

Abstract
Background & Aims
Interactions between C-C chemokine receptor types 2 (CCR2) and 5 (CCR5) and their ligands, including CCL2 and CCL5, mediate fibrogenesis by promoting monocyte/macrophage recruitment and tissue infiltration, as well as hepatic stellate cell activation. Cenicriviroc (CVC) is an oral, dual CCR2/CCR5 antagonist with nanomolar potency against both receptors. CVC’s anti-inflammatory and antifibrotic effects were evaluated in a range of preclinical models of inflammation and fibrosis.
Methods
Monocyte/macrophage recruitment was assessed in vivo in a mouse model of thioglycollate-induced peritonitis. CCL2-induced chemotaxis was evaluated ex vivo on mouse monocytes. CVC’s antifibrotic effects were evaluated in a thioacetamide-induced rat model of liver fibrosis and mouse models of diet-induced non-alcoholic steatohepatitis (NASH) and renal fibrosis. Study assessments included body and liver/kidney weight, liver function test, liver/kidney morphology and collagen deposition, fibrogenic gene and protein expression, and pharmacokinetic analyses.
Results
CVC significantly reduced monocyte/macrophage recruitment in vivo at doses ≥20 mg/kg/day (p < 0.05). At these doses, CVC showed antifibrotic effects, with significant reductions in collagen deposition (p < 0.05), and collagen type 1 protein and mRNA expression across the three animal models of fibrosis. In the NASH model, CVC significantly reduced the non-alcoholic fatty liver disease activity score (p < 0.05 vs. controls). CVC treatment had no notable effect on body or liver/kidney weight.
Conclusions
CVC displayed potent anti-inflammatory and antifibrotic activity in a range of animal fibrosis models, supporting human testing for fibrotic diseases. Further experimental studies are needed to clarify the underlying mechanisms of CVC’s antifibrotic effects. A Phase 2b study in adults with NASH and liver fibrosis is fully enrolled (CENTAUR Study 652-2-203; NCT02217475).
Introduction
Fibrosis results from a sustained inflammatory response to chronic organ injury and is characterized by the deposition of extracellular matrix proteins, including collagen types 1 and 3 [1]. Hepatic fibrosis is associated with chronic liver disease, a significant global burden that contributes to cirrhosis and hepatocellular carcinoma [2]. Likewise, renal fibrosis is a common manifestation of chronic kidney disease [3], associated with elevated risks of morbidity and mortality [4]. Effective, well-tolerated antifibrotic and anti-inflammatory pharmacotherapies that can be integrated into current disease-management approaches are urgently needed.
The inflammatory response to hepatocyte injury plays a key role in hepatic fibrogenesis and involves recruitment of bone marrow-derived monocytes and macrophages to the site of injury, which is triggered by the activation of resident macrophages (i.e. Kupffer cells [KCs]) [1]. In turn, infiltrating monocytes/macrophages amplify this immune response by producing inflammatory cytokines and chemokines, which further promote recruitment of inflammatory cells and upregulate the activation of hepatic stellate cells (HSCs) [1,5]. Fibrogenic cytokines (e.g. transforming growth factor-beta [TGF-beta]), produced by activated macrophages, promote transdifferentiation of HSCs into myofibroblasts, which are the primary source of scar-forming matrix proteins, including fibrillary collagen types 1 and 3, and the contractile protein alpha-smooth muscle actin (alpha-SMA) [1,6–8].
Recruitment of extra-hepatic inflammatory cells to the site of hepatic injury is largely mediated by interactions between chemokines and their receptors. Monocytes, KCs and HSCs can express C-C chemokine receptor types 2 (CCR2) and 5 (CCR5) on their surface [9–12]. Increasing evidence implicates CCR2/CCR5 and their ligands (including C-C chemokine ligand type 2 [CCL2, aka monocyte chemotactic protein-1 (MCP-1)] and type 5 [CCL5, aka Regulated on Activation, Normal T-cell Expressed and Secreted (RANTES)]), in the pathogenesis of liver fibrosis through promotion of monocyte/macrophage recruitment and tissue infiltration, and HSC activation following liver injury [9–15]. Hepatocytes, KCs and infiltrating monocytes/macrophages are the main sources of TGF-beta, a major fibrogenic cytokine promoting collagen production by activated HSCs [12]. Additional evidence substantiates the roles of CCR2/CCR5 and their ligands in renal fibrosis [16–20]. Thus, CCR2 and CCR5 have become attractive targets for antifibrotic therapy.
Cenicriviroc (CVC) is a novel, oral, once-daily (QD) dual CCR2/CCR5 antagonist with nanomolar potency, and a long plasma half-life (30–40 hours in humans) [21–24]. It has a favorable safety profile and was well tolerated in approximately 600 subjects, including those with mild or moderate hepatic impairment (Child–Pugh A and B) [25,26]. CVC is currently under evaluation in a Phase 2b study in 289 adults with non-alcoholic steatohepatitis (NASH) and liver fibrosis, for which it received Fast Track Designation by the Food and Drug Administration (CENTAUR Study 652-2-203; NCT02217475) [27]. An ex vivo study of human peripheral blood mononuclear cells found that CVC leads to receptor occupancies of ~98% for CCR2 on monocytes (at 6 nmol/L) and ≥90% for CCR5 on CD4+ and CD8+ T-cells (at 3.1 and 2.3 nmol/L, respectively) [28]. As a shorter half-life (~2 hours in mice) and a lower potency have been observed for CVC in rodents versus humans, this was considered in dose selection for disease models. An ex vivo study conducted on mouse monocytes and macrophages showed that CVC concentrations of 250 nmol/L or higher achieve >87% CCR2/CCR5 occupancy in these cells [29,30]. Collectively, these findings suggest that rodent models are well suited to evaluate the anti-inflammatory and antifibrotic properties of CVC, resulting from effective CCR2/CCR5 blockade.
A number of in vitro and in vivo models of fibrosis are commonly used to assess recruitment of inflammatory cells and antifibrotic activity of therapeutic agents [31–33]. Multiple models of fibrosis allow assessment of the broad effect of an antifibrotic agent across species and organs, and reduce the likelihood that efficacy is restricted to one model. Here we provide evidence for the antifibrotic effects of CVC, as demonstrated in models that have evaluated: (1) the ex vivo and in vivo effects of CVC on recruitment/migration of monocytes/macrophages; and (2) the in vivo antifibrotic effects of CVC in liver and kidney fibrosis.
Materials and Methods
All animal procedures were approved by each institution’s animal care and use committee (IACUC), and were conducted in accordance with national guidelines. CVC is cenicriviroc mesylate, provided by Tobira Therapeutics, Inc., USA. The vehicle control used in all in vivo studies was 0.5% [w/v] methylcellulose + 1% Tween®-80 (pH ~1.3).
Effect of CVC on recruitment/migration of monocytes/macrophages
In vivo mouse model of peritonitis
A murine thioglycollate (TG)-induced model of peritonitis, where acute inflammation induced by intraperitoneal (IP) injection of TG results in a rapid increase in monocyte/macrophage migration into the peritoneal cavity [34], was employed to assess the effects of CVC on cell recruitment in vivo. The protocol was approved by the IACUC of Charles River Laboratories Preclinical Services, Montreal (PCS-MTL). The care and use of animals was conducted in accordance with the guidelines of the US National Research Council and the Canadian Council on Animal Care.
Male C57BL/6 mice (n = 44; 8–10 weeks of age; Charles River Laboratories, Canada) were allocated to receive treatments via oral gavage (PO) on Days 1–5 in the following groups: non-disease control, vehicle control twice daily (BID), CVC 5 mg/kg/day (CVC5) BID, CVC 20 mg/kg/day (CVC20) BID, CVC 100 mg/kg/day (CVC100) BID, CVC20 QD, and positive control dexamethasone (corticosteroid known to reduce inflammation in a variety of animal models) 1 mg/kg QD (S1 Table). On Day 4, peritonitis was induced via IP injection of TG 3.85% (1 mL/animal) 2 hours post-dose in all groups except non-disease controls. Study endpoints included: peritoneal lavage cell counts and pharmacokinetic (PK) evaluation. Animals were sacrificed 48 hours post-TG injection by isoflurane inhalation, and peritoneal lavage and blood samples (0.7 mL) were collected. Differential cell counts were assessed in peritoneal lavage samples using an Advia® Hematology System (Siemens Healthcare Diagnostics, USA) with multispecies software and an analysis software designed for mouse peritoneal fluid on Advia® 120 (LabThruPut, USA). A 0.3 mL aliquot of the blood sample was processed to plasma for PK analysis.
Ex vivo migration of mouse monocytes
The protocol was approved by the IACUC of the University of Pennsylvania (protocol number 804755) and animals were maintained according to the National Institutes of Health (NIH) guidelines. Animals were euthanized by CO2 inhalation followed by cervical dislocation.
Mouse monocyte migration in response to CVC treatment was assessed ex vivo in triplicate. TG was injected intraperitoneally into male C57BL/6 mice (n = 3; 8–10 weeks of age; Jackson Laboratory, USA) and activated macrophages were collected 48 hours later by peritoneal lavage. Chemotaxis was assayed using a Transwell® Chamber (Costar, USA) with a 5 μm-pore size polycarbonate filter, as previously described [35]. Briefly, cells were incubated for 2 hours in the presence of 1 nM CCL2 and/or 1 μM CVC (dissolved in dimethyl sulfoxide with 0.5% acetic acid and diluted 1:1000 with serum-free Roswell Park Memorial Institute-1640 medium and 0.5% bovine serum albumin). Cells were harvested from the lower compartment and analyzed by flow cytometry to enumerate F4/80+CD11b+ macrophages using a 3-laser BD FACSCanto™ (BD Biosciences, Canada). Results were analyzed using FlowJo software (Tree Star Inc., USA).
Antifibrotic effects of CVC in animal models of fibrosis
Rat model of thioacetamide (TAA)-induced liver fibrosis (TAA model)
The TAA model is commonly used for the evaluation of treatment at various stages of disease, from inflammation to cirrhosis [36]. The protocol was approved by the Mount Sinai IACUC (approval number: LA12-00318) and animals were maintained according to the NIH guidelines. Anesthesia was performed with 1–5% isoflurane through inhalation; surgery was terminal.
Using male Sprague-Dawley rats (n = 72, 10–12 weeks of age; Harlan Laboratories, USA), fibrosis was induced by IP administration of TAA at a dose of 150 mg/kg three times per week for 8 weeks. Rats (n = 4–8/group) received vehicle control, CVC 30 mg/kg/day (CVC30) or CVC100 QD PO during Weeks 0–8 (early intervention), Weeks 4–8 (established fibrosis) or Weeks 8–12 (cirrhosis reversal) and were sacrificed at Weeks 8, 8 or 12, respectively (S1 Table). Study endpoints included: body and liver weights, liver biochemistry (e.g. serum alanine and aspartate aminotransferase [ALT/AST]), extracellular matrix protein expression in liver tissue (collagen type 1 and alpha-SMA), mRNA expression (collagen type 1, alpha-SMA, beta-platelet-derived growth factor-beta receptor, TGF beta-receptor, matrix metalloproteinase 2, tissue inhibitor of metalloproteinases 1 [TIMP1] and 2 [TIMP2]) and liver morphology. For liver-function testing, plasma samples were obtained from blood collected from the vena cava. Livers were sectioned and fixed in 3.7% formalin, embedded in paraffin, cut at 4 mm thickness and stained with hematoxylin and eosin (H&E) for histological examination.
Mouse model of diet-induced NASH (NASH model)
The protocol was approved by Stelic IACUC (approval number: RP-131). All animals were housed and cared for in accordance with the Japanese Pharmacological Society Guidelines for Animal Use.
NASH was induced in male C57BL/6 mice (Charles River Laboratories, Japan) via subcutaneous injection of 200 μg of streptozotocin 2 days post-birth (causing mild islet inflammation and islet destruction) plus a high-fat diet (57 kcal% fat) from 4 weeks of age (sequentially causing fatty changes to the liver, NASH and fibrosis) [37]. From Weeks 6 to 9, three groups (n = 9/group) received vehicle control, CVC20 or CVC100 BID PO (S1 Table). At Week 9, six animals per group were sacrificed for assessment of liver fibrosis and Non-Alcoholic Fatty Liver Disease (NAFLD) Activity Score (NAS). The animals were sacrificed by exsanguination through direct cardiac puncture under ether anesthesia. Study endpoints included: body and liver weights, plasma biochemistry (e.g. ALT and CCL2), extracellular matrix protein in liver tissue (hydroxyproline content), mRNA expression (collagen type 1, tumor necrosis factor-alpha, MCP-1, TIMP1) and histopathological analyses. Blood samples were collected and plasma samples derived for biochemistry assessments. Sections were cut from paraffin blocks of liver tissue, prefixed in Bouin’s solution and stained with H&E for histological examination.
Mouse model of unilateral ureter obstruction (UUO)-induced renal fibrosis (UUO model)
Unilateral ureter obstruction is a commonly used experimental model of kidney injury, in which interstitial inflammation, tubular cell injury/death and fibrosis, ensue [32]. The protocol was approved by Plato BioPharma, Inc. IACUC (PBI; approval number 2013–04). The care and use of animals was conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals (2010), Action against Medical Accidents Guidelines on Euthanasia (2007), Office of Laboratory Animal Welfare Institutional Animal Care and Use Committee Guidebook (2002), Public Health Service Policy on Humane Care and Use of Laboratory Animals (2002), PBI standard operating procedures on a) moribund sacrifice, b) vivarium maintenance and animal care, and c) animal handling and dosing. Anesthesia was performed by isoflurane injection; animals were euthanized by diaphragm laceration followed by heart laceration while under isoflurane anesthesia.
Male CD-1 mice (n = 51; 7–8 weeks of age; Charles River Laboratories, USA) were allocated to weight-matched treatment groups on Day -1 (1 day prior to either sham [one group] or permanent right UUO surgery [five groups] via aseptic laparotomy). From Days -1 to 4, mice received phosphate-buffered saline (PBS) IP QD, apart from a positive-control group, which received 1D11 (anti-TGF-beta1 antibody) 3 mg/kg IP QD. From Days 0 to 5, mice received vehicle control (sham surgery, UUO-control and UUO+positive-control groups) or CVC 7 mg/kg/day (CVC7), CVC20 or CVC100 PO BID (S1 Table). The CVC100 group was terminated due to poor clinical condition (6/9 animals died and 3/9 euthanized prior to end of study). Of note, a similar dose of CVC100 administered for 3 weeks was well tolerated in the mouse NASH model; therefore, it is plausible that the laparotomy and UUO surgery in combination with CVC may have contributed to the loss of animals. Study endpoints included: body and kidney weights, mRNA expression (TGF-beta, connective tissue growth factor, MCP-1, alpha-SMA, collagen 1a1, fibronectin-1 and collagen 3a1), extracellular matrix protein in renal cortical tissue (hydroxyproline content) and histological analyses. Blood and tissue samples were collected from anesthetized mice 4 hours post-dose on Day 5, prior to sacrifice. A mid-transverse section of the right obstructed kidney was collected for histological analysis.
Outcome measures in animal models of fibrosis
Body and liver/kidney weights
Body weight (measured before and during treatment) and sacrificed animal body and liver/kidney/spleen weights were recorded.
Plasma biochemistry
In the TAA model, serum ALT/AST levels were measured using VITROS® 5,1 FS (Ortho Clinical Diagnostics, USA), with plasma diluted with VITROS® 7% bovine serum albumin if needed. In the NASH model, plasma ALT levels were measured by FUJI DRI-CHEM 7000 (Fujifilm, Japan) and plasma CCL2 concentration quantified by the mouse CCL2/JE/MCP-1 immunoassay kit (R&D, USA).
Liver or obstructed kidney morphology
Collagen deposition (the extent of fibrosis) was visualized in liver/kidney sections with picrosirius red staining. In the TAA model, collagen quantification was performed using computerized Life Science morphometry system (BIOQUANT, USA) on a total of 36 images per animal at 100x magnification (four picrosirius red-stained slides per animal, with nine images taken randomly per slide).
In the NASH model, bright field images of picrosirius red-stained sections were captured around the central vein using a digital camera (DFC280; Leica, Germany) at 200x magnification; the ‘positive’ areas in five fields/sections were measured using ImageJ software (National Institutes of Health, USA). Perivascular areas were subtracted from the total positive areas for each field (modified fibrosis area). The NAS, a histological tool developed to assess disease severity in humans, was assessed in a blinded fashion and calculated according to Kleiner’s criteria on H&E-stained sections [38]. It is based on the semi-quantification of steatosis, lobular inflammation and hepatocellular ballooning.
In the UUO model, ten images/depth/kidney were assessed in a blinded fashion using Axio Imager.A2 (Zeiss, USA) light microscopy (at 200x magnification to enable 60–70% sampling of renal cortical area) and quantified by a composite Collagen Volume Fraction (CVF [% total area imaged]) score expressed as the average positive stain across three anatomically distinct (200–250 μM apart) tissue sections, or depths, from the obstructed kidney.
Immunohistochemistry
In the NASH model, liver sections fixed in acetone were incubated with anti-F4/80 antibody (BMA Biomedicals, Switzerland) to assess inflammation. Double immunohistochemical analyses were performed with anti-CD206 (RayBiotec, USA) or anti-CD16/32 (BD Biosciences, USA) antibodies. Cells were counted and the M1/M2 polarization ratio was calculated as mean percentage of F4/80+CD16/32+ cells/mean percentage of F4/80+CD206+ cells.
Extracellular matrix proteins
Extracellular matrix protein content in tissue samples was measured by evaluating collagen type 1 and alpha-SMA protein expression in the TAA model and hydroxyproline content in the NASH and UUO models.
In the TAA model, total protein was extracted from liver cells and expression levels of collagen type 1 and alpha-SMA were assessed by western blotting. Protein expression levels were normalized to the reference protein (glyceraldehyde-3-phosphate dehydrogenase [GAPDH] or calnexin).
In the NASH model, frozen liver samples were subjected to an alkaline-acid hydrolysis, then centrifuged, and the supernatant collected. Hydroxyproline content was quantified against a hydroxyproline standard curve, with a BCA protein assay kit (Thermo Fisher Scientific, USA) used to normalize the calculated hydroxyproline values.
In the UUO model, frozen renal cortical tissue biopsies were hydrolyzed and centrifuged, and the supernatant was analyzed by OD absorbance at 560 nM on a SpectraMax® 190 (Molecular Devices, USA). Standard curves for conversion of ODs to concentrations were generated using linear regression and sample concentrations were determined using SoftMax® Pro5 software (Molecular Devices, USA).
Gene expression of fibrotic or inflammatory biomarkers
In the hepatic-fibrosis models, RNA was extracted from liver tissues and purified, and 1 μg of total mRNA was reverse-transcribed into complementary DNA. Expression levels were determined by quantitative polymerase chain reaction (PCR) (iQ™ SYBR® Green Supermix [Bio-Rad Laboratories, USA] on the LightCycler® 480 Real-Time PCR System [Roche, Switzerland]) in the TAA model at Week 8 (early intervention and established fibrosis) and Week 12 (cirrhosis reversal), and by real-time PCR (PCR Dice® and SYBR® Premix Ex Taq™ [Takara Bio Inc., Japan]) at Week 9 in the NASH model.
In the UUO model, mRNA expression levels in renal cortical tissue were evaluated using the QuantiGene® Plex 2.0 profiling platform (Affymetrix, USA).
Relative mRNA expression levels were normalized to the following reference genes: TAA model, GAPDH; NASH model, 36B4 (gene symbol: Rplp0); UUO model, hypoxanthine phosphoribosyltransferase.
PK analysis in the TG-induced model of peritonitis and the UUO model
CVC plasma levels (minimum and maximum) were determined by KCAS Bioanalytical Services, USA, using a validated liquid chromatography-tandem mass spectrometry (LC/MS/MS) plasma method (50 μL assay, range 10.0−1920.0 ng/mL).
Statistical analysis
In all studies, statistical significance level was set as p < 0.05.
Recruitment/migration of monocytes/macrophages
In the peritonitis mouse model, one-way analysis of variance with post-hoc Dunnett’s test was performed, using GraphPad Prism® (GraphPad Software, Inc., USA), on differential cell counts obtained from lavage in treatment groups versus the vehicle-control group. For the ex vivo migration of mouse monocytes, statistical significance was determined by a Student’s t test.
Animal models of fibrosis
Data are presented as ±standard error of the mean (SEM) in the TAA model. Statistical significance was determined by the two-tailed Student’s t test.
In the NASH model, data are expressed as mean ±standard deviation (SD). Statistical analyses were performed using the Bonferroni multiple comparison test (GraphPad Prism® 4 software).
In the UUO model, data are expressed as mean ±SEM. All statistical analyses were performed using GraphPad Prism® 6 software. Unpaired ‘t’-tests were used to analyze treatment differences between control groups (sham-surgery and UUO+positive-control 1D11 each vs. UUO control). One-way analysis of variance with post-hoc Dunnett’s test was used to compare treatment differences between CVC groups and UUO controls.
Results
Effect of CVC on recruitment/migration of monocytes/macrophages
In vivo mouse model of peritonitis
In the TG-induced model of peritonitis, CVC treatment led to dose-related decreases in monocyte/macrophage recruitment, of similar or greater magnitude than those observed with dexamethasone (positive control), and achieving statistical significance at doses ≥20 mg/kg/day (p < 0.05; Fig 1A; S1 Fig). Compared to the vehicle-control group, peritoneal lavage monocyte/macrophage counts were decreased by: 5.7%, 45.2%, 76.5%, 26.0% and 38.1% for CVC5 BID, CVC20 BID, CVC100 BID, CVC20 QD and dexamethasone, respectively. Exposure to CVC was dose-related and correlated with the decrease in monocyte/macrophage recruitment, with CVC appearing to be more effective when given BID versus QD, in line with the higher plasma concentrations achieved with BID dosing and the known short half-life in mice (~2 hours). Compared to dexamethasone, monocyte/macrophage-count decreases were significantly more pronounced with CVC100 BID (62.1% greater reduction, p < 0.001).
Fig 1. CVC Effect on Monocyte/Macrophage Migration.

(A) Monocyte/macrophage recruitment reduced in TG-induced mouse model of peritonitis after pre-treatment with CVC (PO BID or QD dosing); (B) cenicriviroc (1 μM) inhibits CCL2-mediated chemotaxis of activated murine macrophages (F4/80+/CD11b+) ex vivo. *p < 0.05 vs. TG + vehicle control; **p < 0.01 vs. TG + vehicle control; ***p < 0.001 vs. TG + vehicle control; † p < 0.001 vs. TG + DEX; ‡ p = 0.018 vs. vehicle control. aVehicle control: 0.5% [w/v] methylcellulose + 1% Tween®-80 (pH ~1.3). BID, twice daily; CCL2, C-C chemokine ligand 2; CVC, cenicriviroc; CVC5, CVC 5 mg/kg/day; CVC20, CVC 20 mg/kg/day; CVC100, CVC 100 mg/kg/day; DEX, dexamethasone; PO, oral gavage; QD, once daily; SEM, standard error of the mean; TG, thioglycollate.
Ex vivo migration of mouse monocytes
Migration of mouse monocytes in response to CCL2, the most potent mediator of chemotaxis for activated macrophages, was reduced following pre-treatment with CVC at a concentration of 1 μM (Fig 1B). Compared to untreated and unstimulated cells, the average fold change in migrating cells (±SD) was 4.6±0.9 (p < 0.05), 0.8±0.2 (p > 0.05) and 0.7±0.4 (p > 0.05) for CCL2-stimulated cells, CCL2-stimulated cells treated with CVC and unstimulated cells treated with CVC, respectively.
Antifibrotic effects of CVC in animal models of fibrosis
CVC effect on body weight and liver or kidney weight
Overall, no notable effects on body weight and liver or kidney weight were observed following CVC administration in animal models of liver and kidney fibrosis (S2 Table). A slight decrease in body weight was observed in the UUO model (5%, CVC20 vs. UUO control on Day 5, p < 0.05), and in the liver-to-body weight ratio in the TAA model (established fibrosis, CVC30 vs. vehicle control) (S2 Table).
CVC effect on liver function
In the NASH model, plasma ALT levels were significantly decreased with both CVC doses versus vehicle control (p < 0.05; Fig 2). AST levels were not assessed.
Fig 2. Reduction of Plasma ALT Levels in NASH Model.
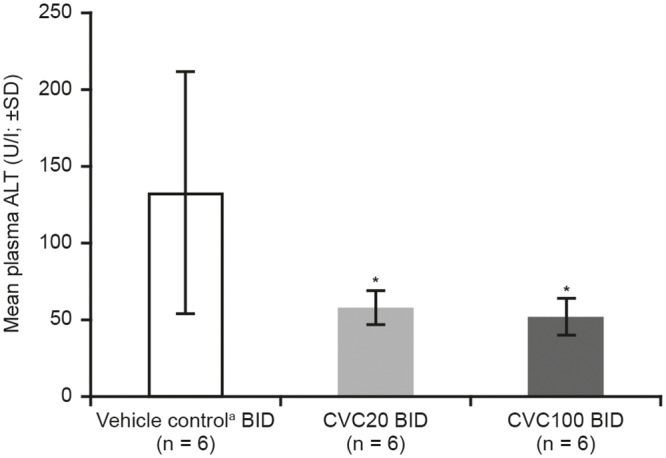
*p < 0.05 vs. vehicle control. aVehicle control: 0.5% [w/v] methylcellulose + 1% Tween®-80 (pH ~1.3). ALT, alanine aminotransferase; BID, twice daily; CVC, cenicriviroc; CVC20, CVC 20 mg/kg/day; CVC100, CVC 100 mg/kg/day; NASH, non-alcoholic steatohepatitis; SD, standard deviation.
In the TAA model, no significant differences in ALT levels were observed between the CVC-treated and vehicle-control groups, whereas AST levels were significantly decreased for CVC30 versus vehicle control (p < 0.05) in the early intervention group (S2 Fig).
CVC effect on plasma CCL2 levels
No significant differences in CCL2 levels measured in the NASH model were observed between the vehicle and CVC20 groups, while a significant increase in plasma CCL2 levels was observed in the CVC100 group compared with the vehicle group (vehicle, 60±4 pg/mL; CVC20, 68±16 pg/mL; CVC100, 91±14 pg/mL).
CVC effect on morphology of liver/obstructed kidney
Liver collagen deposition was reduced following CVC treatment started concurrently with TAA, versus that following vehicle control (48.5% and 37.5% reduction for CVC30 and CVC100, respectively, p < 0.001; Fig 3). When CVC treatment was initiated at the intermediate stage of liver disease (4 weeks after TAA), a significant antifibrotic effect was observed with CVC30 (35.7% reduction vs. vehicle control, p < 0.001), but not CVC100 (Fig 3B). No significant reduction in collagen deposition was observed in the cirrhosis-reversal group when CVC treatment started at Week 8 (S3 Fig). No histologic differences were observed between groups based on examination of H&E-stained liver sections (S4 Fig).
Fig 3. Reduction in Fibrosis.

(A) Representative micrographs of Sirius red-stained liver sections in the rat TAA model (early intervention and established fibrosis; 100x), mouse NASH model (200x) and mouse UUO model (200x). (B) Reduction in collagen deposition in the rat TAA model (early intervention and established fibrosis), mouse NASH modela and mouse UUO modelb. *p < 0.05 vs. sham control; **p < 0.01 vs. vehicle control; ***p < 0.001 vs. vehicle control; †p < 0.05 vs. UUO + vehicle control. aPerivascular area subtracted; bData presented exclude a single outlier from an animal in the CVC20 group, which had a CVF value >2 SDs higher than any other animal in the group. CVF, Collagen Volume Fraction; CVC, cenicriviroc; NASH, non-alcoholic steatohepatitis; SD, standard deviation; SEM, standard error of the mean; TAA, thioacetamide; UUO, unilateral ureteral obstruction.
In the NASH model, collagen deposition in the pericentral region of the liver lobule was markedly reduced in both CVC groups versus vehicle control. Liver sections from the vehicle group exhibited severe micro- and macro-vesicular fat deposition, hepatocellular ballooning and inflammatory cell infiltration (S4 Fig). The CVC20 and CVC100 groups displayed moderate improvements in lobular inflammation and hepatocyte ballooning (S4 Fig), with a significant reduction in NAS versus that in the vehicle group (p < 0.05 and p < 0.01, respectively; Table 1). The percentage of fibrosis area was significantly decreased by CVC treatment versus vehicle control (40.0% and 41.8% reduction for CVC20 and CVC100, respectively, p < 0.01). The modified fibrosis areas (perivascular space subtracted) were also significantly reduced in CVC groups versus those in vehicle controls (52.5% and 67.2% reduction for CVC20 and CVC100, respectively, p < 0.01; Fig 3).
Table 1. Reduction of NAS in NASH Modela.
| Score | Vehicle controlb (n = 6) | CVC20 (n = 6) | CVC100 (n = 6) |
|---|---|---|---|
| Steatosis | |||
| 0 | 0 | 0 | 1 |
| 1 | 4 | 6 | 5 |
| 2 | 2 | 0 | 0 |
| 3 | 0 | 0 | 0 |
| Lobular inflammation | |||
| 0 | 0 | 0 | 0 |
| 1 | 0 | 3 | 3 |
| 2 | 6 | 3 | 3 |
| 3 | 0 | 0 | 0 |
| Hepatocyte ballooning | |||
| 0 | 0 | 0 | 1 |
| 1 | 0 | 3 | 2 |
| 2 | 6 | 3 | 3 |
| NAS, mean (±SD) | 5.3 (±0.5) | 4.0 (±0.6)* | 3.7 (±0.8)** |
*p = 0.0013 vs. vehicle control;
**p = 0.0009 vs. vehicle control.
aInterpretation performed by pathologist blinded by treatment group;
bVehicle control: 0.5% [w/v] methylcellulose + 1% Tween®-80 (pH ~1.3).
CVC, cenicriviroc; CVC20, CVC 20 mg/kg/day; CVC100, CVC 100 mg/kg/day; n, number of animals; NAS, non-alcoholic fatty liver disease activity score; NASH, non-alcoholic steatohepatitis; SD, standard deviation.
Of note, F4/80 immunostaining of liver sections form the vehicle group showed accumulation of F4/80+ cells in the liver lobule, but there were no significant differences in the number and size of F4/80+ cells between the vehicle and either CVC20 or CVC100 group, nor in the percentage of inflammation area (F4/80+ area; vehicle, 4.99±1.10%; CVC20, 4.77±1.02%; CVC-high, 4.96±0.60%; S5 Fig). Additional staining of F4/80+ macrophages with CD16/32 (M1 marker) or CD206 (M2 marker) showed no significant difference in the M1/M2 ratio between the vehicle and the CVC treatment groups (vehicle, 99.6±20.2%; CVC20, 112.3±17.0%; CVC100 125.1±21.9%).
In the UUO model, renal cortical fibrosis, expressed as Collagen Volume Fraction (±SEM), was significantly increased in UUO-obstructed kidneys relative to sham control (11.4-fold±1.0, p < 0.05). 1D11 (positive control) significantly attenuated these UUO-induced increases (50.3%±7.3 reduction vs. UUO control, p < 0.05). Likewise, CVC7 and CVC20 significantly attenuated UUO-induced increases when a single outlier (value >2 SD; 1 animal in CVC20 group) was removed (28.6%±8.8 and 31.8%±6.8 reduction vs. UUO control, respectively, p < 0.05; Fig 3).
CVC effect on extracellular matrix protein content
Compared to vehicle control, protein expression of collagen type 1 in the TAA model was reduced by 29% and 12% for CVC30 and CVC100, respectively, in early intervention, and 30% for CVC30 in established fibrosis. Protein expression of alpha-SMA was reduced following CVC treatment by 17% and 22% for CVC30 and CVC100, respectively, versus vehicle control in early intervention, and 14% for CVC30 in established fibrosis. No reductions were observed in protein expression in the advanced disease cirrhosis group (Fig 4). The differences in collagen type 1 and alpha-SMA protein levels between treatment arms were not statistically significant; however, these results likely reflect the significant decrease of fibrosis observed in histological analyses.
Fig 4. CVC Effects on Extracellular Matrix Protein Content.

Mean expression of (A) collagen type I and (B) alpha-SMA in the TAA model compared to vehicle control; mean expression of hydroxyproline content in (C) NASH model and (D) UUO model. alpha-SMA, alpha-smooth muscle actin; BID, twice daily; CVC, cenicriviroc; NASH, non-alcoholic steatohepatitis; QD, once daily; SD, standard deviation; TAA, thioacetamide; UUO, unilateral ureter obstruction.
In the NASH model, the liver hydroxyproline content tended to decrease in the CVC20 and CVC100 groups compared with the vehicle group (vehicle, 0.75±0.18 μg/mg; CVC20, 0.63±0.05 μg/mg; CVC100, 0.62±0.09 μg/mg; Fig 4).
Compared with sham control, UUO increased hydroxyproline content in obstructed kidneys (1.6±0.2 fold; vehicle control). 1D11 attenuated these increases (37.1±4.9%); however, CVC did not affect these increases in obstructed kidney hydroxyproline content (Fig 4).
CVC effect on gene expression of fibrotic/inflammatory biomarkers
In the TAA model, there were no significant changes between CVC and vehicle-control groups in mRNA expression of the fibrosis markers assessed (S6 Fig).
Collagen type 1 mRNA expression levels in the NASH model decreased with CVC treatment (37% and 27% reduction vs. vehicle control for CVC20 and CVC100, respectively). This reduction was statistically significant only for the CVC20 group (p < 0.05). There were no statistically significant differences in mRNA expression levels between the vehicle-control and CVC groups for the other fibrosis markers tested in this model (S7 Fig).
In the UUO model, UUO significantly increased collagen 1a1, collagen 3a1, fibronectin-1 and TGF-beta1 mRNA expression in vehicle-treated animals versus sham controls, and 1D11 (positive control) significantly attenuated these UUO-induced increases (p < 0.05) (S8 Fig). There was a trend for CVC7 and CVC20 to inhibit UUO-induced increases in the obstructed renal cortical mRNA expression of some fibrogenic genes; however, it did not reach statistical significance relative to UUO control.
CVC PK analysis
In the TG-induced model of peritonitis, following BID dosing, plasma levels of CVC ~14 hours post-dose (trough levels) increased in a dose-dependent manner (Table 2). At CVC20, lower trough plasma levels were seen with QD (n = 5) versus BID dosing (n = 5) (25.7±8.1 ng/mL [26 hours post-dose] vs. 81.2±22.8 ng/mL [14 hours post-dose], respectively).
Table 2. CVC Plasma Trough Levels (Mean±SD).
| Dose | Time post-dose | CVC concentration (mean±SD, ng/mL) |
|---|---|---|
| TG-induced mouse model of peritonitis (Day 6) | ||
| CVC5 BID, n = 2 | ~14 hours | 13.5a |
| CVC20 BID, n = 5 | ~14 hours | 81.2±22.8 |
| CVC100 BID, n = 5 | ~14 hours | 551±253 |
| Mouse model of UUO-induced renal fibrosis (Day 5) | ||
| CVC7 BID, n = 8 | ~4 hours | 251±364 |
| CVC20 BID, n = 8 | ~4 hours | 971±507 |
a3 of 5 samples below the lower limit of quantification (10 ng/mL).
BID, twice-daily; CVC, cenicriviroc; CVC5, CVC 5 mg/kg/day; CVC7, CVC 7 mg/kg/day; CVC20, CVC 20 mg/kg/day; CVC100, CVC 100 mg/kg/day; n, number of animals; SD, standard deviation; TG, thioglycollate; UUO, unilateral ureter obstruction
In the UUO model, all dose groups had exposure to CVC and plasma levels of CVC at the approximate maximum concentration (Cmax) 4 hours post-dose on Day 5, and these increased in a dose-dependent manner (Table 2).
Discussion
CVC displayed significant anti-inflammatory and antifibrotic effects across a range of in vivo models, including liver fibrosis, NASH and kidney fibrosis. CVC reduced monocyte/macrophage recruitment in an in vivo model of peritonitis, and monocyte migration in vitro. A significant decrease in monocyte/macrophage recruitment versus vehicle control (p < 0.05) and the positive control dexamethasone (p < 0.001) was obtained following treatment with CVC100 BID in vivo. These findings support CVC’s anti-inflammatory mode of action, and are further substantiated by results from the NASH model, where a significantly lower NAS score was observed in the CVC treatment groups relative to the vehicle-control group and a greater number of CVC-treated animals had decreased lobular inflammation and prominent hepatocellular ballooning.
In all models, reductions in collagen deposition and production were observed (a significant reduction in collagen deposition, as well as decreased protein and mRNA expression of collagen type 1). While the significant decreases in liver collagen deposition were dose-dependent in the NASH model, where CVC was administered BID given its short half-life in mice (~2 hours), CVC30 in the TAA model lead to the greater reduction in collagen deposition versus CVC100 (48.5% and 37.5% reduction vs. vehicle control, respectively, p < 0.001). The QD administration of CVC in the TAA model and short half-life of CVC in rats (4–5 hours) may have led to increased clearance of CVC, explaining the lack of apparent dose response. In the UUO model, where gavage was performed twice daily in mice, a significant antifibrotic effect was observed at the low dose (CVC7; p < 0.05) suggesting that this could represent the minimum effective dose. The lack of apparent dose response in this model may also be explained by the short treatment duration (5 days) and the fact that adequate and sustained receptor occupancy (necessary to disrupt CCL2- and CCL5-induced migration) may have already been achieved at lower CVC doses. In fact, CCR2 and CCR5 receptor occupancy by CVC was assessed ex vivo on murine cryopreserved peripheral blood mononuclear cells by Lishomwa Ndlhovu and colleagues [39], in which a CVC concentration of 250 nM achieved >90% and 87% occupancy for CCR2 (blood and spleen) and CCR5 (spleen), respectively, in proinflammatory monocytes.
CCR2 and CCR5 have become attractive targets for antifibrotic therapy, as interactions with their ligands, including CCL2 and CCL5, mediate recruitment of inflammatory cells to the site of liver injury, and contribute to fibrosis [9–15,40]. CVC has previously been shown to initiate high CCR2/CCR5 occupancy on mouse and human monocytes [28,29], and to lead to reciprocal increases in CCL2 in both species [25,26]. In this study, an increase in CCL2 was similarly observed in the CVC100 group compared with the vehicle-control group in the NASH model. The findings reported here show that CVC dual CCR2/CCR5 antagonism causes a reduction in the recruitment and migration of pro-inflammatory monocytes/macrophages, ultimately resulting in decreased fibrogenesis, as measured by collagen deposition and gene/protein expression of collagen type 1. In line with this, CVC does not appear to impact fibrolysis, as shown in the TAA model, where reductions in collagen deposition, as well as in collagen type 1 and alpha-SMA protein expression, were observed in the early intervention and established fibrosis groups, but not the cirrhosis-reversal group. These data suggest that CVC may be best suited for preventing fibrosis progression or improving fibrosis regression, rather than reversing well-established cirrhosis.
CVC was also well tolerated in disease models and had no deleterious effects on body and liver/kidney weight, and liver function. Notably, significant decreases in plasma ALT (NASH model) and AST (early intervention in TAA model) occurred, indicating potential reduction in liver damage with CVC.
One of the limitations of this paper is that, while evaluation of CVC in these animal models demonstrated significant antifibrotic activity, the underlying mechanisms that led to these findings (e.g. effects on monocyte/macrophage infiltration, specific monocyte/macrophage subsets and phenotypes in the liver or kidney, effects on Kupffer cells, HSCs, pericytes or fibroblasts) were not fully elucidated. For example, macrophage staining was only performed in the NASH model, and flow cytometry was not conducted on liver tissue to evaluate the effects of CVC on the infiltrating inflammatory monocyte subset characterized by high expression of CCR2, specifically Ly6Chi. Another limitation is the fact that the experiments presented here were conducted by various groups, using different methodologies and on models that are not widely used, which may limit interpretation of findings.
The effects of CVC are currently being evaluated in established animal models of acute and chronic hepatic injury, such as the acetaminophen (APAP), carbon tetrachloride (CCl4) and methionine and choline deficient diet (MCD)-induced models [41–43]. Preliminary data have demonstrated that CVC treatment significantly decreased infiltration of Ly6Chi monocyte-derived macrophages into the liver in all three models [42]. CVC treatment was also associated with a significant protection from acute APAP-induced liver injury, with a significant reduction in ALT levels and in necrotic area relative to vehicle control. A significant reduction in necrotic area was also observed in the acute CCl4 model. In the chronic MCD model of NASH, CVC treatment significantly ameliorated steatohepatitis, as assessed by the histological NAFLD activity score, and reduced hepatic fibrosis, as evidenced by decreased Sirius red staining and hydroxyproline content.
CVC has completed Phase 2b clinical development for treatment of HIV-1 infection in antiretroviral treatment-naïve adults with CCR5-tropic virus (Study 652-2-202; NCT01338883) [26]. CVC provided potent CCR2/CCR5 blockade and showed antifibrotic properties. Indeed, the proportion of subjects with aspartate aminotransferase-to-platelet count ratio index (APRI) score ≥0.5 and non-invasive hepatic fibrosis risk (FIB-4) score ≥1.45 decreased by 75% and 73%, respectively, between Baseline and Week 24; these decreases were maintained at Week 48 [44]. More recently, an independent analysis of study samples showed that CVC treatment led to significant reduction in the enhanced liver fibrosis (ELF) scores at Week 48 (p < 0.0001) [45]. In addition, CVC exhibited a good tolerability profile in humans, including hepatically impaired subjects [21,25,26]. Overall, these results establish proof of principle for human testing of CVC for fibrosis-related conditions, including NASH.
NASH, a hepatic manifestation of the metabolic syndrome typically associated with insulin resistance, is the most serious form of highly prevalent NAFLD [46–48]. Mild to moderate fibrosis is present in 76–100% of patients with NASH, and severe fibrosis in 15–50%; cirrhosis affects 7–16% of adults with the condition [49]. CVC 150 mg QD is in Phase 2b clinical development for the treatment of adults with NASH and liver fibrosis (CENTAUR Study 652-2-203; NCT02217475). No drugs are currently approved to treat NASH but, in addition to CVC, several other agents are under investigation. These have different mechanisms of action to CVC, and include high-dose vitamin E, pioglitazone (peroxisome proliferator-activated receptor [PPAR]-gamma agonist), obeticholic acid (a farnesoid X receptor agonist) and elafibranor (formerly GFT505, a peroxisome proliferator-activated receptor-alpha/delta agonist) [50–52]. Pioglitazone, obeticholic acid and elafibranor have shown amelioration of insulin resistance and/or antifibrotic activity in animal models [53–55] and have undergone Phase 2 clinical evaluation [50,51,56] (NCT01694849). Taken together, these findings provide a strong rationale for targeting both inflammatory and metabolic pathways in NASH, and warrant the need to evaluate combination therapies as a means to further improve treatment outcomes.
In conclusion, the comprehensive and consistent preclinical data demonstrating anti-inflammatory and antifibrotic effects of CVC, existing human safety data, oral availability, and therapeutic targets strongly implicated in experimental and human liver diseases [9–15] provide a strong rationale to further evaluate CVC as a treatment for NASH with liver fibrosis.
Supporting Information
aThe number of animals at the start of the study is indicated, with the number of animals at the end of the study on which analyses were conducted indicated in brackets; bVehicle control: 0.5% [w/v] methylcellulose + 1% Tween®-80. BID, twice daily; CVC, cenicriviroc; DEX, dexamethasone; IP, intraperitoneal; NASH, non-alcoholic steatohepatitis; PBS, phosphate buffer saline; PO, oral gavage; QD, once daily; SC, subcutaneous; STAM, stelic animal model; TAA, thioacetamide; TG, thioglycollate; UUO, unilateral ureter obstruction.
(DOCX)
*p < 0.05 vs. vehicle control; CVC, cenicriviroc; DEX, dexamethasone; NASH, non-alcoholic steatohepatitis; SD, standard deviation; SEM; standard error of mean; TAA, thioacetamide; UUO, unilateral ureter obstruction.
(DOCX)
Total and differential cell counts were assessed in peritoneal lavage samples using an Advia® Hematology System (Siemens Healthcare Diagnostics, USA) with multispecies software and an analysis software designed for mouse peritoneal fluid on Advia® 120 (LabThruPut, New York, USA). The software applies cluster analysis on the two channels (peroxidase and basophil channels) pictured. In the peroxidase channel, eosinophils are shown in yellow, neutrophils in magenta and mononuclear cells (lymphocytes, monocytes and macrophages) in cyan. In the basophil channel, neutrophils and eosinophils are shown in magenta and cellular debris in white. Information from both channels are combined to obtain mononuclear cells and neutrophil counts. The peritoneal fluid white-blood-cell count, and the absolute and differential mononuclear cell, neutrophil and eosinophil counts are then calculated. BID, twice daily; CVC, cenicriviroc; CVC5, CVC 5 mg/kg/day; CVC20, CVC 20 mg/kg/day; CVC100, CVC 100 mg/kg/day; QD, once daily; TG, thioglycollate.
(TIF)
(A) Average ALT levels and (B) Average AST levels in the early intervention, established fibrosis and cirrhosis reversal groups. *p < 0.05 vs. vehicle control; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CVC, cenicriviroc; QD, once daily; SD, standard deviation; TAA, thioacetamide.
(TIF)
CVC, cenicriviroc; QD, once daily; SD, standard deviation; TAA, thioacetamide.
(TIF)
Representative micrographs of H&E-stained liver sections in (A) the rat TAA model (40x) and (B) the mouse NASH model (50x). CVC, cenicriviroc; H&E, hematoxylin and eosin; NASH, non-alcoholic steatohepatitis; TAA, thioacetamide.
(TIF)
CVC, cenicriviroc; NASH, non-alcoholic steatohepatitis.
(TIF)
(A) Collagen type I; (B) alpha-SMA; (C) beta-PDGFR; (D) TGF-beta; (E) MMP2; (F) TIMP1; (G) TIMP2. CVC, cenicriviroc; MMP2, matrix metalloproteinase 2; beta-PDGFR, beta-platelet-derived growth factor-beta receptor; QD, once daily; SE, standard error; alpha-SMA, alpha-smooth muscle actin; TAA, thioacetamide; TGF-beta, transforming growth factor-beta; TIMP, tissue inhibitor of metalloproteinase.
(TIF)
*p < 0.05 vs. vehicle control; BID, twice daily; CVC, cenicriviroc; MCP-1, monocyte chemotactic protein-1; NASH, non-alcoholic steatohepatitis; SD, standard deviation; TIMP, tissue inhibitor of metalloproteinase; TNF, tumor necrosis factor.
(TIF)
(A) Collagen 1a1; (B) Collagen 3a1; (C) alpha-SMA; (D) TGF-beta; (E) MCP-1; (F) Fibronectin; (G) CTFG. *p < 0.05 vs. sham control; †p < 0.05 vs. UUO control; BID, twice daily; CTFG, connective tissue growth factor; CVC, cenicriviroc; MCP-1, monocyte chemotactic protein-1; QD, once daily; SEM, standard error of the mean; alpha-SMA, alpha-smooth muscle actin; TGF-beta, transforming growth factor-beta; UUO, unilateral ureter obstruction.
(TIF)
Acknowledgments
Editorial support was provided by Camille Bonomelli of Alpharmaxim Healthcare Communications. Analyses of the pharmacokinetic samples were performed by KCAS, Shawnee, KS. Charles River Preclinical Services Montreal, InVivo Pharmacology technical team conducted the TG model of peritonitis study.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by Tobira Therapeutics, Inc. (701 Gateway Blvd, Suite 300, South San Francisco, CA 94080, USA; 1 (650) 741-6625; www.tobiratherapeutics.com). The sponsor has been involved in study design, data collection and analysis, decision to publish, and preparation of the manuscript.
References
- 1.Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64: 830–841. 10.1136/gutjnl-2014-306842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim YS, Kim WR. The global impact of hepatic fibrosis and end-stage liver disease. Clin Liver Dis. 2008;12: 733–746. 10.1016/j.cld.2008.07.007 [DOI] [PubMed] [Google Scholar]
- 3.Eddy AA. Overview of the cellular and molecular basis of kidney fibrosis. Kidney Int Suppl. 2014;4: 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al. Chronic kidney disease: global dimension and perspectives. Lancet. 2013;382: 260–272. 10.1016/S0140-6736(13)60687-X [DOI] [PubMed] [Google Scholar]
- 5.Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61: 416–426. 10.1136/gutjnl-2011-300304 [DOI] [PubMed] [Google Scholar]
- 6.Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60: 1090–1096. 10.1016/j.jhep.2013.12.025 [DOI] [PubMed] [Google Scholar]
- 7.Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58: 1461–1473. 10.1002/hep.26429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Minicis S, Seki E, Uchinami H, Kluwe J, Zhang Y, Brenner DA, et al. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology. 2007;132: 1937–1946. [DOI] [PubMed] [Google Scholar]
- 9.Saiman Y, Friedman SL. The role of chemokines in acute liver injury. Front Physiol. 2012;3: 213 10.3389/fphys.2012.00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seki E, De Minicis S, Gwak GY, Kluwe J, Inokuchi S, Bursill CA, et al. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest. 2009;119: 1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seki E, De Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology. 2009;50: 185–197. 10.1002/hep.22952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimmermann HW, Tacke F. Modification of chemokine pathways and immune cell infiltration as a novel therapeutic approach in liver inflammation and fibrosis. Inflamm Allergy Drug Targets. 2011;10: 509–536. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell C, Couton D, Couty JP, Anson M, Crain AM, Bizet V, et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol. 2009;174: 1766–1775. 10.2353/ajpath.2009.080632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. 2012;302: G1310–G1321. 10.1152/ajpgi.00365.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berres ML, Koenen RR, Rueland A, Zaldivar MM, Heinrichs D, Sahin H, et al. Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J Clin Invest. 2010;120: 4129–4140. 10.1172/JCI41732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang YS, Lee MH, Song HK, Ko GJ, Kwon OS, Lim TK, et al. CCR2 antagonism improves insulin resistance, lipid metabolism, and diabetic nephropathy in type 2 diabetic mice. Kidney Int. 2010;78: 883–894. 10.1038/ki.2010.263 [DOI] [PubMed] [Google Scholar]
- 17.Panzer U, Steinmetz OM, Stahl RA, Wolf G. Kidney diseases and chemokines. Curr Drug Targets. 2006;7: 65–80. [DOI] [PubMed] [Google Scholar]
- 18.Segerer S, Mac K M, Regele H, Kerjaschki D, Schlöndorff D. Expression of the C-C chemokine receptor 5 in human kidney diseases. Kidney Int. 1999;56: 52–64. [DOI] [PubMed] [Google Scholar]
- 19.Vielhauer V, Anders HJ, MacK M, Cihak J, Strutz F, Stangassinger M, et al. Obstructive nephropathy in the mouse: progressive fibrosis correlates with tubulointerstitial chemokine expression and accumulation of CC chemokine receptor 2- and 5-positive leukocytes. J Am Soc Nephrol. 2001;12: 1173–1187. [DOI] [PubMed] [Google Scholar]
- 20.Xia Y, Entman ML, Wang Y. CCR2 regulates the uptake of bone marrow-derived fibroblasts in renal fibrosis. PLoS One. 2013;8: e77493 10.1371/journal.pone.0077493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lalezari J, Gathe J, Brinson C, Thompson M, Cohen C, Dejesus E, et al. Safety, efficacy, and pharmacokinetics of TBR-652, a CCR5/CCR2 antagonist, in HIV-1-infected, treatment-experienced, CCR5 antagonist-naive subjects. J Acquir Immune Defic Syndr. 2011;57: 118–125. 10.1097/QAI.0b013e318213c2c0 [DOI] [PubMed] [Google Scholar]
- 22.Marier JF, Trinh M, Pheng LH, Palleja SM, Martin DE. Pharmacokinetics and pharmacodynamics of TBR-652, a novel CCR5 antagonist, in HIV-1-infected, antiretroviral treatment-experienced, CCR5 antagonist-naïve patients. Antimicrob Agents Chemother. 2011;55: 2768–2774. 10.1128/AAC.00713-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baba M, Takashima K, Miyake H, Kanzaki N, Teshima K, Wang X, et al. TAK-652 inhibits CCR5-mediated human immunodeficiency virus type 1 infection in vitro and has favorable pharmacokinetics in humans. Antimicrob Agents Chemother. 2005;49: 4584–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuroshima,K, Inanami M, Kanzaki N, Takeda Chemical Industries, Ltd. Receptor binding properties of TAK-652, a small molecular weight CCR5 antagonist (Report No. TAK-652/00045). 2003.
- 25.Lefebvre E, Gottwald M, Lasseter K, Chang W, Willett M, Smith PF, et al. Pharmacokinetics, Safety, and CCR2/CCR5 Antagonist Activity of Cenicriviroc in Participants With Mild or Moderate Hepatic Impairment. Clin Transl Sci. 2016;9: 139–148. 10.1111/cts.12397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson M, Saag M, Dejesus E, Gathe J, Lalezari J, Landay AL, et al. A 48-week randomized Phase 2b study evaluating cenicriviroc vs. efavirenz in treatment-naive HIV-infected adults with CCR5-tropic virus. AIDS. 2016;30: 869–878. 10.1097/QAD.0000000000000988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friedman SL, Sanyal A, Goodman Z, Lefebvre E, Gottwald M, Fischer L, et al. Efficacy and safety study of cenicriviroc for the treatment of non-alcoholic steatohepatitis in adult subjects with liver fibrosis: CENTAUR Phase 2b study design. Contemp Clin Trials. 2016;47: 356–365. 10.1016/j.cct.2016.02.012 [DOI] [PubMed] [Google Scholar]
- 28.Jalbert E, Byron MM, Shikuma C, Jenkins H, Lefebvre E, Barbour JD. Cenicriviroc achieves high CCR5 receptor occupancy at low nanomolar concentrations. Presented at: 21st Conference on Retroviruses and Opportunistic Infections (CROI 2014); 2014 Mar 3; Boston, MA, USA. 530.
- 29.Byron MM, D'Antoni ML, Premeaux T, Lefebvre E, Ndhlovu LC. Dual CCR2/CCR5 antagonism by cenicriviroc efficiently inhibits both MCP-1 and RANTES induced chemokine receptor internalization in murine pro-inflammatory monocytes. Presented at: Immunology 2015™—The American Association of Immunologists (AAI) Annual Meeting; 2015 May 11; New Orleans, LA, USA.
- 30.Tobira Therapeutics, Inc., Ndhlovu Laboratory. In Vitro CCR2 and CCR5 Receptor Affinity Studies of Cenicriviroc Mesylate CVC (TBR-652), Maraviroc (MVC) or BMS-22, on Murine Blood and Splenic Monocytes (Study Number 652-9-1024). 2015.
- 31.Hayashi H, Sakai T. Animal models for the study of liver fibrosis: new insights from knockout mouse models. Am J Physiol Gastrointest Liver Physiol. 2011;300: G729–G738. 10.1152/ajpgi.00013.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ucero AC, Benito-Martin A, Izquierdo MC, Sanchez-Niño MD, Sanz AB, Ramos AM, et al. Unilateral ureteral obstruction: beyond obstruction. Int Urol Nephrol. 2014;46: 765–776. 10.1007/s11255-013-0520-1 [DOI] [PubMed] [Google Scholar]
- 33.Kanuri G, Bergheim I. In vitro and in vivo models of non-alcoholic fatty liver disease (NAFLD). Int J Mol Sci. 2013;14: 11963–11980. 10.3390/ijms140611963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henderson RB, Hobbs JA, Mathies M, Hogg N. Rapid recruitment of inflammatory monocytes is independent of neutrophil migration. Blood. 2003;102: 328–335. [DOI] [PubMed] [Google Scholar]
- 35.Reshef R, Luger SM, Hexner EO, Loren AW, Frey NV, Nasta SD, et al. Blockade of lymphocyte chemotaxis in visceral graft-versus-host disease. N Engl J Med. 2012;367: 135–145. 10.1056/NEJMoa1201248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Traber PG, Chou H, Zomer E, Hong F, Klyosov A, Fiel MI, et al. Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease. PLoS One. 2013;8: e75361 10.1371/journal.pone.0075361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K, et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol. 2013;46: 141–152. 10.1007/s00795-013-0016-1 [DOI] [PubMed] [Google Scholar]
- 38.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41: 1313–1321. [DOI] [PubMed] [Google Scholar]
- 39.Byron MM, D'Antoni ML, Premeaux T, Lefebvre E, Ndhlovu LC. Dual CCR2/CCR5 antagonism by cenicriviroc efficiently inhibits both MCP-1 and RANTES induced chemokine receptor internalization in murine pro-inflammatory monocytes. J Immunol. 2015;194: 187–8..25416810 [Google Scholar]
- 40.Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology. 2014;147: 577–594. 10.1053/j.gastro.2014.06.043 [DOI] [PubMed] [Google Scholar]
- 41.Krenkel O, Mossanen JC, Ergen C, Heymann F, Lefebvre E, Eulberg D, et al. CCR2+ infiltrating monocytes promote acetaminophen-induced acute liver injury—therapeutic implications of inhibiting CCR2 and CCL2. J Hepatol. 2015;62: S206. [Google Scholar]
- 42.Krenkel O, Püngel T, Mossanen J, Ergen C, Heymann F, Lefebvre E, et al. Dual CCR2/CCR5 anatagonist cenicriviroc leads to potent and significant reduction in proinflammatory CCR2+ monocyte infiltration in experimental acute liver injury. Presented at: The 66th Annual Meeting of the American Association for the Study of Liver Diseases: The Liver Meeting 2015; 2015 Nov 17; San Francisco, CA, USA. 1756.
- 43.Puengel T, Krenkel O, Mossanen J, Longerich E, Lefebvre E, Trautwein C, et al. The dual CCR2/CCR5 antagonist cenicriviroc ameliorates steatohepatitis and fibrosis in vivo by inhibiting the infiltration of inflammatory monocytes into injured liver. J Hepatol. 64: s159–s182. [Google Scholar]
- 44.Thompson M, Chang W, Jenkins H, Flynt A, Gottwald M, Lefebvre E. Improvements in APRI and FIB-4 fibrosis scores correlate with decreases in sCD14 in HIV-1 infected adults receiving cenicriviroc over 48 weeks. Hepatology 2014;60: 424A. [Google Scholar]
- 45.Sherman KE, Abdel-Hameed E, Rouster SD. CCR2/CCR5 antagonism with cenicriviroc decreases fibrosis scores in HIV-infected patients. Presented at: HEP DART 2015, Frontiers in Drug Development for Viral Hepatitis; 2015 Dec 6; Hawaii, USA.
- 46.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120: 1183–1192. [DOI] [PubMed] [Google Scholar]
- 47.Hashimoto E, Taniai M, Tokushige K. Characteristics and diagnosis of NAFLD/NASH. J Gastroenterol Hepatol. 2013;28: 64–70. 10.1111/jgh.12271 [DOI] [PubMed] [Google Scholar]
- 48.Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis—new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. 2013;10: 627–636. 10.1038/nrgastro.2013.149 [DOI] [PubMed] [Google Scholar]
- 49.Sheth SG, Gordon FD, Chopra S. Nonalcoholic steatohepatitis. Ann Intern Med. 1997;126: 137–145. [DOI] [PubMed] [Google Scholar]
- 50.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385: 956–965. 10.1016/S0140-6736(14)61933-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362: 1675–1685. 10.1056/NEJMoa0907929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Staels B, Rubenstrunk A, Noel B, Rigou G, Delataille P, Millatt LJ, et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. 2013;58: 1941–1952. 10.1002/hep.26461 [DOI] [PubMed] [Google Scholar]
- 53.Ding SY, Shen ZF, Chen YT, Sun SJ, Liu Q, Xie MZ. Pioglitazone can ameliorate insulin resistance in low-dose streptozotocin and high sucrose-fat diet induced obese rats. Acta Pharmacol Sin. 2005;26: 575–580. [DOI] [PubMed] [Google Scholar]
- 54.Han JY, Kim YJ, Kim L, Choi SJ, Park IS, Kim JM, et al. PPARgamma agonist and angiotensin II receptor antagonist ameliorate renal tubulointerstitial fibrosis. J Korean Med Sci. 2010;25: 35–41. 10.3346/jkms.2010.25.1.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Higashi K, Oda T, Kushiyama T, Hyodo T, Yamada M, Suzuki S, et al. Additive antifibrotic effects of pioglitazone and candesartan on experimental renal fibrosis in mice. Nephrology (Carlton). 2010;15: 327–335. [DOI] [PubMed] [Google Scholar]
- 56.Ratziu V, Harrison S, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an Agonist of the Peroxisome Proliferator-activated Receptor-ɑ and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology. 2016;150: 1147–1159. 10.1053/j.gastro.2016.01.038 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
aThe number of animals at the start of the study is indicated, with the number of animals at the end of the study on which analyses were conducted indicated in brackets; bVehicle control: 0.5% [w/v] methylcellulose + 1% Tween®-80. BID, twice daily; CVC, cenicriviroc; DEX, dexamethasone; IP, intraperitoneal; NASH, non-alcoholic steatohepatitis; PBS, phosphate buffer saline; PO, oral gavage; QD, once daily; SC, subcutaneous; STAM, stelic animal model; TAA, thioacetamide; TG, thioglycollate; UUO, unilateral ureter obstruction.
(DOCX)
*p < 0.05 vs. vehicle control; CVC, cenicriviroc; DEX, dexamethasone; NASH, non-alcoholic steatohepatitis; SD, standard deviation; SEM; standard error of mean; TAA, thioacetamide; UUO, unilateral ureter obstruction.
(DOCX)
Total and differential cell counts were assessed in peritoneal lavage samples using an Advia® Hematology System (Siemens Healthcare Diagnostics, USA) with multispecies software and an analysis software designed for mouse peritoneal fluid on Advia® 120 (LabThruPut, New York, USA). The software applies cluster analysis on the two channels (peroxidase and basophil channels) pictured. In the peroxidase channel, eosinophils are shown in yellow, neutrophils in magenta and mononuclear cells (lymphocytes, monocytes and macrophages) in cyan. In the basophil channel, neutrophils and eosinophils are shown in magenta and cellular debris in white. Information from both channels are combined to obtain mononuclear cells and neutrophil counts. The peritoneal fluid white-blood-cell count, and the absolute and differential mononuclear cell, neutrophil and eosinophil counts are then calculated. BID, twice daily; CVC, cenicriviroc; CVC5, CVC 5 mg/kg/day; CVC20, CVC 20 mg/kg/day; CVC100, CVC 100 mg/kg/day; QD, once daily; TG, thioglycollate.
(TIF)
(A) Average ALT levels and (B) Average AST levels in the early intervention, established fibrosis and cirrhosis reversal groups. *p < 0.05 vs. vehicle control; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CVC, cenicriviroc; QD, once daily; SD, standard deviation; TAA, thioacetamide.
(TIF)
CVC, cenicriviroc; QD, once daily; SD, standard deviation; TAA, thioacetamide.
(TIF)
Representative micrographs of H&E-stained liver sections in (A) the rat TAA model (40x) and (B) the mouse NASH model (50x). CVC, cenicriviroc; H&E, hematoxylin and eosin; NASH, non-alcoholic steatohepatitis; TAA, thioacetamide.
(TIF)
CVC, cenicriviroc; NASH, non-alcoholic steatohepatitis.
(TIF)
(A) Collagen type I; (B) alpha-SMA; (C) beta-PDGFR; (D) TGF-beta; (E) MMP2; (F) TIMP1; (G) TIMP2. CVC, cenicriviroc; MMP2, matrix metalloproteinase 2; beta-PDGFR, beta-platelet-derived growth factor-beta receptor; QD, once daily; SE, standard error; alpha-SMA, alpha-smooth muscle actin; TAA, thioacetamide; TGF-beta, transforming growth factor-beta; TIMP, tissue inhibitor of metalloproteinase.
(TIF)
*p < 0.05 vs. vehicle control; BID, twice daily; CVC, cenicriviroc; MCP-1, monocyte chemotactic protein-1; NASH, non-alcoholic steatohepatitis; SD, standard deviation; TIMP, tissue inhibitor of metalloproteinase; TNF, tumor necrosis factor.
(TIF)
(A) Collagen 1a1; (B) Collagen 3a1; (C) alpha-SMA; (D) TGF-beta; (E) MCP-1; (F) Fibronectin; (G) CTFG. *p < 0.05 vs. sham control; †p < 0.05 vs. UUO control; BID, twice daily; CTFG, connective tissue growth factor; CVC, cenicriviroc; MCP-1, monocyte chemotactic protein-1; QD, once daily; SEM, standard error of the mean; alpha-SMA, alpha-smooth muscle actin; TGF-beta, transforming growth factor-beta; UUO, unilateral ureter obstruction.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.