistance (R rat) to salt-induced hypertension (Dahl et al., 1962), Goto-Kakizaki rat for type 2 diabetes (Goto et al., 1975), HRSP rats for stroke-prone spontaneous hypertension (Yamori et al., 1976), Tremor rat for Canavan disease (Yamada et al., 1985), and Zitter rat for spongiform encephalopathy (Kondo et al., 1993). Rats have been considered a better model animal than mice for toxicological studies because of size, ease of handling, lower background rates of neoplasia, and other intrinsic factors. Notably, their metabolic pathways appear to share much more similarity to humans than do those of mice. However, one problem that places the rat system behind of the mouse system for molecular toxicology studies is the difficulty to prepare ES cells, which are essential for gene targeting. Until very recently methods to derive and propagate rat ES cells were not available (Li et al., 2008), so that engineering for genome modified rats was not possible. A striking breakthrough was the recent introduction of genome editing technologies, including ZFN, TALEN and CRISPR/Cas, which assure deletion of rat genes or precision knock-in of transgenes at specific sites in the rat genome to produce conditional knockout rats without the use of ES cells (Mashimo et al., 2010).
As systematic or conditional knockout mice of both Nrf2 and Keap1 have been generated, the mouse system has been served as a powerful analytical tool to examine the Keap1-Nrf2 system in vivo. This situation will be sustained into the future. However, some of the limitations of the mouse system will be overcome by the use of other animals, and rats appear to be the front-runner choice. In the toxicological field, aryl hydrocarbon receptor (AhR) (Fujii-Kuriyama and Mimura, 2005) and Nrf2 are 2 important transcription factors that regulate expression of the phase I and phase II detoxication enzymes, respectively. Recently, AhR knockout line of rats was reported by using Sprague Dawley (SD) outbred background using ZFN technology (Harrill et al., 2013). Quite recently, as an independent attempt from this study, Nrf2 knockout line of rats has been generated in SD background using TALEN technology for analysis of the pathophysiology of hypertension (Priestley et al., 2015). Our Nrf2 knockout lines of rats use the F344 strain for which there is a wealth of published toxicological information and foresee these animals as useful tools in toxicological research, especially as a new model animal for Keap1-Nrf2 studies. In Figure 3, we listed the rat genes that were induced by CDDO-Im in wild-type F344 rats, but where the induction was blunted by the Nrf2 mutations. Many of such Nrf2-dependent genes identified in rats were found to be responsible for detoxication and anti-oxidative responses. However, the profile of the Nrf2-dependent genes is fairly divergent when compared with that of mice, conforming the previous observation that the detoxification genes in these species are substantially different (Wild and Turner, 2002).
As a prelude to the broader use of these animals in toxicological research, we have characterized the effect of Nrf2 disruption on the toxicity and disposition of AFB1. Rats, unlike mice, are sensitive to the carcinogenic actions of this mycotoxin. However, there have been limited opportunities to use genetically engineered animals to probe the key determinants of sensitivity or resistance. AFB1 exerts carcinogenic effects through its bioactivation. As presented in Figure 5A, the reactive metabolite AFB1-8,9-epoxide binds to DNA to form AFB1-N7-guanine. Therefore, AFB1-N7-guanine and AFB1-Lys in albumin are biomarkers of AFB1 toxicity. On the other hand, GSTs and AKRs are essential for the detoxication of AFB1 metabolites. A transgenic rat harboring AKR7A1 (human AKR7A3), which enhances detoxication of a reactive metabolite AFB1-dialdehyde, was utilized to examine protection against acute and chronic AFB1 toxicity (Roebuck et al., 2009). Overexpression of AKR7A3 increased formation of AFB1-alcohols in liver and urine. However, the AKR7A3 transgenic rats appeared not to protect against the formation of GST-P-positive preneoplastic foci upon chronic exposure to AFB1, implying that the prevention of protein-adduct formation mediated by AKR was not a critical process for protection against AFB1 tumorigenicity. Therefore, the results of AKR7A3 transgenic mice indicate by inference that a glutathione conjugation of AFB1-8,9-epoxide by GSTs is a primary detoxication reaction.
A Gsta3 knockout mouse was utilized to examine whether glutathione conjugation is essential for detoxication of AFB1 (Kensler et al., 2014). As expected, hepatic AFB1-N7-guanine level and urinary excretion of AFB1-N7-guanine were both elevated in the Gsta3 knockout mice compared with those of wild-type mice. In contrast, urinary excretion of AFB1-NAC was much lower in Gsta3 knockout mouse than in wild-type mice. In addition, Nrf2 activation by CDDO-Im administration or Keap1 deletion did not rescue Gsta3 knockout mouse from the genotoxicity. These results indicate that GSTA3 is responsible for the detoxication of AFB1 in mice. Cross-species comparisons indicate that humans and rats form similar levels of AFB1-Lys in albumin upon AFB1 exposure, but in contrast mice and hamsters form far less AFB1-Lys (Wild et al., 1996). In addition to the intrinsic resistance to the hepatocarcinogenicity of AFB1, these wide ranging results suggest that mouse may not be a suitable animal model for the study of AFB1 toxicity.
It is intriguing to note the recent finding using the “Solt-Farber” protocol for induction of hepatocellular carcinoma in rats (Zavattari et al., 2015). This protocol utilizes a combination of diethyl-nitrosamine and 2-acetaminofluorene, followed by a partial hepatectomy (Solt et al., 1977). In the Solt-Farber model, somatic mutations in either Nrf2 or Keap1 genes are found in 71% of GST-P-positive early preneoplastic lesions (Zavattari et al., 2015). Missense mutations of Nrf2 are more frequent than those of Keap1. Importantly, the Nrf2 somatic mutations are located in DLG and ETGE motifs, which are 2 independent Keap1-binding motifs of Nrf2, consistent with the observations in lung and esophagus cancers (Fukutomi et al., 2014). These observations suggest that Nrf2 is critical for the progression and development of hepatocellular carcinomas in the Solt-Farber model. In fact, Gstp is an Nrf2 target gene, and its product GST-P has been shown as a representative marker for the preneoplastic lesions, both in this model and following treatment with AFB1. However, there remain many unsolved questions in regards to the Nrf2 contribution to hepatocellular carcinogenesis. For instance, how Nrf2 and Keap1 acquire such frequent somatic mutations in the preneoplastic lesions of Solt-Farber model and other hepatocarcinogenic protocols or which oncogenes or anticancer genes are responsible for carcinogenesis under Nrf2 regulation are not known. We believe that the Nrf2 knockout rat may be a very powerful tool for the elucidation of these issues.
In conclusion, in this study we have generated Nrf2 knockout rats by means of a genome editing technology. We report here that the Nrf2 knockout rat is a useful animal model to evaluate the roles that Nrf2 plays in modulating AFB1 detoxication and toxicity and will have broad application to molecular toxicology studies with many agents and processes of interest to the field.
ACKNOWLEDGMENTS
The authors thank Professor John Hayes (Dundee University, United Kingdom) for anti-GSTA3 antibodies and precious advice, Mochida Pharmaceutical Co. Ltd. for generous supply of CDDO-Im, and the Biomedical Research Core of Tohoku University Graduate School of Medicine for technical support.
FUNDING
MEXT/JSPS KAKENHI (24249015, 26111002, and 15H02507 to M.Y., 24790307, 25117703, and 26460384 to K.T.); AMED-CREST, AMED (to M.Y.); MEXT [a research program of the Project for Development of Innovative Research on Cancer Therapeutics (P-Direct)]; the Mitsubishi Foundation, and the Takeda Science Foundation (to M.Y.); the Gushinkai Foundation and Gonryo Medical Foundation (to K.T.); the Naito Foundation (to K.T. and M.Y.); the U.S. National Institutes of Health R35 CA197222 (to T.W.K.).
REFERENCES
- Dahl L. K., Heine M., Tassinari L. (1962). Effects of chronic excess salt ingestion - evidence that genetic factors play an important role in susceptibility to experimental hypertension. J. Exp. Med. 115, 1173–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djordjevic J., Djordjevic A., Adzic M., Mitic M., Lukic I., Radojcic M. B. (2015). Alterations in the Nrf2-Keap1 signaling pathway and its downstream target genes in rat brain under stress. Brain Res. 1602, 20–31. [DOI] [PubMed] [Google Scholar]
- Egner P. A., Groopman J. D., Wang J. S., Kensler T. W., Friesen M. D. (2006). Quantification of aflatoxin-B1-N7-Guanine in human urine by high-performance liquid chromatography and isotope dilution tandem mass spectrometry. Chem. Res. Toxicol. 19, 1191–1195. [DOI] [PubMed] [Google Scholar]
- Egner P. A., Yu X., Johnson J. K., Nathasingh C. K., Groopman J. D., Kensler T. W., Roebuck B. D. (2003). Identification of aflatoxin M1-N7-guanine in liver and urine of tree shrews and rats following administration of aflatoxin B1. Chem. Res. Toxicol. 16, 1174–1180. [DOI] [PubMed] [Google Scholar]
- Espejo R. T., Feijoo C. G., Romero J., Vasquez M. (1998). PAGE analysis of the heteroduplexes formed between PCR-amplified 16S rRNA genes: Estimation of sequence similarity and rDNA complexity. Microbiol-Uk 144, 1611–1617. [DOI] [PubMed] [Google Scholar]
- Fujii-Kuriyama Y., Mimura J. (2005). Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem. Bioph. Res. Commun. 338, 311–317. [DOI] [PubMed] [Google Scholar]
- Fukutomi T., Takagi K., Mizushima T., Ohuchi N., Yamamoto M. (2014). Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell. Biol. 34, 832–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y., Kakizaki M., Masaki N. (1975). Spontaneous diabetes produced by selective breeding of normal wistar rats. Proc. Jpn. Acad. 51, 80–85. [Google Scholar]
- Harrill J. A., Hukkanen R. R., Lawson M., Martin G., Gilger B., Soldatow V., LeCluyse E. L., Budinsky R. A., Rowlands J. C., Thomas R. S. (2013). Knockout of the aryl hydrocarbon receptor results in distinct hepatic and renal phenotypes in rats and mice. Toxicol. Appl. Pharm. 272, 503–518. [DOI] [PubMed] [Google Scholar]
- Hayashi H., Shimamoto K., Taniai E., Ishii Y., Morita R., Suzuki K., Shibutani M., Mitsumori K. (2012). Liver tumor promoting effect of omeprazole in rats and its possible mechanism of action. J. Toxicol. Sci. 37, 491–501. [DOI] [PubMed] [Google Scholar]
- Hirotsu Y., Katsuoka F., Funayama R., Nagashima T., Nishida Y., Nakayama K., Engel J. D., Yamamoto M. (2012). Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 40, 10228–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., et al. (1997). An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236, 313–322. [DOI] [PubMed] [Google Scholar]
- Jacob H. J. (1999). Functional genomics and rat models. Genome Res. 9, 1013–1016. [DOI] [PubMed] [Google Scholar]
- Johnson N. M., Egner P. A., Baxter V. K., Sporn M. B., Wible R. S., Sutter T. R., Groopman J. D., Kensler T. W., Roebuck B. D. (2014). Complete protection against aflatoxin B(1)-induced liver cancer with a triterpenoid: DNA adduct dosimetry, molecular signature, and genotoxicity threshold. Cancer Prev. Res. (Phila) 7, 658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler K. H., Slocum S. L., Chartoumpekis D. V., Dolan P. M., Johnson N. M., Ilic Z., Crawford D. R., Sell S., Groopman J. D., Kensler T. W., and., et al. (2014). Genetic or pharmacologic activation of nrf2 signaling fails to protect against aflatoxin genotoxicity in hypersensitive gsta3 knockout mice. Toxicol. Sci. 139, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler T. W., Egner P. A., Trush M. A., Bueding E., Groopman J. D. (1985). Modification of aflatoxin B1 binding to DNA in vivo in rats fed phenolic antioxidants, ethoxyquin and a dithiothione. Carcinogenesis 6, 759–763. [DOI] [PubMed] [Google Scholar]
- Kensler T. W., Roebuck B. D., Wogan G. N., Groopman J. D. (2011). Aflatoxin: a 50-year odyssey of mechanistic and translational toxicology. Toxicol. Sci. 120 Suppl 1, S28–S48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A., Kang M. I., Okawa H., Ohtsuji M., Zenke Y., Chiba T., Igarashi K., Yamamoto M. (2004). Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo A., Sendoh S., Takamatsu J., Nagara H. (1993). The zitter rat: membranous abnormality in the Schwann cells of myelinated nerve fibers. Brain Res. 613, 173–179. [DOI] [PubMed] [Google Scholar]
- Li P., Tong C., Mehrian-Shai R., Jia L., Wu N., Yan Y., Maxson R. E., Schulze E. N., Song H., Hsieh C. L., et al. (2008). Germline competent embryonic stem cells derived from rat blastocysts. Cell 135, 1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat L. E. (2004). Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell. Biol. 5, 89–99. [DOI] [PubMed] [Google Scholar]
- Maruyama A., Tsukamoto S., Nishikawa K., Yoshida A., Harada N., Motojima K., Ishii T., Nakane A., Yamamoto M., Itoh K. (2008). Nrf2 regulates the alternative first exons of CD36 in macrophages through specific antioxidant response elements. Arch. Biochem. Biophys. 477, 139–145. [DOI] [PubMed] [Google Scholar]
- Mashimo T. (2014). Gene targeting technologies in rats: zinc finger nucleases, transcription activator-like effector nucleases, and clustered regularly interspaced short palindromic repeats. Dev. Growth Differ. 56, 46–52. [DOI] [PubMed] [Google Scholar]
- Mashimo T., Takizawa A., Voigt B., Yoshimi K., Hiai H., Kuramoto T., Serikawa T. (2010). Generation of knockout rats with X-Linked severe combined immunodeficiency (X-SCID) using zinc-finger nucleases. PLoS One 5, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mclellan L. I., Judah D. J., Neal G. E., Hayes J. D. (1994). Regulation of aflatoxin B-1-metabolizing aldehyde reductase and glutathione-S-transferase by chemoprotectors. Biochem. J. 300, 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick B. A., Auerbach S. S., Stockton P. S., Foley J. F., Malarkey D. E., Sills R. C., Irwin R. D., Tice R. R. (2012a). Testing an aflatoxin B1 gene signature in rat archival tissues. Chem. Res. Toxicol. 25, 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick B. A., Auerbach S. S., Stockton P. S., Foley J. F., Malarkey D. E., Sills R. C., Irwin R. D., Ticet R. R. (2012b). Testing an aflatoxin B1 gene signature in rat archival tissues. Chem. Res. Toxicol. 25, 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuishi Y., Taguchi K., Kawatani Y., Shibata T., Nukiwa T., Aburatani H., Yamamoto M., Motohashi H. (2012). Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79. [DOI] [PubMed] [Google Scholar]
- Ohta T., Iijima K., Miyamoto M., Nakahara I., Tanaka H., Ohtsuji M., Suzuki T., Kobayashi A., Yokota J., Sakiyama T., et al. (2008). Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 68, 1303–1309. [DOI] [PubMed] [Google Scholar]
- Okawa H., Motohashi H., Kobayashi A., Aburatani H., Kensler T. W., Yamamoto M. (2006). Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 339, 79–88. [DOI] [PubMed] [Google Scholar]
- Padmanabhan B., Tong K. I., Ohta T., Nakamura Y., Scharlock M., Ohtsuji M., Kang M. I., Kobayashi A., Yokoyama S., Yamamoto M. (2006). Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell. 21, 689–700. [DOI] [PubMed] [Google Scholar]
- Priestley J. R., Kautenburg K. E., Casati M. C., Endres B. T., Geurts A. M., Lombard J. H. (2015). The Nrf2 knockout rat: a new animal model to study ndothelial dysfunction, oxidant stress, and microvascular rarefaction. Am. J. Physiol. Heart Circ. Physiol. 310, H478-87 ajpheart 00586 02015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roebuck B. D., Johnson D. N., Sutter C. H., Egner P. A., Scholl P. F., Friesen M. D., Baumgartner K. J., Ware N. M., Bodreddigari S., Groopman J. D., et al. (2009). Transgenic expression of aflatoxin aldehyde reductase (AKR7A1) modulates aflatoxin B-1 metabolism but not hepatic carcinogenesis in the rat. Toxicol. Sci. 109, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl P. F., McCoy L., Kensler T. W., Groopman J. D. (2006). Quantitative analysis and chronic dosimetry of the aflatoxin B1 plasma albumin adduct Lys-AFB1 in rats by isotope dilution mass spectrometry. Chem. Res. Toxicol. 19, 44–49. [DOI] [PubMed] [Google Scholar]
- Singh A., Misra V., Thimmulappa R. K., Lee H., Ames S., Hoque M. O., Herman J. G., Baylin S. B., Sidransky D., Gabrielson E., et al. (2006). Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 3, e420.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoko J. J., Wakabayashi N., Noda K., Kimura S., Tobita K., Shigemura N., Tsujita T., Yamamoto M., Kensler T. W. (2014). Loss of Nrf2 in mice evokes a congenital intrahepatic shunt that alters hepatic oxygen and protein expression gradients and toxicity. Toxicol. Sci. 141, 112–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt D. B., Medline A., Farber E. (1977). Rapid emergence of carcinogen-induced hyperplastic lesions in a new model for the sequential analysis of liver carcinogenesis. Am. J. Pathol. 88, 595–618. [PMC free article] [PubMed] [Google Scholar]
- Suh J. H., Shenvi S. V., Dixon B. M., Liu H., Jaiswal A. K., Liu R. M., Hagen T. M. (2004). Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA. 101, 3381–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Shibata T., Takaya K., Shiraishi K., Kohno T., Kuitoh H., Tsuta K., Furuta K., Goto K., Hosoda F., et al. (2013). Regulatory nexus of synthesis and degradation deciphers cellular Nrf2 expression levels. Mol. Cell. Biol. 33, 2402–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K., Hirano I., Itoh T., Tanaka M., Miyajima A., Suzuki A., Motohashi H., Yamamoto M. (2014). Nrf2 enhances cholangiocyte expansion in Pten-deficient livers. Mol. Cell. Biol. 34, 900–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K., Maher J. M., Suzuki T., Kawatani Y., Motohashi H., Yamamoto M. (2010). Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol. Cell. Biol. 30, 3016–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K., Motohashi H., Yamamoto M. (2011). Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 16, 123–140. [DOI] [PubMed] [Google Scholar]
- Taguchi K., Yamamoto M. (2015). Keap1-Nrf2 regulatory system and cancer, Chapter 17. In Inoue J and Takekawa M. Protein modifications in pathogenic dysregulation of signaling. Springer Japan, 269-285. [Google Scholar]
- Wakabayashi N., Itoh K., Wakabayashi J., Motohashi H., Noda S., Takahashi S., Imakado S., Kotsuji T., Otsuka F., Roop D. R., et al. (2003). Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 35, 238–245. [DOI] [PubMed] [Google Scholar]
- Wang X. J., Sun Z., Villeneuve N. F., Zhang S., Zhao F., Li Y., Chen W., Yi X., Zheng W., Wondrak G. T., et al. (2008). Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29, 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watai Y., Kobayashi A., Nagase H., Mizukami M., McEvoy J., Singer J. D., Itoh K., Yamamoto M. (2007). Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells 12, 1163–1178. [DOI] [PubMed] [Google Scholar]
- Wild C. P., Hasegawa R., Barraud L., Chutimataewin S., Chapot B., Ito N., Montesano R. (1996). Aflatoxin albumin adducts: a basis for comparative carcinogenesis between animals and humans. Cancer Epidemiol. Biomarkers Prev . 5, 179-189. [PubMed] [Google Scholar]
- Wild C. P., Turner P. C. (2002). The toxicology of aflatoxins as a basis for public health decisions. Mutagenesis 17, 471–481. [DOI] [PubMed] [Google Scholar]
- Yamada J., Serikawa T., Ishiko J., Inui T., Takada H., Kawai Y., Okaniwa A. (1985). Rats with congenital tremor and curled whiskers and hair. Jikken Dobutsu 34, 183–188. [DOI] [PubMed] [Google Scholar]
- Yamashita Y., Ueyama T., Nishi T., Yamamoto Y., Kawakoshi A., Sunami S., Iguchi M., Tamai H., Ueda K., Ito T., et al. (2014). Nrf2-inducing anti-oxidation stress response in the rat liver–new beneficial effect of lansoprazole. PLoS One 9, e97419.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamori Y., Horie R., Handa H., Sato M., Fukase M. (1976). Pathogenetic similarity of strokes in stroke-prone spontaneously hypertensive rats and humans. Stroke 7, 46–53. [DOI] [PubMed] [Google Scholar]
- Yanagawa T., Itoh K., Uwayama J., Shibata Y., Yamaguchi A., Sano T., Ishii T., Yoshida H., Yamamoto M. (2004). Nrf2 deficiency causes tooth decolourization due to iron transport disorder in enamel organ. Genes Cells 9, 641–651. [DOI] [PubMed] [Google Scholar]
- Yates M. S., Kwak M. K., Egner P. A., Groopman J. D., Bodreddigari S., Sutter T. R., Baumgartner K. J., Roebuck B. D., Liby K. T., Yore M. M., et al. (2006). Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole. Cancer Res. 66, 2488–2494. [DOI] [PubMed] [Google Scholar]
- Yeligar S. M., Machida K., Kalra V. K. (2010). Ethanol-induced HO-1 and NQO1 are differentially regulated by HIF-1alpha and Nrf2 to attenuate inflammatory cytokine expression. J. Biol. Chem. 285, 35359–35373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavattari P., Perra A., Menegon S., Kowalik M. A., Petrelli A., Angioni M. M., Follenzi A., Quagliata L., Ledda-Columbano G. M., Terracciano L., et al. (2015). Nrf2, but not beta-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology 62, 851–862. [DOI] [PubMed] [Google Scholar]
- Zhang P., Singh A., Yegnasubramanian S., Esopi D., Kombairaju P., Bodas M., Wu H., Bova S. G., Biswal S. (2010). Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol. Cancer Ther. 9, 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. K., Wu K. C., Klaassen C. D. (2013). Genetic activation of Nrf2 protects against fasting-induced oxidative stress in livers of mice. PLoS One 8, e59122.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., Xu Y., Yu S., Lu L., Ding M., Cheng J., Song G., Gao X., Yao L., Fan D., et al. (2014). An efficient genotyping method for genome-modified animals and human cells generated with CRISPR/Cas9 system. Sci. Rep. 4, 6420.. [DOI] [PMC free article] [PubMed] [Google Scholar]