

Abstract
Dickkopf1 (DKK1) is a secretory protein that antagonizes oncogenic Wnt signaling by binding to the Wnt coreceptor low-density lipoprotein receptor–related protein 6 (LRP6). DKK1 may also regulate its own signaling to promote cancer cell proliferation, but the mechanism is not understood. Here, we identified cytoskeleton-associated protein 4 (CKAP4) as a DKK1 receptor and evaluated CKAP4-mediated DKK1 signaling in cancer cell proliferation. We determined that DKK1 binds CKAP4 and LRP6 with similar affinity but interacts with these 2 receptors with different cysteine-rich domains. DKK1 induced internalization of CKAP4 in a clathrin-dependent manner, further supporting CKAP4 as a receptor for DKK1. DKK1/CKAP4 signaling activated AKT by forming a complex between the proline-rich domain of CKAP4 and the Src homology 3 domain of PI3K, resulting in proliferation of normal cells and cancer cells. Expression of DKK1 and CKAP4 was frequent in tumor lesions of human pancreatic and lung cancers, and simultaneous expression of both proteins in patient tumors was negatively correlated with prognosis and relapse-free survival. An anti-CKAP4 antibody blocked the binding of DKK1 to CKAP4, suppressed AKT activity in a human cancer cell line, and attenuated xenograft tumor formation in immunodeficient mice. Together, our results suggest that CKAP4 is a potential therapeutic target for cancers that express both DKK1 and CKAP4.
Introduction
Dickkopf1 (DKK1) was originally identified as an embryonic head inducer in Xenopus embryos and shown to be a secreted protein that antagonizes Wnt signaling (1, 2). Of the multiple Wnt signaling pathways, including β-catenin–dependent and –independent pathways (3, 4), DKK1 has been thought to modulate the β-catenin–dependent pathway (β-catenin pathway). DKK1 contains 2 characteristic cysteine-rich domains (CRD1 and CRD2) (1) and binds to low-density lipoprotein receptor–related protein 5 (LRP5) or LRP6, which functions as a Wnt coreceptor, through CRD2, thereby suppressing the β-catenin pathway (5–8). Wnt3a induces LRP6 internalization in a caveolin-dependent manner, and the internalization was required for activation of the β-catenin pathway in certain types of cells (9–12), while DKK1 induces LRP6 internalization through a clathrin-mediated route resulting in removal of LRP6 from the plasma membrane (5, 13), thereby inhibiting the β-catenin pathway. Because DKK1 is one of the direct target molecules expressed by the β-catenin pathway (14, 15), it is believed that DKK1 creates a negative-feedback loop for the β-catenin pathway.
Genetic alterations of the β-catenin pathway components, including adenomatous polyposis coli (APC), β-catenin, and AXIN, are frequently observed in various human cancers where the β-catenin pathway is aberrantly activated (16). Given that DKK1 is a downstream target gene of the β-catenin pathway, it is reasonable that DKK1 overexpression was observed in multiple myeloma, hepatocellular carcinoma, and prostate, kidney, lung, pancreatic, and esophageal cancers (17–21), if the β-catenin pathway is aberrantly activated in these cancers. It has also been reported that DKK1 expression was reduced because of DKK1 DNA hypermethylation in colon cancer, and overexpression of DKK1 suppressed intestinal epithelial proliferation and tumorigenicity of colon cancer cells (14, 22, 23). Therefore, DKK1 has been suggested to have tumor suppressor ability. DKK1, however, showed a positive role for cell proliferation in human adult bone marrow cells and human lung cancer A549 cells, and in A549 cells anti-DKK1 antibody suppressed cellular proliferation (18, 24), suggesting that DKK1 has distinct functions independent of the β-catenin pathway. Therefore, the significance of DKK1 expression might vary in different cancer contexts. We hypothesized that DKK1 binds to an unknown cell surface receptor, other than LRP6, to stimulate cellular proliferation, and that DKK1 and the novel receptor are implicated in human cancers.
Using mass spectrometry analyses, we identified cytoskeleton-associated protein 4 (CKAP4, also known as P63, CLIMP-63, and ERGIC-63) as a novel DKK1-binding protein on the cell surface membrane of Madin-Darby canine kidney (MDCK) epithelial cells. CKAP4 is a type II transmembrane protein that is reversibly palmitoylated (25, 26). It was originally discovered as a protein that is localized to the ER and binds to microtubules. Subsequently, CKAP4 was shown to be localized to the cell surface membrane of type II pneumocytes, bladder epithelial cells, and vascular smooth muscle cells, where it functions as a receptor for several ligands, including surfactant protein A (SP-A), tissue plasminogen activator (tPA), and anti-proliferating factor (APF) (27–29).
Here we showed that CKAP4 is a receptor for DKK1 and DKK1/CKAP4 signaling promotes normal and cancer cell proliferation through the PI3K/AKT pathway. In addition, we found that coexpression of DKK1 and CKAP4 is negatively correlated with prognosis and relapse-free survival of patients with pancreatic and lung cancers and that anti-CKAP4 antibody suppresses xenograft tumor formation in immunodeficient mice.
Results
DKK1 is secreted apically in polarized MDCK cells and stimulates cellular proliferation.
Recently, we demonstrated that different Wnts (Wnt3a, Wnt5a, and Wnt11) and Wnt receptors (frizzled 2, frizzled 7, LRP6, ROR2, and RYK) have distinct secretory and trafficking routes in polarized MDCK epithelial cells (30, 31). For instance, Wnt3a was secreted basolaterally and LRP6 was trafficked to the basolateral membrane. In order to compare with the Wnt trafficking routes, the secretory pathway of DKK1 in polarized MDCK cells was examined. MDCK cells expressing DKK1-FLAG (MDCK/DKK1-FLAG) were generated (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI84658DS1), and the mRNA levels of endogenous dog DKK1 and exogenously expressed human DKK1 were quantified by PCR using the same primers. Since MDCK cells expressed little DKK1 mRNA, the exogenous human DKK1 mRNA level was calculated to be about 38,000-fold more than the endogenous dog DKK1 mRNA level (Supplemental Table 1). MDCK/DKK1-FLAG cells were cultured as a monolayer on a filter support to establish 2-dimensional (2D) apical-basolateral polarity, and DKK1-FLAG was predominantly secreted in the apical fractions (Figure 1A). Immunocytochemical study also showed that DKK1-FLAG was more dominantly present at the apical membrane compared with the basolateral membrane of MDCK cells (Figure 1B). The concentration of DKK1-FLAG secreted from MDCK/DKK1-FLAG into conditioned medium was calculated to be 57.2 nM (Figure 1C).
Figure 1. DKK1 promotes MDCK cellular proliferation.

(A) MDCK/DKK1-FLAG cells were subjected to the apical-basolateral sorting assay. Ap, apical side; Bl, basolateral side. (B) MDCK/DKK1-FLAG cells cultured on a Transwell filter were stained with anti-DKK1 (green) and anti–E-cadherin (red, a basolateral marker) antibodies and DRAQ5 DNA Dye (nucleus, blue). (C) Different volumes (1, 2, and 5 μl) of conditioned medium from MDCK and MDCK/DKK1-FLAG cells were probed with anti-DKK1 antibody. (D and E) Outside diameter of MDCK and MDCK/DKK1-FLAG cysts cultured in 3D Matrigel was measured (n = 50) (D). The cells were stained with the indicated antibodies and DRAQ5 DNA Dye (E). Ki-67–positive rate was expressed as the percentage of Ki-67–staining cells in DRAQ5-staining cells per cyst (n = 50). Results are plotted as box and whiskers where the median is represented with a line, the box represents the 25th to 75th percentiles, and error bars show the 5th to 95th percentiles. *P < 0.05; **P < 0.01 (2-tailed Student’s t test). (F) MDCK cells were apically (Ap) or basolaterally (Bl) treated with 250 ng/ml DKK1, and then the cells were stained with anti–Ki-67 antibody (red) and DRAQ5 DNA Dye (blue). Ki-67–positive cells are expressed as the percentage of positively stained cells compared with total cells per field (n = 5 fields). NT, nontreatment. Results are shown as means ± SD of 3 independent experiments. **P < 0.01; NS, not significant (compared with the NT group, 2-tailed Student’s t test). (G) Cell numbers of control MDCK, MDCK/DKK1-FLAG, MDCK/DKK1-FLAG-GPI, or MDCK/DKK1-SPCUT-FLAG cells were counted. SPCUT, signal peptide–deleted DKK1. Results are shown as means ± SD of 3 independent experiments. Scale bars: 20 μm (B, E, and F); 100 μm (D).
In 3D culture conditions with Matrigel, MDCK cells form cysts (32). The size of MDCK cysts was increased and Ki-67–positive cells were concomitantly increased by DKK1 expression (Figure 1, D and E). Furthermore, in a filter-support 2D culture of polarized MDCK cells, addition of recombinant DKK1 to the apical side resulted in an increase in Ki-67–positive cells, but addition to the basolateral side showed no effect (Figure 1F). DKK1 expression in MDCK cells indeed suppressed AXIN2 mRNA expression induced by Wnt3a when the cells were cultured in ordinary 2D culture conditions in which they were not well polarized (Supplemental Figure 1B). Irrespective of the β-catenin signal inhibitory activity, DKK1 promoted MDCK cell proliferation in 2D culture conditions (Figure 1G). As a control, expression of DKK1-SPCUT, in which a signal peptide was removed from DKK1, did not affect cellular proliferation (Figure 1G and Supplemental Figure 1A). DKK1 knockdown reduced the DKK1 concentration in conditioned medium of MDCK/DKK1-FLAG cells to 0.17 nM (Supplemental Figure 1C). Consistently, DKK1-enhanced Ki-67–positive cells were decreased by DKK1 knockdown (Supplemental Figure 1, C and E). Knockdown of LRP6, LRP5, and Kremen1, which are known to bind to DKK1 (5, 33), did not affect DKK1-enhanced Ki-67 staining (Supplemental Figure 1, D and E). Taken together, these results suggest that DKK1 acts on the putative receptor that is located on the apical membranes in MDCK cells and stimulates cellular proliferation.
CKAP4 is a novel DKK1 receptor.
To identify the receptor that mediates DKK1-dependent cellular proliferation, a glycosylphosphatidylinositol (GPI) anchor motif was fused to the C-terminal region of DKK1-FLAG and the construct was expressed in MDCK cells (MDCK/DKK1-FLAG-GPI cells), because proteins with the GPI anchor motif are trafficked to the apical membrane and retained there (34). Expression of DKK1-FLAG-GPI stimulated cellular proliferation to levels similar to those stimulated by DKK1-FLAG expression (Figure 1G and Supplemental Figure 1A). The cell surface proteins of MDCK/DKK1-FLAG-GPI cells were biotinylated and cell lysates were precipitated with anti-FLAG antibody, and the eluates were further purified using NeutrAvidin Agarose beads (Supplemental Figure 2A). The precipitated proteins were stained with silver staining and analyzed by mass spectrometry (Figure 2A and Supplemental Table 2). Among the DKK1-binding proteins, we focused on CKAP4.
Figure 2. CKAP4 is identified as a novel DKK1-interacting protein.

(A) The DKK1-interacting proteins were detected by silver staining. Seventeen proteins that bind to DKK1-FLAG-GPI were analyzed by mass spectrometry. The results are listed in Supplemental Table 2. (B) Polarized MDCK cells were biotinylated apically (Ap) or basolaterally (Bl), and cell surface proteins were precipitated with NeutrAvidin Agarose beads. The precipitates were probed with anti-CKAP4 and anti–transferrin receptor (TfR) antibodies. TfR was used as a basolateral membrane protein marker. (C) Lysates (Input) of control MDCK, MDCK/DKK1-FLAG, or MDCK/DKK1-FLAG-GPI cells were immunoprecipitated with anti-FLAG antibody. The IPs were probed with the indicated antibodies. (D) Lysates (Input) of MDCK cells stably expressing WT or deletion mutants of DKK1-FLAG were immunoprecipitated with anti-FLAG antibody. The IPs were probed with the indicated antibodies. CRD, cysteine-rich domain. *Nonspecific band. (E and F) Various deletion mutants of CKAP4 were transiently expressed in X293T/DKK1-FLAG cells, and cell lysates were immunoprecipitated with anti-FLAG antibody or nonimmune IgG and the IPs were probed with the indicated antibodies. ECD, extracellular domain. (G) Top panel: The indicated concentration of DKK1 was incubated with 0.5 nM GST-CKAP4-ECD or 0.5 nM GST for 2 hours. The precipitates were probed with anti-DKK1 antibody. Bottom panels: The signals of DKK1 bound to GST-CKAP4-ECD (circles) or GST (squares) were quantified using ImageJ software and expressed as arbitrary units. Scatchard analysis of the binding is shown. (H) MDCK cells were stimulated with 10 nM DKK1, and cell surface proteins were biotinylated at each time point and precipitated with NeutrAvidin Agarose beads. The precipitates were probed with anti-CKAP4 and anti-clathrin antibodies. Clathrin was used as a loading control. Results are shown as means ± SD of 3 independent experiments.
CKAP4 was indeed localized to the apical membranes of polarized MDCK cells (Figure 2B). DKK1-FLAG and DKK1-FLAG-GPI indeed formed a complex with endogenous CKAP4 and LRP6 in MDCK cells (Figure 2C). DKK1 contains 2 cysteine-rich domains (CRD1 and CRD2), which are conserved among DKK family members (2), and CRD2 has been shown to play a role in LRP6-binding and Wnt inhibitory activities (6, 7). Using DKK1 deletion mutants, CRD1, but not CRD2, was shown to be required for DKK1 binding to CKAP4 (Figure 2D and Supplemental Figure 2B). The DKK1ΔCRD1-FLAG was secreted from MDCK/DKK1ΔCRD1-FLAG cells more efficiently than DKK1-FLAG was secreted from MDCK/DKK1-FLAG cells, although the reasons are unknown (Supplemental Figure 2C). Under the conditions that DKK1ΔCRD1-FLAG was able to suppress Wnt3a-dependent AXIN2 expression, the DKK1 mutant did not affect MDCK cellular proliferation (Supplemental Figure 1B and Supplemental Figure 2D).
Various deletion mutants of CKAP4-HA were expressed in X293T/DKK1-FLAG cells (Supplemental Figure 3A), and DKK1-FLAG was immunoprecipitated. Extracellular domain (ECD) of CKAP4-HA and CKAP4ΔC1-HA formed a complex with DKK1-FLAG to levels similar to those in WT CKAP4-HA, but CKAPΔC2-HA or CKAP4ΔC3-HA did not (Figure 2E). In addition, DKK1-FLAG did not interact with leucine zipper (LZ) domain deletion mutant (ΔLZ) of CKAP4-HA (Figure 2F). Therefore, the LZ domain of CKAP4 could be necessary for CKAP4 binding to DKK1.
DKK1 bound to CKAP4-ECD directly in a dose-dependent manner, and the KD was calculated to be 0.42 nM (Figure 2G). Biotinylation assay showed that cell surface localization of CKAP4 was reduced and increased by overexpression of DKK1 or depletion of exogenous DKK1, respectively (Supplemental Figure 3B). Also, the addition of recombinant DKK1 decreased CKAP4 from the cell surface membrane in time- and dose-dependent manners (Figure 2H and Supplemental Figure 3C). The concentration of DKK1 required for 50% of maximal response was 0.24 nM (Supplemental Figure 3C), which is similar to the KD value for the binding of DKK1 to CKAP4. Monodansylcadaverine (35) and chlorpromazine, which both inhibit clathrin-dependent receptor endocytosis, inhibited DKK1-dependent internalization of CKAP4 (Supplemental Figure 3D). Taken together, these results suggest that CKAP4 acts as a receptor for DKK1 and DKK1 induces CKAP4 internalization in a clathrin-dependent manner.
DKK1 signaling through CKAP4 activates the PI3K and AKT pathway, resulting in cellular proliferation.
CKAP4 knockdown inhibited DKK1-induced MDCK cellular proliferation but not DKK1-independent cellular proliferation in 2D culture (Figure 3A). To understand the mechanism underlying DKK1-dependent proliferation, the effects of various kinase inhibitors on DKK1-mediated cellular proliferation were examined. Among the inhibitors, SP600125 (a JNK inhibitor), PP2 (a Src inhibitor), Wortmannin (a PI3K inhibitor), and AKT inhibitor VIII all significantly suppressed DKK1-dependent proliferation (Figure 3B). AKT activation, which is detected by anti-AKT (anti–phosphorylated AKT [Ser473]) antibody, was increased to a greater extent in MDCK/DKK1-FLAG and MDCK/DKK1-FLAG-GPI cells compared with control MDCK cells. The activation of other kinases, including ERK, JNK, and Src, was not increased (Figure 3C). CKAP4 knockdown suppressed AKT activity in MDCK/DKK1-FLAG cells to a greater extent than control MDCK cells (Figure 3D). Recombinant DKK1 activated AKT in a time-dependent manner (Figure 3E). PI3K produces phosphatidylinositol-3,4,5-triphosphate (PIP3), which in turn contributes to the activation of AKT (36). Wortmannin, LY294002, and AKT inhibitor VIII or knockdown of p110α, a PI3K catalytic subunit, inhibited AKT activity in MDCK/DKK1-FLAG cells (Figure 3, F and G), indicating that PI3K is involved in DKK1-dependent AKT activation. Classical PI3K consists of p110α (or β) catalytic and p85 regulatory subunits (37, 38). Endogenous CKAP4 formed a complex with endogenous p85α and p110α in a DKK1-dependent manner in MDCK and X293T cells (Figure 4, A and B). In X293T/DKK1-FLAG cells, p85α was not associated with the CKAP4 mutant in which the cytoplasmic region was deleted (CKAP4-ECD-HA) (Figure 4C).
Figure 3. DKK1 signaling through CKAP4 activates the PI3K/AKT pathway, resulting in cellular proliferation.

(A) Control MDCK and MDCK/DKK1-FLAG cells were transfected with siRNA for control or CKAP4, and the cells were then subjected to the 2D cell proliferation assay. Results are shown as the mean ± SD of 3 independent experiments. (B) Control MDCK and MDCK/DKK1-FLAG cells were cultured at densities of 1 × 105 cells in a 35-mm dish 2-dimensionally for 60 hours. The cells were treated for the last 36 hours with 10 μM CHIR99021, 100 μM CKI-7, 10 μM Compound C, 10 μM H-89, 1 μM Y27632, 10 μM U0126, 10 μM SP600125, 10 μM PP2, 200 nM Wortmannin, or 5 μM AKT inhibitor VIII, and then cell numbers were enumerated. Relative cell numbers are shown as fold changes compared with those in DMSO-treated control MDCK cells. Results are shown as the mean ± SD of 3 independent experiments. *P < 0.05; **P < 0.01 (2-tailed Student’s t test). (C) Lysates of control MDCK, MDCK/DKK1-FLAG, or MDCK/DKK1-GPI-FLAG were probed with the indicated antibodies. Clathrin was used as a loading control. (D) Control MDCK or MDCK/DKK1-FLAG cells were transfected with control or CKAP4 siRNA, and cell lysates were probed with the indicated antibodies. Clathrin was used as a loading control. (E) MDCK cells were stimulated with 10 nM DKK1 for the indicated time periods, and cell lysates were probed with the indicated antibodies. (F) Control MDCK or MDCK/DKK1-FLAG cells were treated with 200 nM Wortmannin, 50 μM LY294002, or 5 μM AKT inhibitor VIII for 30 minutes, and cell lysates were probed with the indicated antibodies. (G) Control MDCK or MDCK/DKK1-FLAG cells were transfected with control (scramble) or p110α siRNA, and cell lysates were probed with the indicated antibodies.
Figure 4. CKAP4 forms a complex with PI3K.

(A) Lysates (Input) of control MDCK, MDCK/DKK1-FLAG, control X293T, or X293T/DKK1-FLAG cells were immunoprecipitated with anti-CKAP4 antibody or nonimmune IgG and probed with the indicated antibodies. (B) Lysates (Input) of control MDCK, MDCK/DKK1-FLAG, control X293T, or X293T/DKK1-FLAG cells were immunoprecipitated with anti-CKAP4 antibody or nonimmune IgG and probed with the indicated antibodies. (C) P85α was expressed with CKAP4-HA or CKAP4-ECD-HA in X293T/DKK1-FLAG cells, and cell lysates were immunoprecipitated with anti-HA antibody or nonimmune IgG and probed with anti-p85α and anti-HA antibodies. (D) Amino acid sequence of CKAP4 cytoplasmic region. (E) Lysates of X293T/DKK1-FLAG cells transiently transfected with CKAP4-HA or PA mutant–CKAP4-HA were immunoprecipitated with anti-HA antibody or nonimmune IgG and probed with anti-p85α and anti-HA antibodies. (F) Lysates of X293T/DKK1-FLAG cells transiently transfected with WT or deletion mutant GFP-p85α were immunoprecipitated with anti-CKAP4 antibody or nonimmune IgG and probed with anti-GFP and anti-CKAP4 antibodies. (G) Schematic illustration of human p85α deletion mutants used in this study.
The well-known growth factor–mediated mechanism for PI3K activation is through PI3K recruitment to tyrosine-phosphorylated receptors (38). Since there are no tyrosine residues in the cytoplasmic region of CKAP4 (Figure 4D), theoretically it is impossible for CKAP4 to be tyrosine phosphorylated. Therefore, the novel mechanism by which CKAP4 forms a complex with PI3K was investigated. One possibility is that the Src homology 3 (SH3) domain of p85α is responsible for binding to CKAP4, because CKAP4 contains a proline-rich region, 36PPPAPQQPPPPPAPHPQQHPQQHPQ60 (Figure 4D). Because 2 classes of ligand consensus motifs for the p85 SH3 domain, RXLPPRPXX and XXXPPXPXX, are proposed (39, 40), 3 proline residues (40P, 44P, and 46P) were mutated to alanine (a PA mutant). PA mutant–CKAP4 did not form a complex with p85α in X293T/DKK1-FLAG cells (Figure 4E). In addition, the SH2 domain was not required for the binding of p85α to CKAP4, but the SH3 domain was obligatory (Figure 4, F and G), suggesting that DKK1-induced AKT activation is mediated by the interaction between the proline-rich region of CKAP4 and the SH3 domain of p85α.
DKK1 and CKAP4 are expressed in human cancers.
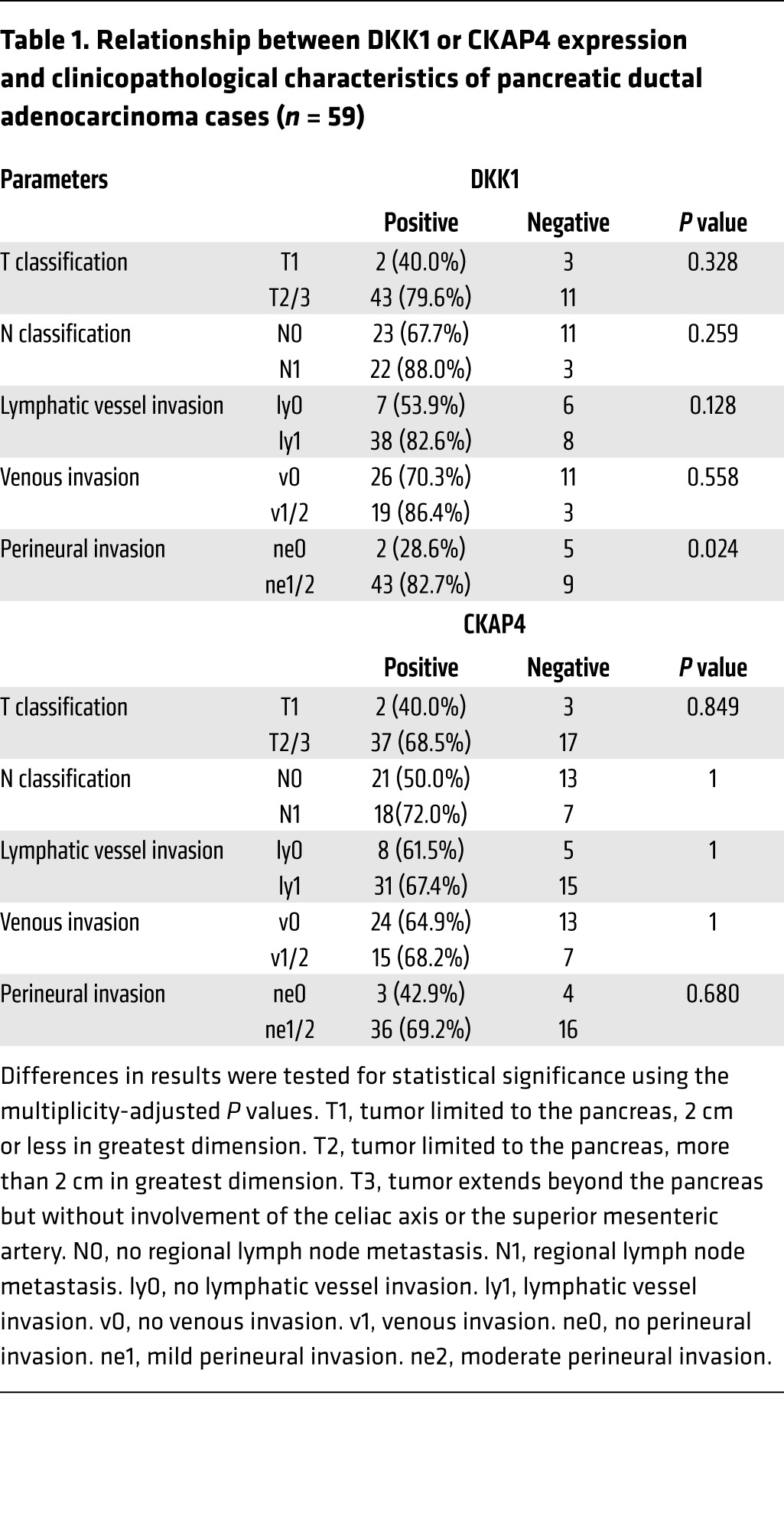
Whether CKAP4 is expressed in human cancers was investigated as well as DKK1. In pancreatic ductal adenocarcinoma, DKK1 and CKAP4 were strongly detected in 45 of 59 (76.3%) and 39 of 59 (66.1%) cases, respectively, while both proteins were minimally detected in nontumor regions of pancreatic ducts under our staining conditions (Figure 5A). Since, in some cases, tumor lesions showed a small area of DKK1 or CKAP4 expression, the stained areas were classified into 4 categories (<5%, 5%–20%, 20%–50%, and 50%–95%) (Figure 5A and Supplemental Figure 4, A and B). The results were considered positive when the total area of a tumor lesion showed greater than 5% staining. The positive expression result and the size of the area showing DKK1 or CKAP4 expression were not correlated with the T grade (tumor size and depth of invasion) or N grade (degree of lymph node metastasis) of the tumor. Positivity of DKK1 was significantly associated with perineural invasion (P = 0.024) of the tumor (Table 1). Positive staining for phosphorylated AKT (pAKT) (S473) was observed more frequently in tumor lesions that were positive for both DKK1 and CKAP4 than in tumor lesions that were only positive for DKK1 (Supplemental Figure 5, A and B). Approximately 86% (18 of 21 cases) of tumor lesions with both DKK1 and CKAP4 positive were positive for pAKT (S473), while 56% (5 of 9 cases) of the tumor lesions with either DKK1 or CKAP4 positive or both negative were positive for pAKT (S473) (Supplemental Figure 5, A and B, and Supplemental Table 3). In addition, the 5-year survival was about 50% in DKK1- and/or CKAP4-negative patients, whereas it was significantly reduced (P = 0.037) in both DKK1- and CKAP4-positive patients (Figure 5B). The expression of CKAP4 and both DKK1 and CKAP4 shortened the duration of relapse-free survival (P = 0.047 and 0.006, respectively) (Figure 5C).
Figure 5. DKK1 and CKAP4 are expressed in human pancreatic cancer.

(A) Pancreatic ductal adenocarcinoma tissues (n = 59) were stained with anti-DKK1 or anti-CKAP4 antibody and hematoxylin. Scale bars: 50 μm. Percentages of DKK1- or CKAP4-positive cases in the tumor lesions are shown in the right panel. (B and C) The relationship between overall survival (B) or relapse-free survival (C) and DKK1 and CKAP4 expression in patients with pancreatic ductal adenocarcinoma was analyzed. The log-rank test was used for statistical analysis.
Table 1. Relationship between DKK1 or CKAP4 expression and clinicopathological characteristics of pancreatic ductal adenocarcinoma cases (n = 59).
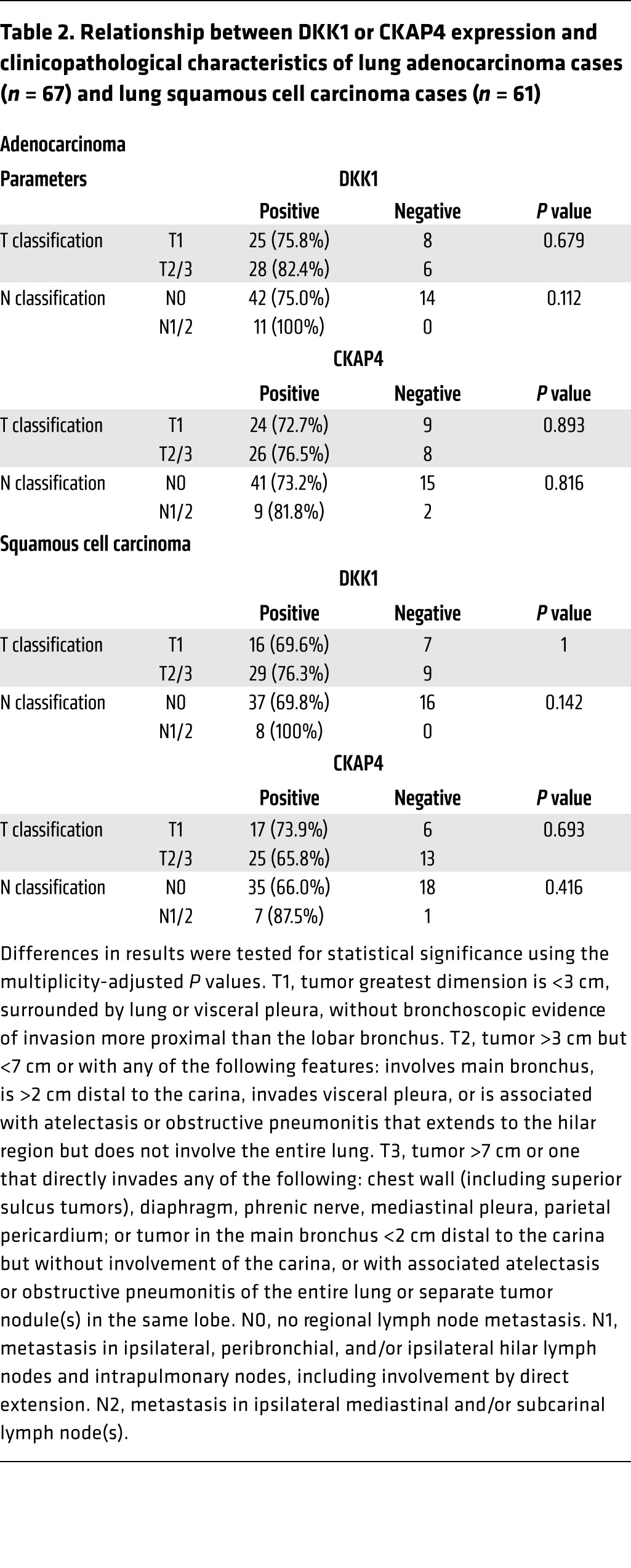
In lung adenocarcinoma specimens, DKK1- or CKAP4-positive cells were minimally detected in nontumor epithelium, whereas tumor lesions showed clear staining for DKK1 and CKAP4 expression (Figure 6A and Supplemental Figure 4, C and D). In total, 53 of 67 (79.1%) and 50 of 67 (74.6%) cases of lung adenocarcinoma were positive for DKK1 and CKAP4 expression, respectively. Among lung squamous cell carcinoma cases, DKK1 and CKAP4 were positive in 45 of 61 (73.8%) and 42 of 61 (74.6%) cases, respectively (Figure 6B and Supplemental Figure 4, E and F). The positive expression result and the size of the DKK1 or CKAP4 expression area in lung cancers were not correlated with the T grade or N grade of the tumor (Table 2). As well as pancreatic cancer, pAKT (S473)–positive staining was observed frequently in lung tumor lesions with both DKK1 and CKAP4 positive (Supplemental Figure 5, C and D, and Supplemental Table 3). In lung cancer cases, the follow-up period was insufficient for examination of overall survival (median follow-up period was 547 days in lung adenocarcinoma cases and 657 days in lung squamous cell carcinoma cases). Therefore, relapse-free survival was examined. Both DKK1 and CKAP4 expression was negatively correlated with relapse-free survival (P = 0.040 in adenocarcinoma and P = 0.043 in squamous cell carcinoma) (Figure 6, C and D). It is notable that DKK1 was positive in 45.5% of patients (5 of 11 cases) with atypical adenomatous hyperplasia (AAH), a putative precursor lesion of adenocarcinoma (Figure 6E). However, CKAP4 was minimally detected in AAH patients under our staining conditions.
Figure 6. DKK1 and CKAP4 are expressed in human lung cancer.

(A and B) Lung adenocarcinoma tissues (A) (n = 67) and squamous cell carcinoma tissues (B) (n = 61) were stained with anti-DKK1 or anti-CKAP4 antibody and hematoxylin. Percentages of DKK1- or CKAP4-positive cases in the tumor lesions are shown in the right panel. (C and D) The relationship between relapse-free survival and DKK1 and CKAP4 expression in patients with lung adenocarcinoma (C) and squamous cell carcinoma (D) was analyzed. The log-rank test was used for statistical analysis. (E) Lung tissues showing atypical adenomatous hyperplasia (AAH) (n = 11) were stained with anti-DKK1 or anti-CKAP4 antibody and hematoxylin. Percentages of DKK1- or CKAP4-positive cases in the AAH lesions are shown in the right panel. Scale bars: 100 μm (A, B, and E).
Table 2. Relationship between DKK1 or CKAP4 expression and clinicopathological characteristics of lung adenocarcinoma cases (n = 67) and lung squamous cell carcinoma cases (n = 61).
CKAP4 is involved in cancer cell proliferation.
CKAP4 was expressed in all cancer and noncancer cell lines we examined (Supplemental Figure 6A). While DKK1 was highly expressed in some cancer cell lines, including A549 lung, SUIT-2 and S2-CP8 pancreatic, KKLS gastric, and KYSE-70 esophageal cancer cells, and HepG2 hepatoblastoma cells, DKK1 was not well expressed in other cancer cell lines, including Calu-6 lung, HeLaS3 cervical, and AGS gastric cancer cells (Supplemental Figure 6A). In X293T, Eph4, and MDCK cells DKK1 was hardly detected (Supplemental Figure 6A and Supplemental Table 1).
CKAP4 was localized to the cell surface membrane in A549 and S2-CP8 cells (Supplemental Figure 6B). DKK1 formed a complex with CKAP4 at endogenous levels in A549 cells (Supplemental Figure 6C). Endogenous p85α formed a complex with endogenous CKAP4 in S2-CP8 cells, and the complex formation was suppressed by DKK1 knockdown (Supplemental Figure 6D). The association of endogenous p110α with CKAP4 depended on p85α (Supplemental Figure 6E). When DKK1 or CKAP4 was depleted in S2-CP8 and A549 cells, AKT activity and cellular proliferation in 2D culture were suppressed, and the inhibition was rescued by expression of DKK1-FLAG or CKAP4-HA (Figure 7, A and B, and Supplemental Figure 6, F and I). Expression of CKAP4-HA in DKK1 knockdown cells (S2-CP8/shDKK1/CKAP4-HA and A549/shDKK1/CKAP4-HA cells) or that of DKK1-FLAG in CKAP4 knockdown cells (S2-CP8/shCKAP4/DKK1-FLAG and A549/shCKAP4/DKK1-FLAG cells) did not rescue the inhibition of cellular proliferation (Supplemental Figure 6, G, H, J, and K). Knockdown of either DKK1 or CKAP4 in S2-CP8 and A549 cells inhibited cell proliferation in 3D culture, and expression of DKK1 and CKAP4 rescued the phenotypes of DKK1 and CKAP4 knockdown cancer cells, respectively (Figure 7, C and D). Furthermore, DKK1 or CKAP4 knockdown reduced migration activity of S2-CP8 and A549 cells (Supplemental Figure 7, A and B).
Figure 7. DKK1-CKAP4 signaling is required for cancer cell proliferation in vitro.

(A and B) S2-CP8 (A) and A549 (B) cells stably expressing control shRNA; DKK1 shRNA; DKK1 shRNA and DKK1-FLAG; CKAP4 shRNA; or CKAP4 shRNA and CKAP4-HA were subjected to the 2D cell proliferation assay. Results are shown as the mean ± SD of 3 independent experiments. (C and D) Left panels: The same cell lines used in A and B were cultured for 5 days in 3D Matrigel. Right panel: The number of spheres per field (n = 5) was counted, and results are expressed as the ratio compared with that of control spheres. **P < 0.01; NS, not significant (compared with the shControl group, 2-tailed Student’s t test). Scale bars: 200 μm.
HeLaS3 cells expressed CKAP4, but DKK1 expression in HeLaS3 cells was little compared to expression in S2-CP8 and A549 cells (Supplemental Figure 6A). Knockdown of CKAP4 in HeLaS3 cells did not affect cellular proliferation in 2D and 3D culture (Supplemental Figure 8, A and B). Taken together, these results suggest that expression of both proteins is necessary for cellular proliferation. SP-A and tPA were known to act as ligands for CKAP4 (27, 28). SP-A was expressed little in S2-CP8 and A549 cells (Supplemental Figure 9A), and tPA was expressed in S2-CP8 but not A549 cells (Supplemental Figure 9B). tPA knockdown did not affect SP-CP8 cell proliferation in 2D culture under the conditions that DKK1 knockdown inhibited SP-CP8 cell proliferation (Supplemental Figure 9, C and D). Therefore, other CKAP4 ligands, such as SP-A and tPA, could not be involved in cellular proliferation of these cancer cells.
The role of CKAP4 expression in tumorigenesis in vivo was investigated by s.c. implantation of cancer cells into the flanks of immunodeficient mice. The volumes and weights of the xenograft tumors derived from S2-CP8 and A549 cells stably expressing CKAP4 shRNA were less than those of control tumors (Figure 8, A and B). Knockdown of CKAP4 reduced the number of Ki-67–positive cells and AKT activity in xenograft tumors (Figure 8, C and D). Expression of CKAP4 rescued the phenotypes induced by CKAP4 knockdown (Figure 8, A–D). In addition, knockdown of DKK1 in S2-CP8 and A549 cells suppressed xenograft tumor formation, Ki-67 staining, and AKT activity (Supplemental Figure 10), which showed the same phenotypes as CKAP4 knockdown cancer cells. DKK1ΔCRD1-FLAG expression did not rescue AKT activity and tumor formation, which were decreased in DKK1 knockdown S2-CP8 cells (Supplemental Figure 11), supporting that DKK1 functionally interacts with CKAP4 in tumor formation in vivo. These results suggest that CKAP4 is a potential molecular target for cancer therapy.
Figure 8. DKK1-CKAP4 signaling is required for cancer cell proliferation in vivo.

(A and B) S2-CP8 cells (A) and A549 cells (B) stably expressing control shRNA (n = 10 for S2-CP8 cells and n = 8 for A549 cells), CKAP4 shRNA (n = 6 for S2-CP8 cells and n = 4 for A549 cells), or CKAP4 shRNA and CKAP4-HA (n = 4) were implanted s.c. into immunodeficient mice. Top panels: Representative appearance of 1 mouse (left picture) and extirpated xenograft tumors (right picture) are shown. Dashed lines show the outline of xenograft tumors. The volumes (bottom left panel) and weights (bottom right panel) of the xenograft tumors were measured. Results are plotted as box and whiskers where the median is represented with a line, the box represents the 25th to 75th percentile, and error bars show the 5th to 95th percentile. Scale bars: 10 mm. (C and D) Sections prepared from xenograft tumors of S2-CP8 cells (C) and A549 cells (D) were stained with hematoxylin and anti–Ki-67 (top panel) or anti-pAKT (bottom panel) antibody. Ki-67–positive cells are expressed as the percentage of positively stained cells compared with total cells per field (n = 5 fields) in the right panel. Percentages of pAKT (S473)–positive tumors in the total xenograft tumors tested are shown in the right panel. Results are shown as means ± SD of 3 independent experiments. *P < 0.05; **P < 0.01; NS, not significant (compared with the shControl group, 2-tailed Student’s t test). Scale bars: 100 μm.
Anti-CKAP4 antibody suppressed tumor formation.
Rabbit anti-CKAP4 polyclonal antibody was generated using the extracellular domain of CKAP4 as an antigen. The antibody recognized endogenous CKAP4 in S2-CP8 cells, and its validity was confirmed by overexpression and knockdown of CKAP4 (Supplemental Figure 12A). When intact S2-CP8 cells, without permeabilization, were stained with anti-CKAP4 antibody, the cell surface was visible (Figure 9A). This antibody inhibited the in vitro binding of DKK1 to CKAP4 (Figure 9B) and suppressed DKK1-induced, but not insulin-induced, AKT activation in MDCK cells (Figure 9C). The anti-CKAP4 antibody also decreased AKT activity in S2-CP8 and A549 cells (Figure 9D) and inhibited cellular proliferation in 3D culture (Figure 9E). As other examples of cancer cells in which both DKK1 and CKAP4 are expressed, SUIT-2 and HepG2 cells were examined. The anti-CKAP4 antibody also decreased AKT activity and 3D cellular proliferation of these cancer cells (Supplemental Figure 12, B and C).
Figure 9. Antiproliferative effect of anti-CKAP4 antibody on DKK1- and CKAP4-expressing cancer cells in vitro.

(A) Nonpermeabilized S2-CP8 cells were stained with anti-CKAP4 antibody (green), phalloidin (red), and DRAQ5 DNA Dye (blue). (B) After 2 nM GST-CKAP4-ECD was treated with 20 nM anti-CKAP4 or anti-GST antibodies, the mixtures were incubated with 2.5 nM DKK1 for 2 hours. The protein mixtures were precipitated with glutathione agarose, and the precipitates were probed with anti-DKK1 antibody. Bottom panel: The band intensity was analyzed by ImageJ software, and the level was expressed as arbitrary units compared with nontreatment (GST Ab [–] and CKAP4 Ab [–]). (C) Lysates of MDCK cells stimulated with 10 nM DKK1 or 1 nM insulin for 30 minutes in the presence of 167 nM anti-CKAP4 or anti-GST antibody were probed with the indicated antibodies. Bottom panel: pAKT and AKT band intensities were analyzed by ImageJ software, and the pAKT level normalized to AKT level was expressed as arbitrary units compared with buffer and GST antibody treatment. Clathrin was used as a loading control. (D) Lysates of S2-CP8 cells and A549 cells treated with 33 nM anti-CKAP4 or anti-GST antibody for 4 hours were probed with the indicated antibodies. Right panel: pAKT and AKT band intensities were analyzed by ImageJ software, and the pAKT level normalized to AKT level was expressed as arbitrary units compared with GST antibody treatment. Clathrin was used as a loading control. (E) The number of anti-CKAP4 antibody–treated S2-CP8 and A549 spheres per field (n = 5) was expressed as the ratio of that of anti-GST antibody–treated spheres. Results are shown as means ± SD of 3 independent experiments. *P < 0.05; **P < 0.01 (2-tailed Student’s t test). Scale bars: 20 μm (A); 200 μm (E).
Finally, whether the anti-CKAP4 antibody demonstrates efficacy as an anticancer agent for cancer therapy was examined. S2-CP8 and A549 cells were implanted into immunodeficient mice, and tumor volumes were allowed to develop to 40–60 mm3. The anti-CKAP4 or anti–glutathione-S-transferase antibody was injected i.p. twice per week, and tumor development was continued for an additional 14 days in mice implanted with S2-CP8 cells and 25 days in mice implanted with A549 cells. The anti-CKAP4 antibody reduced the volumes and weights of both tumor types (Figure 10, A and B). Ki-67–positive cell number and AKT activity in tumors were decreased in mice injected with anti-CKAP4 antibody (Figure 10, C and D). As primary cultured cancer cells from patient tumors, cancer tissue-originated spheroids (CTOSs) were prepared (41, 42). Among CTOSs from lung cancer patients, LC189 expressed both DKK1 and CKAP4, while LB95 expressed neither DKK1 nor CKAP4 (Supplemental Figure 12D). Anti-CKAP4 antibody inhibits spheroid growth of LC189 but not LB95 (Supplemental Figure 12E).
Figure 10. Antiproliferative effect of anti-CKAP4 antibody on DKK1- and CKAP4-expressing cancer cells in vivo.

(A and B) S2-CP8 cells (A) and A549 cells (B) were implanted s.c. into immunodeficient mice. Anti-CKAP4 (150 μg/body) (n = 6) or GST antibody (150 μg/body) (n = 6) was injected into the intraperitoneal cavity twice per week. Left panels: Representative appearance of 1 mouse (top picture) and extirpated xenograft tumors (bottom picture) are shown. The volume (middle panel) and weight (right panel) of the xenograft tumors were measured. (C and D) Sections prepared from xenograft tumors of S2-CP8 cells (C) and A549 cells (D) were stained with hematoxylin and anti–Ki-67 (top panel) or anti-pAKT (bottom panel) antibody. Ki-67–positive cells were counted, and the number is expressed as the percentage of total cells per field (n = 5 fields) in the right panel. Percentages of pAKT (S473)–positive tumors in the total xenograft tumors tested are shown in the right panel. Results are shown as means ± SD of 3 independent experiments. *P < 0.05; **P < 0.01 (2-tailed Student’s t test). Scale bars: 10 mm (A and B); 100 μm (C and D).
Antibody treatment for about 4 weeks did not cause histopathological damage to organs, including the lung, heart, kidney, liver, or bladder (Supplemental Figure 13A). The anti-CKAP4 antibody did not affect cellular proliferation of HeLaS3 cells in both 3D culture and xenograft tumor formation assays (Supplemental Figure 13, B and C). Taken together, these results suggest that anti-CKAP4 antibody suppresses proliferation of cancer cells expressing both DKK1 and CKAP4.
Discussion
CKAP4 functions as a novel receptor for DKK1 and mediates DKK1-induced cellular proliferation.
Although DKK1 was originally identified as a negative regulator of Wnt signaling, it is now suggested that DKK1 stimulates proliferation of noncancerous and cancerous cells (2, 17–19); however, the mechanisms remain largely unknown. In this study, we identified CKAP4 as a novel DKK1 receptor and showed that DKK1 signaling through CKAP4 promotes cellular proliferation through activation of the PI3K/AKT pathway. The following results support CKAP4 functioning as a DKK1 receptor: (a) CKAP4 was localized to the cell surface membrane and internalized by DKK1 in a clathrin-dependent manner; (b) DKK1 directly bound to the extracellular domain of CKAP4 with a KD of 0.42 nM, which is comparable to the KD (about 0.34–0.5 nM) of DKK1 binding to LRP6 (5, 6); (c) the DKK1 mutant that lacked CKAP4-binding activity did not affect proliferation of MDCK and S2-CP8 cells; (d) DKK1 induced complex formation between the intracellular domains of CKAP4 and PI3K; (e) DKK1 knockdown–induced phenotypes in cancer cells were the same as CKAP4 knockdown–induced phenotypes; and (f) AKT activation was detected efficiently in tumor lesions where DKK1 and CKAP4 were simultaneously expressed. These results support that the interaction of DKK1 and CKAP4 is important for signaling in vivo.
DKK1 binds to LRP6 through CRD2 (6, 7). As CRD1 of DKK1 was required for binding to CKAP4, it is likely that DKK1 interacts with LRP6 and CKAP4 through different domains. Among DKK family members, DKK3 contains CRD2, which possesses low homology with that of other DKKs, and DKK3 does not show inhibitory activity for Wnt signaling (2). In contrast, CRD1 is highly conserved among the DKK family members. Therefore, DKKs other than DKK1 might also bind to CKAP4.
CKAP4 was originally discovered as a protein that mainly localized to the ER (25) and later shown to function as a ligand-specific receptor (27–29). SP-A blocks surfactant secretion from lamellar bodies in lung type II pneumocytes (28, 43). SP-A binds to CKAP4 to regulate surfactant clearance in pneumocytes, and CKAP4 knockdown suppresses SP-A–mediated inhibition of surfactant secretion (44). SP-A also induces CKAP4 translocation from the ER to cell surface membrane through AKT activation (45). tPA binds to CKAP4 on rat vascular smooth muscle cells, and anti-CKAP4 antibody inhibits tPA binding and decreased tPA activity (27). In addition, APF, which is produced from the bladder cells of interstitial cystitis patients, binds to CKAP4 and induces the nuclear localization of CKAP4 (29). In contrast to DKK1, APF inhibits AKT activity and cellular growth (46). Thus, CKAP4 acts as a receptor for different ligands in certain types of cells where ligands may decide the intracellular localization of CKAP4. DKK1 and SP-A expression levels were very low in MDCK cells (Supplemental Figure 5A and Supplemental Figure 8A). Although tPA was expressed in MDCK cells, knockdown of tPA did not affect apical localization of CKAP4 (Supplemental Figure 14). Therefore, it is unlikely that the apical localization of CKAP4 in polarized MDCK cells is regulated by these ligands.
Our results suggest that the LZ domain of CKAP4 is required for binding to DKK1. Although the regions of CKAP4 that interact with other ligands are not known, it has been hypothesized that the negatively charged amino acid cluster region of CKAP4 (the region of amino acids 318–328 containing 5 negatively charged amino acids) binds to the positively charged region of SP-A (44). Therefore, DKK1 and SP-A would interact with different regions on CKAP4. It is important to clarify whether these ligands share a common 3D structural domain that binds to CKAP4.
Since there is no tyrosine residue in the cytoplasmic region of CKAP4, we proposed that the proline-rich region of CKAP4 interacted with the SH3 domain of p85α. Although we are not sure how CKAP4 interacts with p85α in a DKK1-dependent manner, it is intriguing to speculate that in the absence of DKK1, the proline-rich region of CKAP4 is masked and the closed region is opened by DKK1, which may result in binding with p85α. However, there are several mechanisms to activate PI3K, which also functions downstream of Ras (38). We are currently investigating the proteins that interact with the intracellular domain of CKAP4 to identify another possible mechanism for DKK1/CKAP4 signaling to activate PI3K.
A novel cancer therapy using CKAP4 as a molecular target.
CKAP4 was detected in tumor lesions of 66%–74% of pancreatic and lung cancer cases, whereas positive signal (expression) was minimally detected in nontumor regions under our staining conditions. Therefore, CKAP4 may be a specific marker for these types of cancer. DKK1 was also detected in the same tumor lesions of these cancers with similar frequency to CKAP4. Because the positivity of both DKK1 and CKAP4 was correlated with poor prognosis and worse relapse-free survival, their simultaneous expression plays critical roles in cancer aggressiveness. It is notable that DKK1 staining but not CKAP4 staining was detectable in lung AAH. AAH cells proliferate along the preexisting alveolar epithelium and show increased cell size and prominent nucleoli. The grade of atypia in lung AAH is usually milder than that in adenocarcinoma (47). Therefore, if CKAP4 is expressed in normal and AAH legions to similar levels, it is intriguing to speculate that CKAP4 overexpression might be involved in the transition from the precancerous state to the cancerous state. However, it was reported that high expression of CKAP4 is correlated with longer overall survival in cholangiocarcinoma and hepatocellular carcinoma patients (48, 49), although the expression levels of DKK1 were not examined in these cases. CKAP4 might function in a positive or negative role for cellular proliferation in cancer contexts.
The serum DKK1 levels are higher in patients with lung, esophageal, pancreatic, gastric, bile duct, breast, and cervical cancers compared with healthy populations, and the high serum DKK1 levels correlate with DKK1-positive staining in primary tumors of lung and pancreatic cancer patients (17, 18). Aberration of the PI3K/AKT signaling pathway has been implicated in human cancers (36). A rabbit anti-DKK1 polyclonal antibody inhibited xenograft tumor formation activity of DKK1-overexpressing A549 cells in immunodeficient mice by inducing apoptosis (18). Interestingly, the antibody was generated by immunization of the N-terminal 120 amino acids of DKK1, including CRD1, which is consistent with our findings that showed that CRD1 is responsible for binding to CKAP4. Since DKK1 functions as a tumor suppressor in colon cancer (23), an anti-DKK1 antibody might promote tumor formation as a side effect.
Our rabbit anti-CKAP4 polyclonal antibody inhibited DKK1-dependent AKT activity, migration, and proliferation ability of S2-CP8, A549, SUIT-2, and HepG2 cells in vitro. The anti-CKAP4 antibody also suppressed xenograft tumor formation by S2-CP8 and A549 cells. In addition, the antibody inhibited spheroid formation of primary cultured lung cancer cells expressing both DKK1 and CKAP4. In contrast, the anti-CKAP4 antibody did not affect HeLaS3 cell proliferation. Because HeLaS3 cells express low levels of DKK1, anti-CKAP4 antibody effects could be specific for cancer cells expressing both DKK1 and CKAP4. Although about 4-week administration of the anti-CKAP4 antibody did not cause tissue damage in normal tissues, we have to carefully observe effects of anti-CKAP4 antibody on normal tissues by long-term administration. Since CKAP4 functions as a receptor for several ligands, effects of anti-CKAP4 antibody on other signaling in vivo also need to be clarified. Nevertheless, an anti-CKAP4 antibody may have an advantage for inhibiting DKK1/CKAP4 signaling in cancer therapy compared with an anti-DKK1 antibody.
Taken together, these results suggest that the DKK1/CKAP4 axis promotes at least pancreatic and lung cancer cell proliferation through the PI3K/AKT signaling pathway. In contrast, in mice that develop pancreatic cancer induced by expression of oncogenic KRas, forced expression of DKK1 was shown to inhibit tumor formation by suppressing Wnt signaling (50). It is necessary to examine CKAP4 expression on the cell surface membrane of pancreatic cancer cells in the model mice. Analyzing the novel DKK1 pathway that we proposed in this study could contribute to the development of clinically useful biomarkers and immunotherapy for cancer.
Methods
Supplemental Methods are available online with this article; doi:10.1172/JCI84658DS1.
Materials and chemicals.
MDCK type I (MDCK) dog renal tubule cells and Eph4 mouse mammary epithelial cells were provided by S. Tsukita (Osaka University, Suita, Japan) in January 2013 and October 2011, respectively. S2-CP8 and SUIT-2 pancreatic cancer cells were purchased from Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University, in April 2014. A549 and NCI-H1579 lung adenocarcinoma cells were provided by Y. Shintani (Osaka University, Suita, Japan) in January 2014. HeLaS3 cervical cancer cells were provided by K. Matsumoto (Nagoya University, Aichi, Japan) in May 2002. AGS and KKLS gastric cancer cells were provided by M. Hatakeyama (Tokyo University, Tokyo, Japan) in April 2014 and by W. Yasui (Hiroshima University, Hiroshima, Japan) in August 2006, respectively. HepG2 hepatoblastoma cells were provided by Y. Matsuura (Osaka University, Osaka, Japan) in July 2004. Lenti-X 293T (X293T) cells were purchased from Takara Bio Inc. in October 2011. Calu-6 lung adenocarcinoma cells and KYSE-70 and TE-11 esophageal cancer cells were provided by Shionogi Pharmaceutical Research Center in November 2011 and July 2012, respectively. P110α in pcDNA3mycTEVFLAG and p85α in pCAGGS were provided by T. Asano (Hiroshima University).
A549, NCI-H1579, MDCK, S2-CP8, SUIT-2, HeLaS3, KYSE-70, TE-11, and Calu-6 cells were grown in DMEM supplemented with 10% FBS. HepG2 cells were grown in DMEM supplemented with 10% FBS and 25 mM glucose. KKLS and AGS cells were grown in RPMI-1640 supplemented with 10% FBS. Stably transfected cells with plasmids were maintained in both growth media above supplemented with 10 μg/ml blasticidin S, 1 μg/ml puromycin, or 800 μg/ml G418. Cell line authentication was not performed.
Anti-CKAP4 polyclonal antibody was generated in rabbits by immunization with synthetic peptides corresponding to amino acid residues 128–602 of human CKAP4.
The following drugs were used: CHIR99021 and Y27632 (Wako Pure Chemical Industries); U0126 (Promega Corp.); H-89, PP2, chlorpromazine, monodansylcadaverine, Compound C, and SP600125 (Sigma-Aldrich); AKT inhibitor VIII (Calbiochem); and LY294002 (Cell Signaling Technology).
Isolation of DKK1-interacting protein.
MDCK cells transfected with the DKK1-FLAG-GPI construct were selected using 10 μg/ml blasticidin S to generate MDCK/DKK1-FLAG-GPI cells. Confluent MDCK/DKK1-FLAG-GPI cells in 3 dishes (10-cm culture dish) were incubated with 0.5 mg/ml sulfo-NHS-LC-biotin for 30 minutes at 4°C and quenched by washing 3 times with 5 ml of PBS containing 50 mM NH4Cl. Cells were then harvested and lysed in 1 ml of NP-40 buffer (20 mM Tris-HCl pH 8.0, 10% glycerol, 137 mM NaCl, and 1% NP-40) with protease inhibitors (10 μg/ml leupeptin, aprotinin, and 1 mM PMSF). After 10 minutes of centrifugation, the supernatant was precleaned with 20 μl of 50% slurry of protein G Sepharose beads for 30 minutes at 4°C. The precleaned lysates were incubated with anti-FLAG antibody (Novus Biologicals) for 30 minutes at 4°C and further incubated with 40 μl of 50% slurry of protein G Sepharose beads for 1 hour at 4°C. Beads were washed 3 times with 1 ml of NP-40 buffer and 3 times with TBS (25 mM Tris-HCl pH 7.4 and 150 mM NaCl). Recovered beads were incubated 3 times with FLAG peptide (0.2 mg/ml) to elute proteins in 50 μl of TBS for 30 minutes at 4°C. Then, 40 μl of 50% slurry of NeutrAvidin Agarose beads was added to eluted protein samples (~150 μl), and the mixture was further incubated for 2 hours at 4°C. The beads were washed twice with 1 ml of NP-40 buffer and once with 1 ml of 10 mM Tris-HCl pH 7.5, and bound complexes were dissolved in 20 μl of Laemmli’s sample buffer. The DKK1-FLAG-GPI–interacting proteins were detected by silver staining. Seventeen bands (arrowheads in Figure 2A) were cut from the gel and analyzed by mass spectrometry.
Patients and cancer tissues.
The present study involved following presurgical untreated patients, who underwent surgical resection at Osaka University between April 2001 and April 2015: 59 pancreatic ductal adenocarcinoma patients with ages ranging from 47 to 83 years (median, 70 years), 67 lung adenocarcinoma patients with ages ranging from 39 to 85 years (median, 68 years), 61 lung squamous cell carcinoma patients with ages ranging from 38 to 82 years (median, 71 years), and 11 lung AAH patients with ages ranging from 58 to 78 years (median, 67 years).
Tumors were staged according to the Union for International Cancer Control TNM staging system. Resected specimens were macroscopically examined to determine the location and size of a tumor, and specimens for histology were fixed in 10% (vol/vol) formalin and processed for paraffin embedding. Hospital specimens for examination were sectioned at 4 μm thickness and stained with H&E or immunoperoxidase for independent evaluations.
IHC studies of DKK1, CKAP4, and pAKT in pancreatic and lung cancer tissues.
IHC studies were performed as previously described (51) with modification. Briefly, all tissue sections were stained using a DakoReal EnVision Detection System (Dako) in accordance with the manufacturer’s recommendations. Tissue peroxidase activity was blocked with 3% H2O2-methanol for 15 minutes, and the sections were then incubated with goat serum for 1 hour to block nonspecific antibody binding sites. Tissue specimens were treated with anti-DKK1 (1:100), anti-CKAP4 (1:100), or anti–pAKT (S473) (1:50) antibody for 16 hours at 4°C. Then, the specimens were detected by incubating with goat anti-mouse IgG-HRP for 1 hour and subsequently with DAB (Dako). The tissue sections were then counterstained with 0.1% (wt/vol) hematoxylin. Tumors in which the positively stained area covered more than 5% were classified as DKK1- or CKAP4-positive. Four researchers, including 2 pathologists, evaluated the positivity.
Xenograft tumor assay.
Xenograft tumor assay was performed as previously described (51, 52) with modification. Six-week-old male BALB/cAnNCrj-nu immunodeficient mice (Charles River Laboratory Japan Inc.) were anesthetized with a combination of medetomidine (0.3 mg/kg body weight) and midazolam (4 mg/kg). The mice then received a dorsal s.c. injection of S2-CP8 cells (5 × 106 cells) or A549 cells (5 × 106 cells) suspended in 100 μl of PBS. The immunodeficient mice were then sacrificed at 14 or 21 days (for S2-CP8 cells) or 28 days (for A549 cells) after transplantation.
To examine effects of anti-CKAP4 antibody, when the average tumor size reached 50 mm3, mice were divided into 2 groups. Anti-CKAP4 (150 μg/body) or anti–glutathione-S-transferase (GST) antibody (150 μg/body) was injected into the intraperitoneal cavity twice per week for 2 weeks (at days 0, 4, 8, and 12 for S2-CP8 cells) and for 3.5 weeks (at days 0, 4, 9, 12, 16, 18, and 22 for A549 cells; at days 0, 3, 8, 10, 14, 17, and 21 for HeLaS3 cells). The nude mice were then sacrificed at 14 days for S2-CP8 cells and at 25 days for A549 and HeLaS3 cells after antibody treatment.
The areas containing transplanted cells were measured, weighed, and processed for IHC analyses. Tumor volumes were calculated using the following formula: (major axis) × (minor axis) × (minor axis) × 0.5 (53).
Statistics.
All experiments were repeated at least 3 times, and the results were expressed as means ± SD. Statistical analyses were performed using JMP software (SAS Institute Inc.). Means of continuous outcome variables were tested with 2-tailed Student’s t test. Cumulative probabilities of overall survival and relapse-free survival were computed with the Kaplan-Meier method; and log-rank test was used to assess their statistical significance. In order to prevent inflation of type I error by multiple comparisons, P values for experiments shown in Figure 5, B and C; Figure 6, C and D; and Tables 1 and 2 were adjusted by multiplication of unadjusted P values by type I inflation parameters. The probability of at least 1 type I error rate being less than 5% among multiple tests was divided by 0.05 in order to compute the type I inflation parameters. Each type I error variable was generated through Monte Carlo simulation by consideration of observed correlation coefficients among DKK1, CKAP4, and the combination of DKK1 and CKAP4, and among clinicopathological parameters. P values less than 0.05 were considered statistically significant. Western blotting data are representative of at least 3 independent experiments.
Study approval.
The protocol for human specimens was approved by the ethical review board of the Graduate School of Medicine, Osaka University, Japan (no. 13455). All protocols used for animal experiments in this study were approved by the Animal Research Committee of Osaka University, Japan (no. 21-048-1).
Author contributions
HK performed the majority of the experiments and coauthored the manuscript with the assistance of KF. YO carried out some in vitro experiments. KS, SN, and EM analyzed the expression of DKK1 and CKAP4 in cancer specimens with the assistance of YS, MO, HT, H Eguchi, and YD. H Endo and MI acquired data of CTOSs. AK designed experiments, interpreted results, and coauthored the manuscript.
Supplementary Material
Acknowledgments
We would like to thank the Center of Medical Research and Education, Graduate School of Medicine, Osaka University, for In-gel digestion and liquid chromatography–tandem mass spectrometry analysis. We also would like to thank S. Tsukita, M. Hatakeyama, Y. Matsuura, T. Asano, K. Matsumoto, and A. Shintani for donating cells and plasmids and helping statistical analyses. This work was supported by Grants-in-Aid for Scientific Research to A. Kikuchi (25250018; 2013–2015) and for Scientific Research on Innovative Areas to A. Kikuchi (23112004; 2011–2015) from the Ministry of Education, Science and Culture of Japan and by grants from the Uehara Memorial Foundation.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(7):2689–2705. doi:10.1172/JCI84658.
See the related Commentary beginning on page 2419.
Contributor Information
Hirokazu Kimura, Email: h.kimura@molbiobc.med.osaka-u.ac.jp.
Katsumi Fumoto, Email: ka_fumoto@molbiobc.med.osaka-u.ac.jp.
Kensaku Shojima, Email: dream.of.electric.sheep.japan@gmail.com.
Satoshi Nojima, Email: s_nojima@molpath.med.osaka-u.ac.jp.
Yoshihito Osugi, Email: yosugi@molbiobc.med.osaka-u.ac.jp.
Hideo Tomihara, Email: htomihara@gesurg.med.osaka-u.ac.jp.
Hidetoshi Eguchi, Email: heguchi@gesurg.med.osaka-u.ac.jp.
Yasushi Shintani, Email: yshintani@thoracic.med.osaka-u.ac.jp.
Hiroko Endo, Email: endou-hi@mc.pref.osaka.jp.
Masahiro Inoue, Email: minoue0112@gmail.com.
Yuichiro Doki, Email: ydoki@gesurg.med.osaka-u.ac.jp.
Meinoshin Okumura, Email: meinosin@thoracic.med.osaka-u.ac.jp.
Eiichi Morii, Email: morii@molpath.med.osaka-u.ac.jp.
Akira Kikuchi, Email: akikuchi@molbiobc.med.osaka-u.ac.jp.
References
- 1.Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature. 1998;391(6665):357–362. doi: 10.1038/34848. [DOI] [PubMed] [Google Scholar]
- 2.Niehrs C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 2006;25(57):7469–7481. doi: 10.1038/sj.onc.1210054. [DOI] [PubMed] [Google Scholar]
- 3.Kikuchi A, Yamamoto H, Sato A, Matsumoto S. New insights into the mechanism of Wnt signaling pathway activation. Int Rev Cell Mol Biol. 2011;291:21–71. doi: 10.1016/B978-0-12-386035-4.00002-1. [DOI] [PubMed] [Google Scholar]
- 4.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 5.Mao B, et al. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature. 2001;411(6835):321–325. doi: 10.1038/35077108. [DOI] [PubMed] [Google Scholar]
- 6.Semënov MV, Tamai K, Brott BK, Kühl M, Sokol S, He X. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr Biol. 2001;11(12):951–961. doi: 10.1016/S0960-9822(01)00290-1. [DOI] [PubMed] [Google Scholar]
- 7.Brott BK, Sokol SY. Regulation of Wnt/LRP signaling by distinct domains of Dickkopf proteins. Mol Cell Biol. 2002;22(17):6100–6110. doi: 10.1128/MCB.22.17.6100-6110.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahn VE, Chu ML, Choi HJ, Tran D, Abo A, Weis WI. Structural basis of Wnt signaling inhibition by Dickkopf binding to LRP5/6. Dev Cell. 2011;21(5):862–873. doi: 10.1016/j.devcel.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto H, Komekado H, Kikuchi A. Caveolin is necessary for Wnt-3a-dependent internalization of LRP6 and accumulation of b-catenin. Dev Cell. 2006;11(2):213–223. doi: 10.1016/j.devcel.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Sakane H, Yamamoto H, Kikuchi A. LRP6 is internalized by Dkk1 to suppress its phosphorylation in the lipid raft and is recycled for reuse. J Cell Sci. 2010;123(pt 3):360–368. doi: 10.1242/jcs.058008. [DOI] [PubMed] [Google Scholar]
- 11.Jiang Y, He X, Howe PH. Disabled-2 (Dab2) inhibits Wnt/β-catenin signalling by binding LRP6 and promoting its internalization through clathrin. EMBO J. 2012;31(10):2336–2349. doi: 10.1038/emboj.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demir K, et al. RAB8B is required for activity and caveolar endocytosis of LRP6. Cell Rep. 2013;4(6):1224–1234. doi: 10.1016/j.celrep.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto H, Sakane H, Yamamoto H, Michiue T, Kikuchi A. Wnt3a and Dkk1 regulate distinct internalization pathways of LRP6 to tune the activation of β-catenin signaling. Dev Cell. 2008;15(1):37–48. doi: 10.1016/j.devcel.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 14.González-Sancho JM, et al. The Wnt antagonist DICKKOPF-1 gene is a downstream target of beta-catenin/TCF and is downregulated in human colon cancer. Oncogene. 2005;24(6):1098–1103. doi: 10.1038/sj.onc.1208303. [DOI] [PubMed] [Google Scholar]
- 15.Niida A, et al. DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene. 2004;23(52):8520–8526. doi: 10.1038/sj.onc.1207892. [DOI] [PubMed] [Google Scholar]
- 16.Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17(1):45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Yamabuki T, et al. Dikkopf-1 as a novel serologic and prognostic biomarker for lung and esophageal carcinomas. Cancer Res. 2007;67(6):2517–2525. doi: 10.1158/0008-5472.CAN-06-3369. [DOI] [PubMed] [Google Scholar]
- 18.Sato N, et al. Wnt inhibitor Dickkopf-1 as a target for passive cancer immunotherapy. Cancer Res. 2010;70(13):5326–5336. doi: 10.1158/0008-5472.CAN-09-3879. [DOI] [PubMed] [Google Scholar]
- 19.Makino T, et al. Dickkopf-1 expression as a marker for predicting clinical outcome in esophageal squamous cell carcinoma. Ann Surg Oncol. 2009;16(7):2058–2064. doi: 10.1245/s10434-009-0476-7. [DOI] [PubMed] [Google Scholar]
- 20.Shen Q, et al. Serum DKK1 as a protein biomarker for the diagnosis of hepatocellular carcinoma: a large-scale, multicentre study. Lancet Oncol. 2012;13(8):817–826. doi: 10.1016/S1470-2045(12)70233-4. [DOI] [PubMed] [Google Scholar]
- 21.Han SX, et al. Serum dickkopf-1 is a novel serological biomarker for the diagnosis and prognosis of pancreatic cancer. Oncotarget. 2015;6(23):19907–19917. doi: 10.18632/oncotarget.4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinto D, Gregorieff A, Begthel H, Clevers H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003;17(14):1709–1713. doi: 10.1101/gad.267103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aguilera O, et al. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene. 2006;25(29):4116–4121. doi: 10.1038/sj.onc.1209439. [DOI] [PubMed] [Google Scholar]
- 24.Gregory CA, Singh H, Perry AS, Prockop DJ. The Wnt signaling inhibitor dickkopf-1 is required for reentry into the cell cycle of human adult stem cells from bone marrow. J Biol Chem. 2003;278(30):28067–28078. doi: 10.1074/jbc.M300373200. [DOI] [PubMed] [Google Scholar]
- 25.Schweizer A, Ericsson M, Bächi T, Griffiths G, Hauri HP. Characterization of a novel 63 kDa membrane protein. Implications for the organization of the ER-to-Golgi pathway. J Cell Sci. 1993;104(pt 3):671–683. doi: 10.1242/jcs.104.3.671. [DOI] [PubMed] [Google Scholar]
- 26.Vedrenne C, Hauri HP. Morphogenesis of the endoplasmic reticulum: beyond active membrane expansion. Traffic. 2006;7(6):639–646. doi: 10.1111/j.1600-0854.2006.00419.x. [DOI] [PubMed] [Google Scholar]
- 27.Razzaq TM, Bass R, Vines DJ, Werner F, Whawell SA, Ellis V. Functional regulation of tissue plasminogen activator on the surface of vascular smooth muscle cells by the type-II transmembrane protein p63 (CKAP4) J Biol Chem. 2003;278(43):42679–42685. doi: 10.1074/jbc.M305695200. [DOI] [PubMed] [Google Scholar]
- 28.Gupta N, Manevich Y, Kazi AS, Tao JQ, Fisher AB, Bates SR. Identification and characterization of p63 (CKAP4/ERGIC-63/CLIMP-63), a surfactant protein A binding protein, on type II pneumocytes. Am J Physiol Lung Cell Mol Physiol. 2006;291(3):L436–L446. doi: 10.1152/ajplung.00415.2005. [DOI] [PubMed] [Google Scholar]
- 29.Conrads TP, et al. CKAP4/p63 is a receptor for the frizzled-8 protein-related antiproliferative factor from interstitial cystitis patients. J Biol Chem. 2006;281(49):37836–37843. doi: 10.1074/jbc.M604581200. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto H, et al. The apical and basolateral secretion of Wnt11 and Wnt3a in polarized epithelial cells is regulated by different mechanisms. J Cell Sci. 2013;126(pt 13):2931–2943. doi: 10.1242/jcs.126052. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto H, et al. Basolateral secretion of Wnt5a in polarized epithelial cells is required for apical lumen formation. J Cell Sci. 2015;128(5):1051–1063. doi: 10.1242/jcs.163683. [DOI] [PubMed] [Google Scholar]
- 32.O’Brien LE, Zegers MM, Mostov KE. Opinion: Building epithelial architecture: insights from three-dimensional culture models. Nat Rev Mol Cell Biol. 2002;3(7):531–537. doi: 10.1038/nrm859. [DOI] [PubMed] [Google Scholar]
- 33.Mao B, et al. Kremen proteins are Dickkopf receptors that regulate Wnt/β-catenin signalling. Nature. 2002;417(6889):664–667. doi: 10.1038/nature756. [DOI] [PubMed] [Google Scholar]
- 34.Lisanti MP, Sargiacomo M, Graeve L, Saltiel AR, Rodriguez-Boulan E. Polarized apical distribution of glycosyl-phosphatidylinositol-anchored proteins in a renal epithelial cell line. Proc Natl Acad Sci U S A. 1988;85(24):9557–9561. doi: 10.1073/pnas.85.24.9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlegel R, Dickson RB, Willingham MC, Pastan IH. Amantadine and dansylcadaverine inhibit vesicular stomatitis virus uptake and receptor-mediated endocytosis of a2-macroglobulin. Proc Natl Acad Sci U S A. 1982;79(7):2291–2295. doi: 10.1073/pnas.79.7.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4(4):257–262. doi: 10.1016/S1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 37.Klippel A, Escobedo JA, Hirano M, Williams LT. The interaction of small domains between the subunits of phosphatidylinositol 3-kinase determines enzyme activity. Mol Cell Biol. 1994;14(4):2675–2685. doi: 10.1128/MCB.14.4.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta. 1998;1436(1–2):127–150. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- 39.Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW, Schreiber SL. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell. 1994;76(5):933–945. doi: 10.1016/0092-8674(94)90367-0. [DOI] [PubMed] [Google Scholar]
- 40.Rickles RJ, et al. Identification of Src, Fyn, Lyn, PI3K and Abl SH3 domain ligands using phage display libraries. EMBO J. 1994;13(23):5598–5604. doi: 10.1002/j.1460-2075.1994.tb06897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kondo J, et al. Retaining cell-cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. Proc Natl Acad Sci U S A. 2011;108(15):6235–6240. doi: 10.1073/pnas.1015938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Endo H, et al. Spheroid culture of primary lung cancer cells with neuregulin 1/HER3 pathway activation. J Thorac Oncol. 2013;8(2):131–139. doi: 10.1097/JTO.0b013e3182779ccf. [DOI] [PubMed] [Google Scholar]
- 43.Bates SR, et al. Role of P63 (CKAP4) in binding of surfactant protein-A to type II pneumocytes. Am J Physiol Lung Cell Mol Physiol. 2008;295(4):L658–L669. doi: 10.1152/ajplung.90233.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bates SR. P63 (CKAP4) as an SP-A receptor: implications for surfactant turnover. Cell Physiol Biochem. 2010;25(1):41–54. doi: 10.1159/000272062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kazi AS, Tao JQ, Feinstein SI, Zhang L, Fisher AB, Bates SR. Role of the PI3-kinase signaling pathway in trafficking of the surfactant protein A receptor P63 (CKAP4) on type II pneumocytes. Am J Physiol Lung Cell Mol Physiol. 2010;299(6):L794–L807. doi: 10.1152/ajplung.00372.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shahjee HM, Koch KR, Guo L, Zhang CO, Keay SK. Antiproliferative factor decreases Akt phosphorylation and alters gene expression via CKAP4 in T24 bladder carcinoma cells. J Exp Clin Cancer Res. 2010;29: doi: 10.1186/1756-9966-29-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mori M, Rao SK, Popper HH, Cagle PT, Fraire AE. Atypical adenomatous hyperplasia of the lung: a probable forerunner in the development of adenocarcinoma of the lung. Mod Pathol. 2001;14(2):72–84. doi: 10.1038/modpathol.3880259. [DOI] [PubMed] [Google Scholar]
- 48.Li MH, et al. Expression of cytoskeleton-associated protein 4 is related to lymphatic metastasis and indicates prognosis of intrahepatic cholangiocarcinoma patients after surgery resection. Cancer Lett. 2013;337(2):248–253. doi: 10.1016/j.canlet.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 49.Li SX, et al. Prognostic significance of cytoskeleton-associated membrane protein 4 and its palmitoyl acyltransferase DHHC2 in hepatocellular carcinoma. Cancer. 2014;120(10):1520–1531. doi: 10.1002/cncr.28593. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, et al. Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Res. 2013;73(15):4909–4922. doi: 10.1158/0008-5472.CAN-12-4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujii S, Matsumoto S, Nojima S, Morii E, Kikuchi A. Arl4c expression in colorectal and lung cancers promotes tumorigenesis and may represent a novel therapeutic target. Oncogene. 2015;34(37):4834–4844. doi: 10.1038/onc.2014.402. [DOI] [PubMed] [Google Scholar]
- 52.Shojima K, et al. Wnt5a promotes cancer cell invasion and proliferation by receptor-mediated endocytosis-dependent and -independent mechanisms, respectively. Sci Rep. 2015;5: doi: 10.1038/srep08042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mitsuishi Y, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22(1):66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.