

Abstract
A titanium-based catalytic enantioselective dichlorination of simple allylic alcohols is described. This dichlorination reaction provides stereoselective access to all common dichloroalcohol building blocks used in syntheses of chlorosulfolipid natural products. An enantioselective synthesis of ent-(–)-deschloromytilipin A and a concise, eight-step synthesis of ent-(–)-danicalipin A are executed and employ the dichlorination reaction as the first step. Extension of this system to enantioselective dibromination and its use in the synthesis of pentabromide stereoarrays relevant to bromosufolipids is reported. The described dichlorination and di-bromination reactions are capable of exerting diastereocontrol in complex settings allowing X-ray crystal structure analysis of natural and unnatural diastereomers of polyhalogenated stereohexads.
Graphical Abstract
1. INTRODUCTION
Although the addition of molecular chlorine to carbon–carbon double bonds is one of the oldest known organic reactions,1a the ability to control the enantioselectivity of alkene dichlorination has largely eluded chemists.1b–d Meanwhile, in recent years there has been a growing interest in the stereoselective synthesis of polychlorinated sulfolipid natural products. Due to the scarcity of methods for enantioselective dichlorination of alkenes, syntheses of molecules within this class of natural products rely on multistep routes for establishing chlorine-bearing chiral centers in an enantioselective fashion. To date there is a single report of using an enantioselective dichlorination in the setting of complex molecule synthesis by Snyder, which employs a super-stoichiometric chiral auxiliary.2a A practical, catalytic, enantioselective dichlorination would be of great value to the synthetic chemist.
Slow progress in the area of enantioselective dichlorination is attributable to a number of factors. First, the highly reactive nature of most commonly employed chlorinating reagents makes achieving catalysis over nonselective background a challenge. Second, even if selectivity is achieved in the formation of an enantiopure chloronium ion (e.g. 1, Scheme 1), regiochemical control in chloride delivery is still necessary. This is because non-regioselective chloride delivery results in a racemic product, since dichlorides formed from the two possible chloride deliveries to non-C2-symmetric chloronium ions are enantiomers (e.g. 2 and ent-2, Scheme 1).2b
Scheme 1.

A challenge in enantioselective dichlorination.
In 2011, Nicolaou reported the first catalytic enantioselective dichlorination of allylic alcohols.2c In fact, Nicolaou’s dichlorination constitutes the first catalytic enantioselective dihalogenation of any kind.2d–f,3 This system relies on the use of styrenyl olefins as substrates to electronically bias chloride delivery. Consequently, the selectivity of this system is poor for non-styrenyl substrates which limits its potential synthetic application and precludes its use in the synthesis of the chlorosulfolipids.
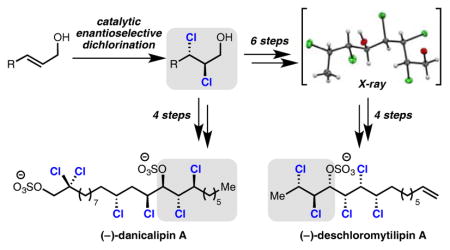
The enantioselective dichlorination of simple, alkyl disubstituted allylic alcohols has been a long sought-after transformation and would be a powerful means of entry to the chlorosulfolipids, a class of acyclic, polychlorinated natural products (Figure 1).4 In this area, dichlorinated allylic alcohols have been identified as highly enabling intermediates in the synthesis of mytilipin A (4), danicalipin A (5), and malhamen-silipin A (6).5,6 Highlighted in Figure 1 are specific dichloroalcohols that have been targeted and employed in syntheses of these molecules. Given the lack of enantioselective dichlorination methods, these motifs are either generated in racemic fashion6a,b or, as in the case of danicalipin A (5), prepared via an enriched epoxide in 4 steps and 80% ee.6c The allylic alcohol precursors to these dichlorides represent desirable, but challenging, settings for catalytic enantioselective dichlorinations.
Figure 1.
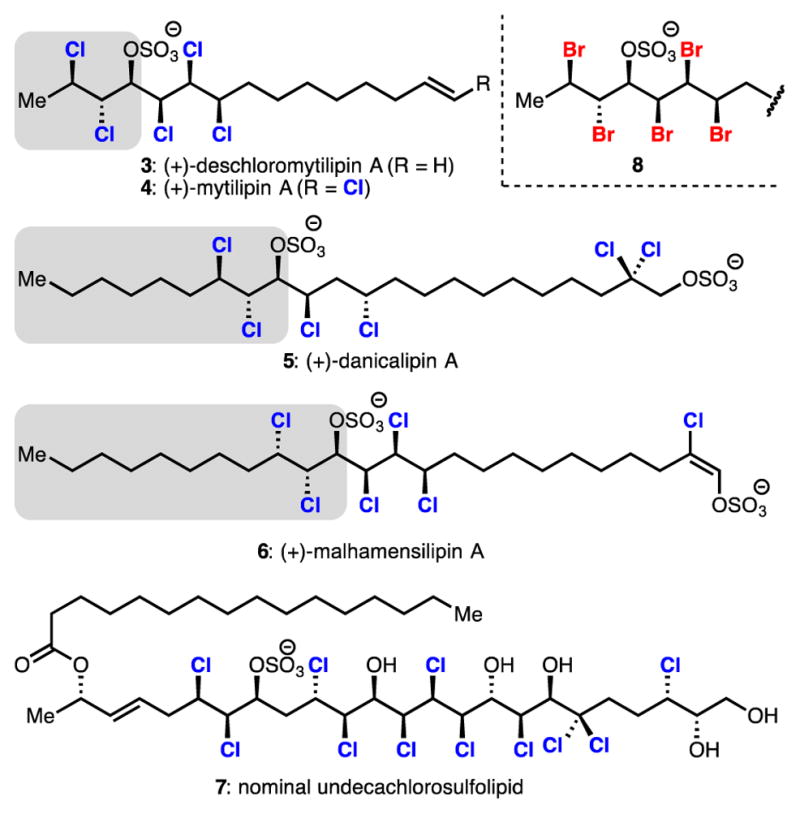
Structures of chlorosulolipid natural products (3–7) and an analogous bromosulfolipid stereohexad (8). Highlighted structures represent corresponding simple dichlorinated allylic alcohols that have been targeted in synthesis.
Described herein is a catalytic enantioselective method for the dichlorination of simple linear aliphatic allylic alcohols. Specifically, our reported Schiff base-catalyzed dihalogenation reaction is capable of accessing the enantioenriched dichloro-alcohols involved in all syntheses of the chlorosulfolipids (Table 2). The utility of this method is demonstrated in a synthesis of the unnatural enantiomer of deschloromytilipin A (3). X-ray crystallography is used to confirm the relative configuration of the stereohexad of this natural product. The ability of this method to provide new diastereoselectivities in complex settings, as well as access to enriched polybromides, allows comparison of the conformations of natural and unnatural polychlorinated and polybrominated stereohexads in solution and solid phase. A concise eight-step synthesis of the unnatural enantiomer of danicalipin A (5) utilizing an enantioenriched dichloride is additionally reported.
Table 2.
Dichlorination Substrate Scopea
| ||||||
|---|---|---|---|---|---|---|
| entry | substrate | yield (%) | ee (%) | mol % 11 | product | prior art or use in synthesis |
| 1b |
16 |
83 | 91 | 10 |
|
Nicolaou (2011): 58%, +61% ee ArlCl2, 20 mol % (DHQD)2PHAL |
| 2 |
18 |
64c | 80 | 15 |
|
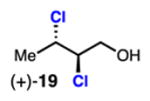
(deschloro)mytilipin A Vanderwal (2013): (±)-19 Burns (this work): (+)-19 |
| 3 |
20 |
86 | 83 | 15 |
|
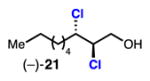
danicalipin A Yoshimitsu (2011): (+)-21 Vanderwal (2014): (±)-21 Burns (this work): (–)-21 |
| 4d |
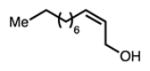 22 |
64 | 81 | 20 |
|
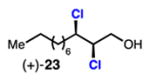
malhamensilipin A Vanderwal (2014): (±)-23 |
| 5d |
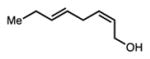 24 |
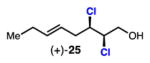
45 | 85 | 30 |
|
nominal undecachlorosufolipid (yet to be utilized) |
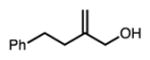
| 6d |
 26 |
61 | 90 | 30 |
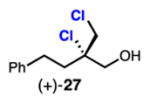
|
Burns (2015) |
Conditions unless otherwise noted: 1.1–1.5 equiv t-BuOCl, 1.1–1.6 equiv ClTi(Oi-Pr)3, 10–30 mol % (R,S)-11, hexanes, −20 °C, 4–12 hours;
3:1 hexanes/CCl4;
isolated by distillation;
absolute configuration unconfirmed.
2. RESULTS AND DISCUSSION
2.1 Catalytic Enantioselective Dichlorination
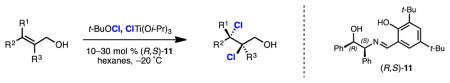
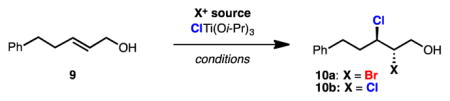
Recently, we reported a titanium-mediated enantioselective bromochlorination of allylic alcohols catalyzed by Schiff base 11 (Table 1).7 Trisubstituted, 1,1-disubsituted, and cis-disubstituted allylic alcohols were bromochlorinated under the described conditions with excellent regio- and enantioselectivity. Unfortunately, application of this system to disubstituted trans-allylic alcohol 9 provided bromochloride 10a and its corresponding bromochloride constituational isomer in a 1.7 to 1.0 ratio, respectively. Reduced temperatures provided a slight improvement in bromochloride product rations (3.0:1.0, Table 1, entry 2). As depicted in Scheme 1, one would expect that if a 1.7 to 1.0 or a 3.o to 1.0 product ratio persisted for the case of di-chlorination, the enantioselectivity of dichloride products would be limited to a modest 26% or 50% ee, respectively, assuming the intermediacy of a discrete enantioenriched chloronium ion.
Table 1.
Development of an Enantioselective Dichlorination of Allylic Alcoholsa
| ||||
|---|---|---|---|---|
| entry | X+ source | conditions | yield (%) | ee (%) |
| 1 |
|
30 mol% 11, hexanes, r.t. | 90 | 66 (1.7 : 1.0)b |
| 2 |
|
30 mol% 11, hexanes, −20 °C | 81 | 90 (3.0 : 1.0)b |
| 3 |
|
30 mol% 11, hexanes, r.t. | 52 | 62 |
| 4 | 13 | 30 mol% 11, hexanes, r.t. | 70 | 65 |
| 5 |
|
30 mol% 11, hexanes, r.t. | 12 | 6 |
| 6 | 14 | 30 mol% 11, hexanes, r.t. | trace | – |
| 7 | 15 | 30 mol% 11, hexanes, r.t. | 66 | 58 |
| 8 |
|
30 mol% 11, hexanes, r.t. | 56 | 72 |
| 9 |
|
30 mol% 12, hexanes, r.t. | 73 | 5 |
| 10 |
|
30 mol% 11, CH2Cl2, r.t. | 62 | 20 |
| 11 |
|
30 mol% 11, Et2O, r.t. | 75 | 67 |
| 12 |
|
30 mol% 11, hexanes, −20 °C | 77 | 78 |
| 13 |
|
10 mol% 11, hexanes, −20 °C | 82 | 76 |
|
| ||||
Reactions were conducted on 0.1 mmol scale with 1.1 equiv X+ source and 1.1 equiv ClTi(Oi-Pr)3. Yields were assessed by 1H-NMR spectroscopy with 1,4-dinitrobenzene as an internal standard;
Ratio of bromochloride constitutional isomers. Yields given for entries 1 and 2 are for both constitutional isomers and ee is reported for the major isomer.
The viability of a selective dichlorination of trans-disubstituted allylic alcohols was initially investigated by replacing N-bromosuccinimide (NBS) with N-chlorosuccinimide (NCS) as a source of electrophilic chlorine in our titanium-mediated dihalogention reaction (Table 1, entry 3). This provided dichloride product 10b in fair yield and higher-than-predicted enantioselectivity (i.e. greater than 26% ee). Such enantioselectivity could originate from an increased regioselectivity in chloride delivery in the case of trapping chloronium intermediates by chlorotitanium species. Another possible explanation is that diastereomeric chloronium-containing titanium complexes are attacked by chloride with different regioselectivity.
Subsequent investigation revealed tert-butylhypochlorite (t-BuOCl) as the most selective source of electrophilic chlorine (Table 1, entries 4–8). This is surprising given the highly reactive nature of t-BuOCl, which exhibits substantial background reactivity in the absence of a chiral catalyst. Mechanistically, we speculate that a chiral titanium complex acts as an oxophilic Lewis acid on t-BuOCl, accelerating transfer of the electrophilic chlorine to the olefin of the titanium-bound allylic alcohol. The higher selectivity for dichlorinations with t-BuOCl compared with NCS could arise from the closer proximity of the oxygen and chlorine in t-BuOCl, situating the chlorine nearer the chiral ligand which is presumably also titanium-bound. Use of ligands other than chiral iminodiol 11 or solvents other than hexanes resulted in diminished selectivity (Table 1, entries 9–11). Decreased temperatures improved both the yield of and selectivity for dichloride 10b and permitted reduction in catalyst loading (Table 1, entries 12–13). Further improvements in selectivity were observed in larger scale reactions employing more controlled drop-wise additions of t-BuOCl solutions.
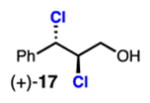
As a benchmark substrate, cinnamyl alcohol 16 was subjected to the dichlorination conditions and produced dichloride (+)-17 in excellent yield and selectivity (Table 2, entry 1). Previously, enantioenriched (+)-17 and (–)-17 were prepared by Nicolaou in +61% and –81% ee, respectively, using 20 mol % pseudoenantiomeric (DHQD)2PHAL or (DHQ)2PHAL.2c
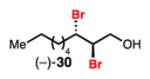
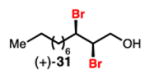
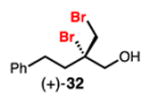
Linear, aliphatic allylic dichloroalcohols 19, 21, and 23 are enantioenriched versions of intermediates that have been used in syntheses of mytilipin A, danicalipin A, and malhamensilipin A, respectively (Table 2, entries 2–4).6a–c Dichlorination of allylic alcohols 18, 20, and 22 under our developed conditions produced the aforementioned dichloroalcohols on practical scale and in synthetically useful yields and enantioselectivities. The low yield in the dichlorination of 18 is likely due to the volatility of dichloride 19, which is isolated via distillation. Dichloroalcohol 21, which is used in our synthesis of ent-(–)-danicalipin A (–)-5 (see Section 2.6), was prepared on 9.1 gram-scale in a single run. The skipped diene 24 presents an interesting chemoselectivity challenge and contains allylic protons which could be acidic within a chloronium intermediate. Our system can dichlorinate the more electron-deficient olefin of 24 to produce 25 in fair yield and good enantioselectivity (Table 2, entry 5); compound 25 maps on to nominal undecachlorosulfolipid 7 (Figure 1). As previously reported, our conditions can dihalogenate 1,1-disubstituted allylic alcohol 26 to furnish 27 selectively (Table 2, entry 6).7 Trisubstituted allylic alcohols are converted to dichlorides with low yield using this method due to facile deprotonation of trisubstituted chloronium ions to give allylic chloride products.
2.2 Catalytic Enantioselective Dibromination
Endeavors to extend our system for dichlorination to the dibromination of allylic alcohols was next undertaken. Like dichlorination, alkene dibromination has been a longstanding challenge in the area of enantioselective catalysis. From a practical perspective, enantioenriched dibromides could have great value as stereodefined building blocks for further functionalization. A palladium-catalyzed enantioselective alkene dibromination was reported in 2003 by Henry,3a but a subsequent report by Denmark3b questioned these results. In their reinvestigation, all attempts to reproduce this chemistry returned racemic dibromides. In 2013 our lab reported a TADDOL-catalyzed enantioselective dibromination reaction which was enantioselective only for electronically biased cinnamyl alcohols.2d We envisioned that our success in the enantioselective dichlorination of simple allylic alcohols could translate back to enantioselective dibromination.
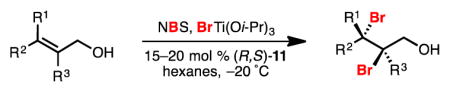
When NBS and BrTi(Oi-Pr)3 were used in place of t-BuOCl and ClTi(Oi-Pr)3 in the halogenation reaction, dibromides were formed in high yield and selectivity (Table 3). As in the case of dichlorination, the origins of this higher-than-predicted selectivity are unclear; possible explanations parallel those discussed earlier for dichlorination (Section 2.1).
Table 3.
Dibromination Substrate Scopea
| |||||
|---|---|---|---|---|---|
| entry | substrate | product | yield (%) | ee (%) | mol % 11 |
| 1b | 16 |
|
75 | 87 | 15 |
| 2 | 18 |
|
82 | 80 | 20 |
| 3c | 20 |
|
86 | 80 | 15 |
| 4c | 22 |
|
79 | 86 | 20 |
| 5c | 26 |
|
84 | 81 | 20 |
Conditions unless otherwise noted: 1.1 1.2 equiv NBS, 1.1 1.3 equiv BrTi(Oi-Pr)3, 15–20 mol % (R,S)-11, hexanes, −20 °C, 4–12 hours;
3:1 hexanes/CCl4;
absolute configuration unconfirmed.
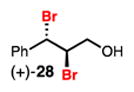
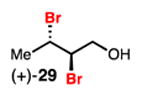
Dibromination of electronically biased allylic alcohol 16 proceeded smoothly, providing 28 in good yield and selectivity2d (Table 3, entry 1). Electronically unbiased allylic alcohols 18, 20, and 22 were also selectively dibrominated providing 29, 30, and 31 in good yields (Table 3, entries 2–4). 1,1-Disubstituted olefins such as 26 could be brominated as well, producing 32 efficiently and with good selectivity (Table 3, entry 5). In contrast to dichlorination, trisubstituted allylic alcohols are also viable substrates for di bromination.7 It is anticipated that these enriched dibromides could serve as useful chiral building blocks.
2.3 Synthesis of (–)-Deschloromytilipin A
With >10 grams of enantioenriched 19, a synthesis of the enantiomer of natural product (+)-(3), herein refered to as deschloromytilipin A, was executed (Scheme 2). This chlorosulfolipid was isolated in 20104e and lacks the vinyl choride found in mytilipin A. Using a strategy similar to that of Yoshimitsu6c and Vanderwal,6a,b oxidation and chloroallylation8 of 19 provided known trichloroalcohol 34 as a single diastereomer. Although never previously elaborated to a chlorosulfolipid, alcohol 34 has been prepared from crotyl alcohol in five chemical transformations in 66% ee using a kinetic resolution.6a,b Acryloylation of 34 and subsequent ring-closing metathesis yielded lactone 35, which was then reduced9 to enediol 36. The application of a Z-selective cross metathesis between 1,4-butenediol and chlorohydrin 34 to directly prepare diol 36 was attempted, but no reaction was observed.6a,10 In the course of this synthesis, it was found that diol 36 is prone to undergo facile intramolecular cyclization via an SN2′ displacement of the allylic chloride by the secondary alcohol. Hence, 36 was used directly after preparation to prevent this unwanted decomposition. Dichlorination of 36 with Et4NCl311 furnished diol 37 with high diastereoselectivity. Silylation and triflation of 37 provided pentachloride 38. Copper-catalyzed alkyl–alkyl cross coupling12 of 38 followed by sulfation completed the first synthesis of (–)-deschloromytilipin A (–)-3 in 11 steps from crotyl alcohol. This route provides access to the unnatural enantiomer of the natural product, which was targeted in order to demonstrate the power and utility of enantioselective catalysis when both ligand antipodes are readily available. This work also represents the first use of an enantioselective dihalogenation in the synthesis of a chlorosulfolipid.13
Scheme 2. Synthesis of (–)-Deschloromytilipin Aa.

aReagents and conditions: (a) Dess–Martin periodinane (1.3 equiv), NaHCO3 (3.0 equiv), CH2Cl2, 0 °C; (b) (Z)-chloroallylpinacolborinate 33 (1.0 equiv), n-BuLi (1.2 equiv), trifluoroacetic anhydride (1.3 equiv), THF, −78 °C, 51% over two steps; (c) acryloyl chloride (2.0 equiv), i-Pr2NEt (1.8 equiv), DMAP (0.2 equiv), CH2Cl2, 0 °C, 80%; (d) Hoveyda–Grubbs II (2 mol %), toluene, 90 °C, 86%; (e) CeCl3·7H2O (2.4 equiv), NaBH4 (2.4 equiv), EtOH, rt, 97%; (f) Et4NCl3 (1.o equiv), 10:1 CH2Cl2/EtOH, −78 °C, 65% (18:1 dr); (g) Tf2O (1.2 equiv), 2,6-lutidine (1.2 equiv), CH2Cl2, 0 °C, 82%; TESOTf (1.7 equiv), 2,6-di-tert-butylpyridine (1.7 equiv), CH2Cl2, 0 °C, 85%; (h) CuCl (0.1 equiv.), N,N,N′,N′-tetramethylethylenediamine (0.3 equiv), 6-hepten-1-yl magnesium bromide (1.2 equiv), THF, 0 °C to rt; 1 M aq. HCl, MeOH, rt, 28%; (i) SO3·pyridine (3.o equiv), THF, rt, 94%.
2.4 Synthesis of a Polybrominated Precursor Relevant to Bromosulfolipids
When the danicalipin A-producing organism, O. danica, is grown in bromide-rich media, the corresponding bromosulfolipid is purportedly made.14a This curiosity raises questions about the pathways and molecular machinery responsible for halogenation in these phytoflagellates, as well as fundamental structural questions about these bromosulfolipids. The salient barrier to the study of these peculiar molecules is the limited data that exists for their characterization. Indeed, isolation reports for most bromosulfolipids identify these structures solely by mass spectrometry.14 Prompted by a desire to provide structural and characterization data for relevant polybrominated motifs, selective syntheses of such stereoarrays were carried out.6k
The synthesis of the olefin precursor to pentabromodiols proceeded in an analogous manner to the synthesis of 36 (Scheme 3). Oxidation of enantioenriched dibromide 29 provided dibromoaldehyde 39, which was particularly capricious relative to its dichloroaldehyde counterpart and rapidly eliminated HBr upon standing at room temperature. Dibromoaldehyde 39, however, could be combined immediately with preformed bromoaluminum reagent 406a,b,15 to provide multigram quantaties of bromohydrin 41. Allylic bromide 41 could be further elaborated by acroylation and ring closing metathesis to provide lactone 42. Cerium borohydride reduction9 afforded diol 43 on gram scale. The relatively high efficiencies of these transformations were surprising given the generally perceived sensitivity of allylic bromide, dibromide, and bromohydrin functionalities.6k
Scheme 3. Synthesis of Stereotetrad 43a.

aReagents and conditions: (a) Dess–Martin periodinane (1.3 equiv.), NaHCO3 (3.0 equiv.), CH2Cl2, 0 °C; (b) 2,2,6,6-tetramethylpiperidine (2.2 equiv.), n-BuLi (2.1 equiv.), allyl bromide (2.1 equiv.), AlEt2Cl (4.0 equiv.), THF, −78 °C, 57%; (c) acryloyl chloride (1.5 equiv.), i-Pr2NEt (1.0 equiv.), DMAP (0.2 equiv.), CH2Cl2, 0 °C, 61%; (d) HG-II (2.5 mol%), toluene, 90 °C, 82%; (e) CeCl3·7H2O (2.4 equiv.), NaBH4 (2.4 equiv.), EtOH, 0 °C, 66%.
2.5 Synthesis and Solid State Conformation of Polychloride and Polybromide Stereohexads
Characterization and conformational data on stereohexad structures of type 37 and its polybrominated analogues could prove practically useful and academically insightful. First, synthesis and characterization of polybromide stereohexads would help confirm the existence of putative bromosulfolipids.6k Second, conformational data on 37 could provide insight in to the manner in which these molecules assemble in a lipid membrane. Third, investigation of unnatural diastereomers of 37 would expand the repertoire of characterization data for complex polyhalostereoarrays. This could be particularly useful in the stereochemical characterization of molecules like nominal undecachlorosulfolipid 7. For these reasons the syntheses of chlorinated and brominated stereohexads was undertaken.
With pentachloride 37 already made, the completion of the synthesis of the analogous pentabromide was conducted (Scheme 4). This was effected through a diastereoselective dibromination of alkene 43 with pyridinium tribromide and resulted in the predominant formation of a stereohexad with the same relative configuration as pentachloride 37. Such diastereoselectivity in dihalogenation of 36 and 43 using trihalide reagents is in accordance with previous reports concerning the dichlorination of similar substrates.6a,b,h The high diastereoselectivity (18:1) observed in the dichlorination suggests that strong substrate bias exists for the halogenation of substrates of this type.
Scheme 4. Synthesis of Stereohexads 44–47a.

aX-ray crystal structures of the natural and unnatural diastereomers of the polychloride stereohexad found within mytilipin A, along with their polybrominated analogues. The aryl carboxylate esters have been omitted in the crystal structures for clarity. Fcc = ferrocene carboxylate and R = p-bromobenzoate.
Efforts were made to obtain X-ray quality crystals of stereohexad 37 and its brominated analogue to compare the conformations of these polyhalides in the solid state. This presented a clear challenge due to the non-crystalline nature of such lipids. After intensive efforts, it was found that ferrocene carboxylate esters 44 and 46 were crystalline solids. Interestingly, 44 and 46 possess a “bent” conformation in the solid state (Scheme 4, top). This conformation is in agreement with that previously predicted by computation and observed in NMR studies for 3.4k Conversion of 37 to (–)-deschloromytilipin A (–)-3 (Scheme 2) confirms its absolute and relative configuration. This represents a unique case wherein the absolute and relative configuration of the complete stereoarray within a chlorosulfolipid natural product has been confirmed by X-ray crystallography.16
During our studies on polyhalogenated stereohexads, it was found that our reported catalytic system can overturn inherent substrate bias to provide access to unnatural diastereomers (36 to 45, and 43 to 47, Scheme 4). This selectivity is particularly impressive considering the number of halogen atoms that could participate in anchimeric assistance.17 Treating tribromoenediol 43 with the titanium-based dihalogenation conditions in the absence of ligand resulted in a reversal of inherent diastereoselectivity to 1:6 favoring diastereomer 47. Interestingly, the use of ligand (S,R)-11 resulted in the formation of 47 as the major diastereomer but in a lower diastereomeric ratio (2.3:1). On the other hand, the use of ligand enantiomer (R,S)-11 resulted in the formation of 47 with higher diastereoselectivity (11:1 dr). This suggests a matched/mismatched diastereoselectivity scenario between the ligand and the titanium-bound substrate – the inherent selectivity of titanium-based dibromination favors diastereomer 47, which is either enhanced or counteracted by the appropriate chiral ligand. Application of our dichlorination conditions to 36 with matched ligand enantiomer (R,S)-11 provided selective access to unnatural 45 in >20:1 dr. Prior to this work, there was no way to overturn the inherent diastereoselectivity of a given substrate in a dihalogenation without changing the substrate itself.
These unnatural polychloride and polybromide stereohexads could be crystallized as their p-bromobenzoates, 45 and 47. In contrast to the natural diastereomers, 45 and 47 possess a linear conformation in the solid state (Scheme 4, bottom). The implications of the structural differences between these polyhalide diastereomers for biological systems are intriguing but at this point unclear. Both pairs of crystal structures for the same diastereomers of pentachlorides and pentabromides (i.e. 44 and 46, and 45 and 47, Scheme 4) show remarkable conformational similarity at the unit cell level. This is unexpected given the differences in Van der Waals radii, polarizability, C–X bond length and electronegativity between chlorine and bromine.18 Pentachlorides 44 and 45 also provide characterization data for unnatural diastereomers of the stereohexad contained within the putatively assigned stereooctad of undecachlorosulfolipid 7.6h,i
2.6 Synthesis of (–)-Danicalipin A
The chlorosulfolipid danicalipin A (5) is estimated to comprise >90% of all polar lipids in the flagellar membrane of freshwater Ochromonas danica algae.19 It possesses a unique structure for a lipid,4b containing two anionic sulfate moieties, one at the terminus and one near the center of its otherwise hydrophobic linear core. Owing to this unusual structural feature and its complex halogenation pattern, danicalipin A has attracted much synthetic interest. To date there have been five reported syntheses of danicalipin A, the most efficient of which are 9 and 12 linear steps by Vanderwal6a and Carreira,6j respectively. Both of these syntheses commence from allylic alcohol starting materials. In the Vanderwal synthesis, access to an enantioenriched dichlorinated intermediate is achieved via a racemic dichlorination followed by resolution of a later intermediate. In the Carreira synthesis, the vicinal dichloride motif is established through diastereoselective epoxidation followed by chlorinolysis of the epoxide. We envisioned that our chemistry could be used to streamline the synthesis of danicalipin by providing one-step, enantioselecitve access to the requisite dichloride building block 21.
Strategically, it was thought that the unnatural enantiomer of danicalipin A might be prepared through a formal hydrochlorination of trans alkene 48, followed by sulfation (Scheme 5). Enantioenriched chlorohydrin alkene 48 might then be synthesized through an enantioselective chloroallylation reaction involving enantioenriched dichloroaldehyde 49 and a discrete, enantiopure chloroallylboronate of general structure 50. Aldehyde 49 would arise from dichlorination and oxidation of allylic alcohol 20. It was surmised that highly functionalized chloroallylboronate 50 might be prepared stereoselectively from a (Z)-chlorovinylboron reagent 51 and organometallic species 52 and 53 via the homologation chemistry of Matteson.20
Scheme 5.
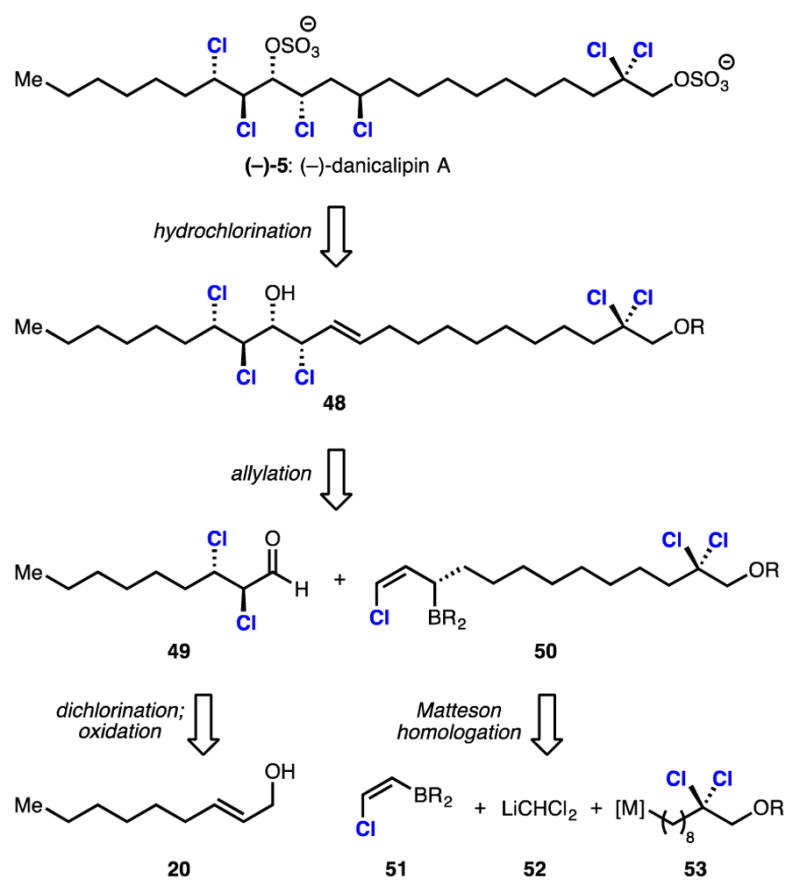
Retrosynthesis of (–)-Danicalipin A
The forward synthesis commenced with the development of a scalable synthesis of an appropriate (Z)-chlorovinylboron reagent (Scheme 6). The preparation of an analogous (Z)-(2-bromovinyl)-MIDA boronate has been accomplished previously by Burke.21 Following a similar strategy it was found that (Z)-(2-chlorovinyl)-MIDA boronate could be synthesized efficiently in one step by treatment of commercially available 54 with iodine monochloride in the presence of 2,6-lutidine. The pure product could be easily isolated by precipitation from diethyl ether. Subsequent treatment with aqueous hydroxide in the presence of one equivalent of commercial enantiopure diol 55 provided chromatographically stable chlorovinylboronic ester 56.20a
Scheme 6.

Concise Synthesis of (–)-Danicalipin Aa
aReagents and conditions: (a) iodine monochloride (2.0 equiv), 2,6-lutidine (3.0 equiv), CH2Cl2, 0 °C to rt, 74%; (b) (R,R)-1,2-dicyclohexyl-1,2-ethanediol 55 (1.1 equiv), aq. NaOH, THF, 0 °C, 85%; (c) CH2Cl2 (1.5 equiv), n-butyllithium (1.1 equiv), THF, –100 °C; 56, ZnCl2 (1.0 equiv), –100 °C to rt; (d) 59 (1.0 equiv), t-butyllithium (1.1 equiv), Et2O/hexanes, –78 °C; magnesium bromide ethyl etherate (1.2 equiv); then 57 (1.1 equiv), THF, –78 °C to rt, 24% from 56; (e) Dess–Martin periodinane (1.3 equiv), NaHCO3 (3.0 equiv), CH2Cl2, 0 °C to rt, 88%; (f) n-butyllithium (1.9 equiv), trifluoroacetic anhydride (1.9 equiv), THF, –78 °C; 49 (1.1 equiv), –78 °C to rt, 75% (g) methanol-d4, rt; concentrate; tetramethylammonium dichlorobromate (1.1 equiv), CH3CN, rt; (h) tributyltin hydride (1.1 equiv), triethylborane (0.2 equiv), air, toluene, –78 °C, 22% over two steps; (i) chlorosulfonic acid, CH2Cl2, rt, 93%.
Homologation to 1,3-dichloroallyl boronic ester 57 was next accomplished by treatment of 56 with dichloromethyllithium followed by zinc chloride according to Matteson’s procedure.20b,c Boronic ester 57 was unstable to chromatographic purification and prolonged storage, but solutions of crude 57 could be advanced to fully functionalized chloroallyl boronic ester 58 via a reaction involving the Grignard reagent of alkyl iodide 59. Boronic ester 58 was of >99% ee as determined by Mosher analysis after oxidation of 58 to its corresponding alcohol. This selectivity is particularly impressive as α-chloroallyl boronic esters have a high propensity to epimerize and decompose.20a,d Addition of magnesium bromide diethyl etherate to the organolithiate was crucial, as solutions of the intermediate organolithiate were found to be unstable and rapidly undergo lithium-halogen exchange on the gem-dichloride moiety.
With chloroallyl boronic ester 58 in hand, allylation reactions were attempted with aldehyde 49 which was obtained through oxidation of enantioenriched dichloroalcohol (–)-21. Initial attempts were low yielding and required excess dichloroaldehyde due to its propensity to decompose under the reaction conditions. Gratifyingly, it was found that chloroallyl boronic ester 58 could be induced to react efficiently with aldehyde 49 at low temperature by first activating it in situ as the borinate. This was accomplished through the addition of n-butyllithium and trifluoroacetic anhydride following a procedure developed by Aggarwal.22 Accordingly, this reaction afforded gram-quantaties of chlorohydrin alkene 60 as a single enantio- and diastereomer in 75% yield. Serendipitously, it was discovered that a kinetic resolution occurs during this chloroallylation reaction, with the minor enantiomer of dichloroaldehyde 49 being essentially unreactive.
After failed attempts to directly hydrochlorinate 60,23 a formal hydrochlorination was achieved through a bromochlorination–radical debromination sequence.24 Initial attempts to bromochlorinate 60 provided the desired bromochloride in low yield due to competitive bromoetherification arising from capture of an intermediate bromonium by the secondary alcohol of 60. Investigation of bromochlorination conditions identified Me4N(Cl2Br)25 as the best source of “BrCl,” providing the desired bromochloride 63 in a mere 21% yield along with 65% yield of bromoether 64, both as single diastereomers (Scheme 7). Although low yielding, the clean reactivity was encouraging as previous reports concerning the iodochlorination of similar substrates gave complex mixtures of products.6a Protection of the alcohol in 60 completely shut down any desired reactivity and underscored the need for having a free alcohol for this dihalogenation reaction. This led us to hypothesize that the ratio of desired bromochloride to undesired bromoether might be influenced by O-deuteration of 60 as this should alter the nature of hydrogen bonding between substrate and halogenating agent. In practice it was found that pretreatment of alcohol 60 with methanol-d4 followed by solvent removal and subsequent reaction with Me4N(Cl2Br) in acetonitrile increased the yield of the bromochlorination to 44%.26
Scheme 7.
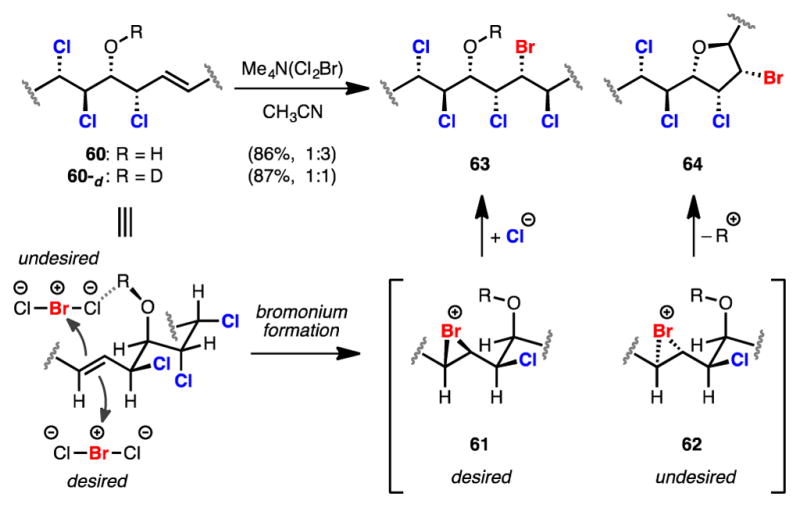
Rationale for the Effect of O-Deuteration
Rationale for this observed change in product ratio is depicted in Scheme 7. Based on the subsequent transformation of bromochloride 63 to (–)-danicalipin A (–)-5 (Scheme 6) and NOE analysis of bromoether 64 (see Supporting Information), it was determined that these products likely arise from diastereomeric bromonium intermediates 61 and 62. Assuming bromonium formation occurs on the conformer for 60 displayed in Scheme 7, the undesired bromonium formation could be directed through hydrogen bonding of the secondary alcohol; deuteration may then render this direction less effective, thus reducing the amount of bromoether. Another possible rationalization is that bromonium formation is reversible and deprotonation of the alcohol during bromoetherification is selectivity determining. Accordingly, the stronger oxygen-deuterium bond would be harder to heterolytically cleave, effectively disfavoring the bromoetherification pathway.
Our synthesis of the unnatural enantiomer of danicalipin A was completed by radical debromination of 63 followed by sulfation (Scheme 6). It is noteworthy that radical debromination can be carried out chemoselectively (74% yield based on recovered starting material) in the presence of alkyl chloride and geminal dichloride functionalities. The described synthesis is 8 linear steps and provides access to a single isomer of (–)-danicalipin A (–)-5.
3. CONCLUSION
The enantioselective dichlorination and dibromination of electronically unbiased, aliphatic allylic alcohols has been demonstrated. The developed method is practical, scalable, and provides access to valuable dichloroalcohols used in the preparation of chlorosulfolipid natural products. Enantioselective syntheses of two chlorosulfolipids, ent-(–)-deschloromytilipin (–)-3 and ent-(–)-danicalipin A (–)-5, using this chemistry as the first step has been described, highlighting the utility and practicality of the system. We anticipate that this method could enable the selective syntheses of all members of the chlorosulfolipid family. Conversion of the dihalo-alcohol products to the relevant stereoarry of naturally occurring stereohexads has provided insight into the absolute stereochemistry of these molecules as well as their conformation. The tactics reported here may be applied to both the synthesis and structural assignment of undecachlorosulfolipid 7 whose structure remains ambiguous.6h,i
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by Stanford University, the National Institutes of Health (R01 GM114061), and the National Science Foundation (DGE-114747 GRF to MLL and DXH).
We are grateful to Dr. A. Oliver (University of Notre Dame) for X-ray crystallographic analysis and Dr. S. Lynch (Stanford University) for assistance with NMR spectroscopy.
Footnotes
Notes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
Experimental procedures, characterizations, spectral data, and CIF files.
The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.(a) Atterberg A, Widman O. Ber Dtsch Chem Ges. 1877;10:1841–1844. [Google Scholar]; (b) Denmark SE, Kuester WE, Burk MT. Angew Chem Int Ed. 2012;51:10938–10953. doi: 10.1002/anie.201204347. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cresswell AJ, Eey STC, Denmark SE. Angew Chem Int Ed. 2015;54:15642–15682. doi: 10.1002/anie.201507152. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chung W-j, Vanderwal CD. Angew Chem Int Ed. 2016 doi: 10.1002/anie.201506388. [DOI] [Google Scholar]
- 2.Snyder SA, Tang ZY, Gupta R. J Am Chem Soc. 2009;131:5744–5745. doi: 10.1021/ja9014716.Monaco MR, Bella M. Angew Chem Int Ed. 2011;50:11044–11046. doi: 10.1002/anie.201104843.Nicolaou KC, Simmons NL, Ying Y, Heretsch PM, Chen JS. J Am Chem Soc. 2011;133:8134–8137. doi: 10.1021/ja202555m.For a catalytic enantioselective dibromination of cinnamyl alcohols, see: Hu DX, Shibuya GM, Burns NZ. J Am Chem Soc. 2013;135:12960–12963. doi: 10.1021/ja4083182.Recently a catalytic, stereo-specific, syn-dichlorination of alkenes was reported: Cresswell AJ, Eey STC, Denmark SE. Nature Chem. 2015;7:146–152. doi: 10.1038/nchem.2141.For a newly reported method for the synthesis of alkyl halides, see: Iwasaki K, Wan KK, Oppedisano A, Crossley SWM, Shenvi RA. J Am Chem Soc. 2014;136:1300–1303. doi: 10.1021/ja412342g.
- 3.(a) El-Qisairi AK, Qaseer HA, Katsigras G, Lorenzi P, Trivedi U, Tracz S, Hartman A, Miller JA, Henry PM. Org Lett. 2003;5:439–441. doi: 10.1021/ol0273093. [DOI] [PubMed] [Google Scholar]; (b) Denmark SD, Carson N. Org Lett. 2015;17:5728–5731. doi: 10.1021/acs.orglett.5b02650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Elovson J, Vagelos PR. Proc Natl Acad Sci USA. 1969;62:957–963. doi: 10.1073/pnas.62.3.957. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Haines TH, Pousada M, Stern B, Mayers GL. Biochem J. 1969;113:565–566. doi: 10.1042/bj1130565. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mayers GL, Pousada M, Haines TH. Biochemistry. 1969;8:2981–2986. doi: 10.1021/bi00835a045. [DOI] [PubMed] [Google Scholar]; (d) Elovson J, Vagelos PR. Biochemistry. 1970;9:3110–3126. doi: 10.1021/bi00818a002. [DOI] [PubMed] [Google Scholar]; (e) Haines TH. Prog Chem Fats Lipids. 1971;11:297–345. [Google Scholar]; (f) Chen JL, Proteau PJ, Roberts MA, Gerwick WH, Slate DL, Lee RH. J Nat Prod. 1994;57:524–527. doi: 10.1021/np50106a015. [DOI] [PubMed] [Google Scholar]; (g) Ciminiello P, Fattorusso E, Forino M, Magno S, Di Rosa M, Ianaro A, Poletti R. J Org Chem. 2001;66:578–582. doi: 10.1021/jo001437s. [DOI] [PubMed] [Google Scholar]; (h) Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, Di Rosa M, Ianaro A, Poletti R. J Am Chem Soc. 2002;124:13114–13120. doi: 10.1021/ja0207347. [DOI] [PubMed] [Google Scholar]; (i) Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, Magno S, Di Meglio P, Ianaro A, Poletti R. Tetrahedron. 2004;60:7093–7098. [Google Scholar]; (j) Kawahara T, Kumaki Y, Kamada T, Ishii T, Okino T. J Org Chem. 2009;74:6016–6024. doi: 10.1021/jo900860e. [DOI] [PubMed] [Google Scholar]; (k) Chao C-H, Huang H-C, Wang G-H, Wen Z-H, Wang W-H, Chen I-M, Sheu J-H. Chem Pharm Bull. 2010;58:944–946. doi: 10.1248/cpb.58.944. [DOI] [PubMed] [Google Scholar]
- 5.(a) Umezawa T, Matsuda F. Tetrahedron Lett. 2014;55:3003–3012. [Google Scholar]; (b) Chung WJ, Vanderwal CD. Acc Chem Res. 2013;47:718–728. doi: 10.1021/ar400246w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nilewski C, Carreira EM. Eur J Org Chem. 2012:1685–1698. [Google Scholar]; (d) Bedke DK, Vanderwal CD. Nat Prod Rep. 2011;28:15–25. doi: 10.1039/c0np00044b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Chung W-J, Carlson JS, Vanderwal CD. J Org Chem. 2014;79:2226–2241. doi: 10.1021/jo5000829. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chung WJ, Carlson JS, Bedke DK, Vanderwal CD. Angew Chem Int Ed. 2013;52:10052–10055. doi: 10.1002/anie.201304565. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yoshimitsu T, Nakatani R, Kobayashi A, Tanaka T. Org Lett. 2011;13:908–911. doi: 10.1021/ol1029518. [DOI] [PubMed] [Google Scholar]; (d) Umezawa T, Shibata M, Kaneko K, Okino T, Matsuda E. Org Lett. 2011;13:904–907. doi: 10.1021/ol102882a. [DOI] [PubMed] [Google Scholar]; (e) Bedke DK, Shibuya GM, Pereira AR, Gerwick WH, Vanderwal CD. J Am Chem Soc. 2010;132:2542–2543. doi: 10.1021/ja910809c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yoshimitsu T, Fukumoto N, Nakatani R, Kojima N, Tanaka T. J Org Chem. 2010;75:5425–5437. doi: 10.1021/jo100534d. [DOI] [PubMed] [Google Scholar]; (g) Bedke DK, Shibuya GM, Pereira A, Gerwick WH, Haines TH, Vanderwal CD. J Am Chem Soc. 2009;131:7570–7572. doi: 10.1021/ja902138w. [DOI] [PubMed] [Google Scholar]; (h) Nilewski C, Geisser RW, Carreira EM. Nature. 2009;457:573–576. doi: 10.1038/nature07734. [DOI] [PubMed] [Google Scholar]; (i) Nilewski C, Deprez NR, Fessard TC, Li DB, Geisser RW, Carreira EM. Angew Chem Int Ed. 2011;50:7940–7943. doi: 10.1002/anie.201102521. [DOI] [PubMed] [Google Scholar]; (j) Bailey AM, Wolfrum S, Carreira EM. Angew Chem Int Ed. 2016;55:639–643. doi: 10.1002/anie.201509082. [DOI] [PubMed] [Google Scholar]; (k) Fischer S, Huwyler N, Wolfrum S, Carreira EM. Angew Chem Int Ed. 2016;55:2555–2558. doi: 10.1002/anie.201510608. [DOI] [PubMed] [Google Scholar]
- 7.Hu DX, Seidl FJ, Bucher C, Burns NZ. J Am Chem Soc. 2015;137:3795–3798. doi: 10.1021/jacs.5b01384. [DOI] [PubMed] [Google Scholar]
- 8.(a) Hoffman RW, Wolff JJ. Chem Ber. 1991;124:563–569. [Google Scholar]; (b) Hoffman RW, Dresely S, Lanz JW. Chem Ber. 1988;121:1501–1507. [Google Scholar]; (c) Rathke MW, Chao E, Wu G. J Organometal Chem. 1976;122:145–149. [Google Scholar]
- 9.Hale KJ, Hummersone MG, Bhatia GS. Org Lett. 2000;2:2189–2192. doi: 10.1021/ol005850y. [DOI] [PubMed] [Google Scholar]
- 10.Koh MJ, Khan RKM, Torker S, Yu M, Mikus MS, Hoveyda AH. Nature. 2015;517:181–186. doi: 10.1038/nature14061.. (b) Also see ref. 6a and references therein.
- 11.Schlama T, Gabriel K, Gouverneur V, Mioskowski C. Angew Chem Int Ed. 1997;36:2342–2344. [Google Scholar]
- 12.(a) Tamura M, Kochi J. Synthesis. 1971:303–305. [Google Scholar]; (b) Yang CT, Zhang ZQ, Liang J, Liu JL, Lu XY, Chen HH, Liu L. J Am Chem Soc. 2012;134:11124–11127. doi: 10.1021/ja304848n. [DOI] [PubMed] [Google Scholar]
- 13.For other syntheses featuring enantioselective dihalogenation, see: Bucher C, Deans RM, Burns NZ. J Am Chem Soc. 2015;137:12784–12787. doi: 10.1021/jacs.5b08398.. (b) See also ref. 2a.
- 14.(a) Haines TH. Annu Rev Microbiol. 1973;27:403–411. doi: 10.1146/annurev.mi.27.100173.002155. [DOI] [PubMed] [Google Scholar]; (b) Bruckstein AH. Baskerville Chem J. 1970;18:3–4. [Google Scholar]
- 15.Hosomi A, Kohra S, Tominaga Y, Ando M, Sakurai H. Chem Pharm Bull. 1987;35:3058–3061. [Google Scholar]
- 16.Nilewski C, Geisser RW, Ebert MO, Carreira EM. J Am Chem Soc. 2009;131:15866–15876. doi: 10.1021/ja906461h. [DOI] [PubMed] [Google Scholar]
- 17.Shemet A, Sarlah D, Carreira EM. Org Lett. 2015;17:1878–1881. doi: 10.1021/acs.orglett.5b00558.. (b) See also ref. 6h.
- 18.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; Sausalito, CA: 2006. [Google Scholar]
- 19.Chen LL, Pousada M, Haines TH. J Biol Chem. 1976;251:1835–1842. [PubMed] [Google Scholar]
- 20.(a) Hoffman RW, Ditrich K, Köster G, Stürmer R. Chem Ber. 1989;122:1783–1789. [Google Scholar]; (b) Matteson DS, Majumdar D. J Am Chem Soc. 1980;102:7588–7590. [Google Scholar]; (c) Matteson DS. J Org Chem. 2013;78:10009–10023. doi: 10.1021/jo4013942. [DOI] [PubMed] [Google Scholar]; (d) Hoffmann RW, Dresely S, Lanz JW. Chem Ber. 1988;121:1501–1507. [Google Scholar]
- 21.Woerly EM, Sruble JR, Palyam N, O’Hara SP, Burke MD. Tetrahedron. 67:4333–4343. doi: 10.1016/j.tet.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Chen JLY, Scott HK, Hesse MJ, Willis CL, Aggarwal VK. J Am Chem Soc. 2013;135:5316–5319. doi: 10.1021/ja401564z. [DOI] [PubMed] [Google Scholar]; (b) Chen JLY, Aggarwal VK. Angew Chem Int Ed. 2014;53:10992–10996. doi: 10.1002/anie.201407127. [DOI] [PubMed] [Google Scholar]
- 23.(a) Gaspar B, Carreira EM. Angew Chem Int Ed. 2008;120:5842–5844. [Google Scholar]; (b) Leggans EK, Barker TJ, Duncan KK, Boger DL. Org Lett. 2012;14:1428–1431. doi: 10.1021/ol300173v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kropp PJ, Daus KA, Crawford SD, Tubergen MW, Kepler KD, Craig SL, Wilson VP. J Am Chem Soc. 1990;112:7433–7434. [Google Scholar]
- 24.A similar iodochlorination–deiodination on the corresponding cis olefin has been used by Vanderwal: see ref. 6a
- 25.(a) Negoro T, Ikeda Y. Bull Chem Soc Jpn. 1984;57:2111–2115. [Google Scholar]; (b) Popov AI, Buckles RE. Inorg Synth. 1957;5:167–175. [Google Scholar]
- 26.For other uses of deuterium isotope effects in natural product total synthesis, see: Vedejs E, Little J. J Am Chem Soc. 2002;124:748–749. doi: 10.1021/ja0120835.Clive DLJ, Cantin M, Khodabocus A, Kong X, Tao Y. Tetrahedron. 1993;49:7917–7930.Clive DLJ, Tao Y, Khodabocus A, Wu Y-J, Angoh AG, Bennett SM, Boddy CN, Bordeleau L, Kellner D, Gleiner G, Middleton DS, Nichols CJ, Richardson SR, Vernon PG. J Am Chem Soc. 1994;116:11275–11286.Miyashita M, Sasaki M, Hattori I, Sakai M, Tanino K. Science. 2004;305:495–499. doi: 10.1126/science.1098851.Quasdorf KW, Huters AD, Lodewyk MW, Tantillo DJ, Garg NK. J Am Chem Soc. 2012;134:1396–1399. doi: 10.1021/ja210837b.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.