

Abstract
BACKGROUND. Treatment of B cell malignancies with adoptive transfer of T cells with a CD19-specific chimeric antigen receptor (CAR) shows remarkable clinical efficacy. However, long-term persistence of T cells targeting CD19, a pan–B cell marker, also depletes normal B cells and causes severe hypogammaglobulinemia. Here, we developed a strategy to target B cell malignancies more selectively by taking advantage of B cell light Ig chain restriction. We generated a CAR that is specific for the κ light chain (κ.CAR) and therefore recognizes κ-restricted cells and spares the normal B cells expressing the nontargeted λ light chain, thus potentially minimizing humoral immunity impairment.
METHODS. We conducted a phase 1 clinical trial and treated 16 patients with relapsed or refractory κ+ non-Hodgkin lymphoma/chronic lymphocytic leukemia (NHL/CLL) or multiple myeloma (MM) with autologous T cells genetically modified to express κ.CAR (κ.CARTs). Other treatments were discontinued in 11 of the 16 patients at least 4 weeks prior to T cell infusion. Six patients without lymphopenia received 12.5 mg/kg cyclophosphamide 4 days before κ.CART infusion (0.2 × 108 to 2 × 108 κ.CARTs/m2). No other lymphodepletion was used.
RESULTS. κ.CART expansion peaked 1–2 weeks after infusion, and cells remained detectable for more than 6 weeks. Of 9 patients with relapsed NHL or CLL, 2 entered complete remission after 2 and 3 infusions of κ.CARTs, and 1 had a partial response. Of 7 patients with MM, 4 had stable disease lasting 2–17 months. No toxicities attributable to κ.CARTs were observed.
CONCLUSION. κ.CART infusion is feasible and safe and can lead to complete clinical responses.
TRIAL REGISTRATION. ClinicalTrials.gov NCT00881920.
FUNDING. National Cancer Institute (NCI) grants 3P50CA126752 and 5P30CA125123 and Leukemia and Lymphoma Society (LLS) Specialized Centers of Research (SCOR) grant 7018.
Introduction
Chimeric antigen receptors (CARs) most commonly combine the antigen-recognizing portion of a mAb with the signaling machinery of T cells. When these molecules are genetically grafted into T lymphocytes, they allow these cells to bind to tumor surface antigens in an MHC-independent fashion and promote T cell costimulation, thereby activating their cytotoxic pathways against the malignant cells (1). Autologous T cells expressing CD19-directed CARs (CD19.CARTs) have shown remarkable activity in patients with acute lymphoblastic leukemia (ALL) (2–6) and some activity in patients with chronic lymphocytic leukemia or small lymphocytic lymphoma (CLL/SLL) (7–9) or non-Hodgkin lymphoma (NHL) (10–13). Nonetheless, sustained clinical responses due to CD19.CARTs usually require long-term persistence and invariably are associated with B cell aplasia; CD19 is a pan–B cell marker, and normal B lymphocytes are also a target for CD19.CART cytotoxicity (5).
If it were possible to effectively target antigens that have a more restricted distribution on malignant B cells versus normal B lymphocytes, then sufficient numbers of normal B lymphocytes might be spared to avoid profound hypogammaglobulinemia. We therefore took advantage of the surface expression of Igs on mature B lymphocytes and mature B lymphoid malignancies, since each cell expresses either κ or λ light chains, but not both. We reasoned that targeting the light chain subtype expressed by monoclonal malignant NHL and CLL/SLL B cells should efficiently kill tumor cells, while sparing normal B cells expressing the reciprocal subtype. This concept is also potentially applicable to multiple myeloma (MM). Although plasma cells no longer express surface Igs, several groups have described an MM-initiating population that expresses surface Igs (14). We have previously developed a CAR that targets the κ light chain (κ.CAR) (15), and here we show results of a phase 1 clinical trial using autologous T cells genetically modified to express the κ.CAR (κ.CARTs). We demonstrate that this approach is feasible and safe and can lead to complete clinical responses.
Results
Patients’ characteristics.
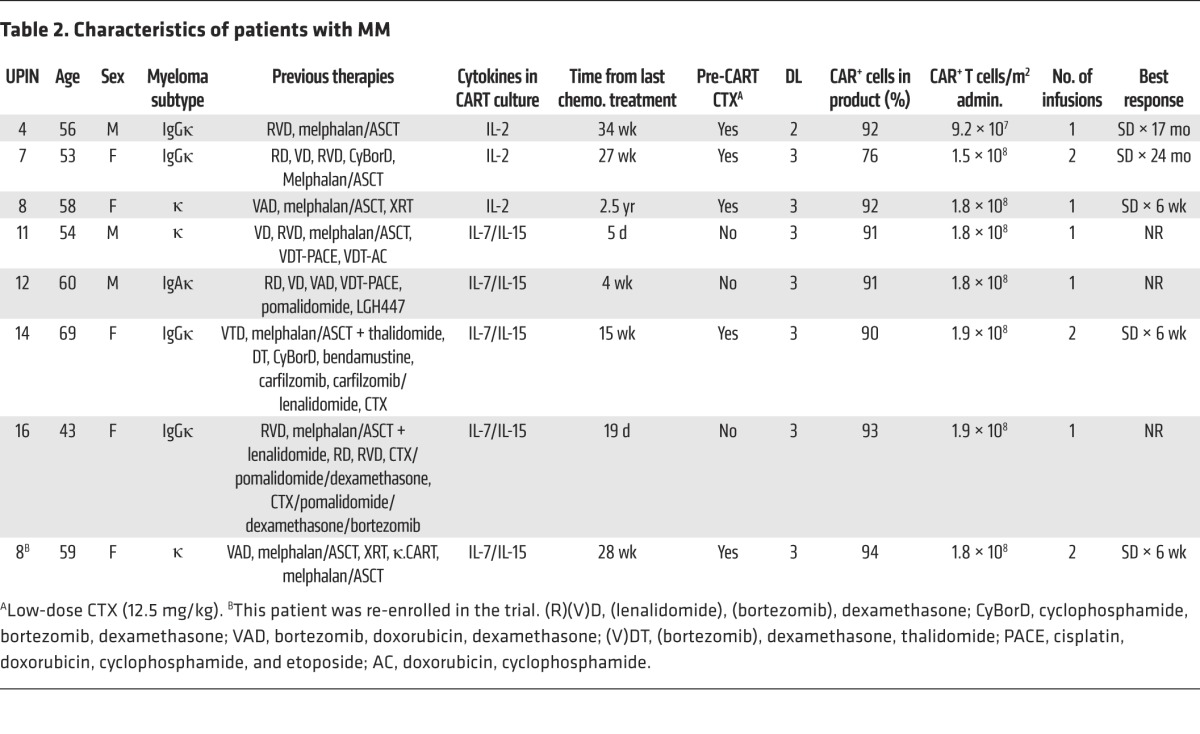
We administered κ.CARTs to 16 patients, 9 with NHL (including CLL/SLL) and 7 with MM (Figure 1). Their characteristics are summarized in Tables 1 and 2. All patients had active disease at the time of κ.CART infusion, and all had relapsed after one or more lines of chemotherapy. Six patients who had neither received other chemotherapy within 4 weeks of κ.CART infusion nor had absolute lymphocyte counts (ALCs) above 500/μl received 12.5 mg/kg cyclophosphamide (CTX) 4 days before the first infusion of κ.CARTs. Five patients received 1 final cycle of chemotherapy within 4 weeks of κ.CART infusion, as documented in Tables 1 and 2. These chemotherapeutic regimens had been previously chosen by the referring physicians as salvage treatment and were not specifically selected for their lymphodepleting potential.
Figure 1. Flow chart of clinical trial NCT00881920 (CHARKALL).

Table 1. Characteristics of patients with NHL or CLL.
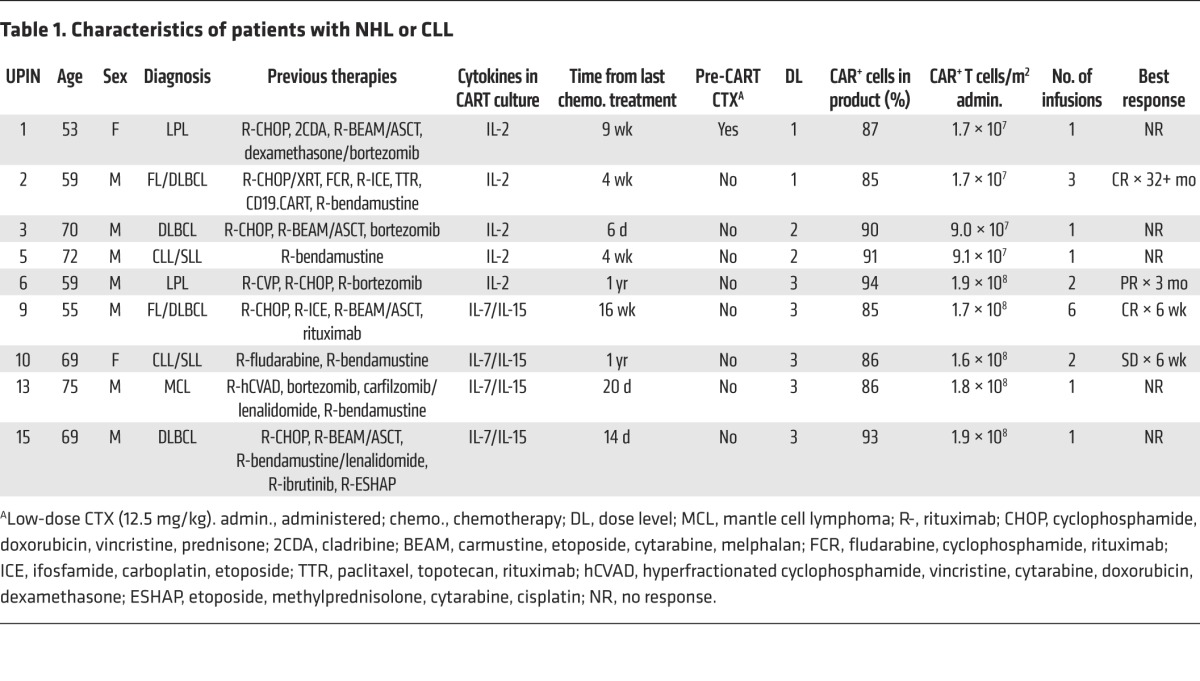
Table 2. Characteristics of patients with MM.
Characteristics of infused CARTs.
We manufactured 27 κ.CART lines from peripheral blood mononuclear cells (PBMCs) isolated from 24 patients (Figure 1). One patient had 2 cell lines and another patient had 3 cell lines made to allow repeat infusions. CAR transduction levels were adequate (>80%) in all manufactured T cell products, regardless of the cytokine(s) used for the expansion (Supplemental Figure 1B; supplemental material available online with this article; doi:10.1172/JCI86000DS1). All lines were composed of more than 99% CD3+ T cells, and each contained a variable ratio of CD4+ and CD8+ cells, with an overall CD8 predominance in the T cell products expanded in IL-2 (78% ± 11%) compared with those expanded in IL-7 and IL-15 (56% ± 12%; P < 0.001) (see Methods and Supplemental Figure 1C). The majority of κ.CARTs were CD45RO+ and lacked CCR7, but a small fraction expressed the central memory–associated phenotypic markers CD62L, CD27, and CD28 (Supplemental Table 1). T cell products grown with IL-7 and IL-15 had approximately twice as many CD45RA+ cells as those grown in IL-2 (4.9% ± 4.1% vs. 10.7% ± 7.1%, P = 0.03) (Supplemental Table 1). NK cells (CD3–CD56+) were not detectable. All products demonstrated cytotoxicity against κ+ targets in vitro, as assessed by 51Cr-release assays (Supplemental Figure 1D). Cytotoxic activity against κ– targets was negligible. Eight patients did not receive their cell lines (Figure 1), because they pursued alternative treatment outside our institution (6 patients), had progressive disease (PD) during cell manufacture (1 patient), or had no evidence of disease at the time of planned infusion (1 patient).
κ.CART expansion and persistence.
Molecular signals for κ.CARTs were consistently detected in the peripheral blood at low levels 3 hours after the first κ.CART infusion but increased to peak at 1 to 2 weeks after the first infusion (94 ± 186 copies/μg of PBMC DNA and 248 ± 750 copies/μg of DNA at weeks 1 and 2, respectively, for all patients). The signals then declined to lower levels by week 6 (26 ± 49 copies/μg of DNA) (Figure 2). Seven patients received a second infusion. The difference in expansion between the first and second infusions was not statistically significant (AUCs for 2 and 6 weeks after infusion: P = 0.109 and P = 0.219, Wilcoxon signed-rank test). Transgene levels were always below the threshold needed to detect a distinct CART population by flow cytometry and became almost undetectable 6 months after the last infusion (3 ± 4 copies/μg of PBMC DNA). There was no correlation between transgene levels in the peripheral blood and preinfusion ALC, disease type, cell dose, cytokines used in culture (IL-2 vs. IL-7 and IL-15), or clinical response (data not shown).
Figure 2. In vivo expansion and persistence of infused κ.CARTs.

In vivo expansion and persistence of infused κ.CARTs was assessed by qPCR of peripheral blood from patients with NHL or CLL (A) and from patients with MM (B). Data points represent critical post-infusion intervals after the first infusion of κ.CARTs. There was no significant difference in the AUC for additional infusions (Supplemental Figure 2). Dotted lines denote each patient (color key shows universal patient identifier numbers [UPINs]), and the continuous line summarizes the mean expansion and persistence (error bars represent the standard deviation).
Acute and long-term toxicities.
All infusions were well tolerated. Apart from a patient with MM who had a grade 3 lymphopenia that was deemed possibly related, none of the adverse events reported (most frequently anemia, leukopenia, fatigue, hyper- or hypokalemia, and elevated aspartate aminotransferase [AST]) were considered to be related to κ.CART infusion. The vast majority of patients had baseline B cell lymphopenia and polyclonal hypogammaglobulinemia as a result of prior treatment, the biology of their underlying disease, or both. With the exception of the 2 CLL patients, B cell lymphopenia was especially pronounced in the NHL patient group (with 1.5% ± 2% of CD19+ cells gated on CD45+ lymphocytes at the time of infusion). This made assessment of new-onset B lymphopenia due to κ.CARTs challenging. However, in patients with MM, who consistently had more circulating B cells at the time of infusion (6% ± 4.9%), we observed an overall 50% reduction in the number of B cells within the first 2 weeks after infusion (3.2% ± 2.5%) (Figure 3A), with a transient preferential elimination of κ+ B cells, as documented by the decrease in the κ/λ ratio (P = 0.009) (Figure 3A) and the reduced mean fluorescence intensity of κ light chain expression (Figure 3B) during the first 2 weeks after infusion.
Figure 3. Biological effects observed after κ.CART infusion.

After κ.CART infusion, a transient decline in total B cell numbers and in the κ/λ ratio in MM patients (n = 8, paired Student’s t test) was observed (A). In general, there was a decrease in the mean fluorescence intensity of κ light chain expression during the first 2 weeks after κ.CART infusion; data from 1 representative patient are shown in B, with κ.CART expansion data in the top panel and κ light chain expression data in the bottom panels. Numbers in the dot plots correspond to the percentage of CD19+ (upper left quadrant) and CD3+ (lower right quadrant) PBMCs; histogram shows the distribution of the intensity of surface κ light chain staining in CD19+ PBMCs, highlighting the κ– (left peak) and κ+ (right peak) population, at each of the time points depicted. Inflammatory cytokines levels (shown for IL-6) increased modestly after κ.CART infusion (C), in both NHL (n = 17 infusions) and MM (n = 10 infusions) patients (paired Student’s t test), and no clinical evidence of CRS was observed. There was no significant generation of HAMAs after κ.CART infusion (D).
Although no patient had symptoms consistent with severe cytokine release syndrome (CRS), because CARTs have been associated with severe CRS, we nonetheless measured cytokine levels serially in serum samples. Although we were able to detect elevation of inflammatory cytokines, such as IL-6 (Figure 3C and Supplemental Figure 3), which generally coincided with peak CART expansion, the fold increase from baseline (20-fold average) was much lower than that observed in patients who experience severe CRS (usually >75-fold) (3).
We investigated whether the treated patients developed an Ab response to the transgene. Human anti-mouse Ab (HAMA) levels almost always remained below the threshold of detection and always below levels likely to be biologically active (maximum level 12.5 U/ml; a negative HAMA assay is defined as ≤74 U/ml) (Figure 3D). Tregs were also monitored and, although NHL patients had a higher frequency of circulating FOXP3+ cells gated on CD4+ cells (17.3% ± 7.9%) compared with that detected in MM patients (9.8% ± 4.6%), their frequency did not correlate with expansion or persistence of CARTs or with clinical response (data not shown).
Clinical outcomes following κ.CART infusion.
We observed clinical responses in 4 of 9 patients with NHL or CLL: 2 with complete responses (CR), 1 with a partial response (PR), and 1 with stable disease (SD). The 2 patients (nos. 2 and 9) who had a CR had a history of transformed follicular NHL and did not receive preinfusion lymphodepletion. Of note, patient 2 had had no response to CD19.CARTs (13), which were administered approximately 2.5 years before being enrolled in the current study and were no longer detectable by PCR analysis of peripheral blood. While awaiting κ.CAR manufacture, the patient received 1 dose of bendamustine because of active disease. Four weeks later, he had evidence of residual disease in the neck by PET/CT scan. He received one κ.CART infusion, followed by a second infusion 8 weeks later, without intervening chemotherapy. The patient entered a CR after the second infusion, and this response has been sustained for almost 3 years (Figure 4). Patient 9 had progressive improvement of pelvic lymphadenopathy and achieved a CR after the third infusion of κ.CARTs (Figure 5). Seven weeks later, however, he had recurrence of disease (in the axillary lymph node) despite a fourth infusion. His disease has, however, remained with minimal progression for more than 1 year. Patient 6 (lymphoplasmacytic lymphoma [LPL]) and patient 11 (CLL) had transient PR and SD, respectively. The remaining 5 of 9 NHL or CLL patients (including the 3 who had received a final cycle of chemotherapy within 4 weeks of κ.CART infusion) had PD.
Figure 4. Patient 2 achieved a durable CR after 2 infusions of κ.CARTs.

Patient 2 had transformed follicular lymphoma (FL) with active disease in the neck 4 weeks after receiving 1 dose of bendamustine, as documented in the preinfusion PET/CT scan, which showed a hypermetabolic 7.2 × 3.4-cm left anterior mass (circled) and a hypermetabolic 1.7 × 1-cm left paratracheal lymph node. Six weeks after the first infusion of κ.CARTs, a PET/CT scan showed a PR, with a decrease in the activity and size of the mass (2.4 × 0.8 cm) and lymph node (1.3 × 0.6 cm). Another PET scan obtained after a second infusion was consistent with a CR. This patient has been without evidence of disease for almost 3 years.
Figure 5. Patient 9 achieved a transient CR after 3 infusions of κ.CARTs.

Patient 9 had transformed FL with active disease in the retroperitoneum and pelvis, including a 2.1 × 1.2-cm left external iliac lymph node near the anterior aspect of the left acetabulum, with a standard uptake value (SUV) of 8.5 (bottom row) and a 1 × 1-cm posterior left common iliac chain lymph node, with an SUV of 4.4 (top row). Six weeks after the first infusion of κ.CARTs, a repeat PET/CT scan showed interval improvement in uptake and, to a lesser degree, in the size of multiple lymph nodes, including mild interval improvement in uptake (SUV 5.5) and size (2 × 0.9-cm) in the external iliac lymph node and improved uptake (SUV 3.4) and a slight decrease in the size of the common iliac lymph node. Six weeks after the second infusion of κ.CARTs, we observed a significant response with what appeared to be residual disease within the left pelvic lymph nodes, with the external iliac lymph node that had remained stable in size but had decreased to an SUV of 2.9 and the common iliac lymph node with an SUV of 2.2. Six weeks after the third infusion of κ.CARTs, there was a decrease in the size (the external iliac lymph node was 1.7 × 0.9 cm) and resolution of the previous uptake within the left pelvic lymph nodes and normal uptake elsewhere, consistent with a CR.
Four of seven MM patients had responses. Patients 4 and 7 had prolonged SD for 17 and 24 months. Patient 4 had only minimal residual disease (MRD) after high-dose melphalan and autologous stem cell transplantation (ASCT) (positive immunofixation with unmeasurable monoclonal protein) and maintained stable MRD for 17 months after a single κ.CART infusion. Patient 7 had a progressive decline in her paraprotein levels (2,240 to 1,730 mg/dl IgG) and improvement in anemia (11.6 to 14.4 g/dl hemoglobin), which was sustained for 2 years (Figure 6). Neither of these 2 patients was on lenalidomide (or any other immunomodulatory drugs) at the time of κ.CART infusion, either because they had previously progressed on the drug or were intolerant of it. Two other patients (nos. 8 and 14) met the criteria for transient SD, namely a less than 50% reduction and a less than 25% increase in paraprotein levels (38% free κ light chain reduction in patient 8 and 11% increase in IgGκ in patient 14). Patient 8 was re-treated with κ.CARTs 1.5 years later, after receiving additional conventional therapy, again with transient SD. Three of seven patients, including the two who had received a final cycle of chemotherapy within four weeks of κ.CART infusion, had no response to κ.CARTs.
Figure 6. Patient 7 had stabilization of disease after 2 infusions of κ.CARTs.

Patient 7 had active IgG MM at the time of κ.CART infusion. Afterward, she had a slow decrease (SD) in her monoclonal protein levels and improvement in her hemoglobin levels, which continued for 2 years, when she had evidence of disease progression. κLC, light chain.
We investigated whether the patients’ responses correlated with numbers of κ.CARTs infused, blood cell counts at infusion (total white blood cell, absolute lymphocyte, and absolute neutrophil counts), circulating Ig and κ light chain levels, and plasma cytokine levels. We found no significant association between these parameters and response.
We further analyzed the antitumor immune responses in the recipients to seek evidence for newly emergent immunity to additional tumor-associated antigens. No evidence for such “epitope spreading” was obtained, as we found no differences in the frequency of precursor T cells responding to known tumor-associated antigens (TAAs), such as NY-ESO, P53, PRAME, or MAGE, in peripheral blood collected before and after κ.CART therapy (Supplemental Figure 4).
Discussion
We report the first clinical trial to our knowledge involving the treatment of B cell–derived malignancies with CARTs targeting a clonally restricted B cell marker intended to limit B cell aplasia while maintaining antitumor activity. We found that adoptive transfer of CARTs that target the κ light chain of Igs is feasible and safe at all dose levels studied and that the κ light chain is a validated target for NHL, since κ.CARTs can induce clinical responses, including sustained CRs, in patients with NHL.
The US Food and Drug Administration (FDA) has granted the “breakthrough” designation to CD19.CARTs for the treatment of refractory or relapsed ALL on the basis of the high rate of clinical responses achieved in different clinical studies (3, 5, 6). In contrast, the efficacy of CD19.CARTs in patients with refractory or relapsed CLL or SLL and other NHLs appears less striking. For example, the overall response rate in a study of CD19.CART treatment of CLL is currently 57% (16), while in patients with diffuse large B cell lymphoma (DLBCL), the rates of CRs to CD19.CARTs correlate with the intensity of the preconditioning chemotherapy before CD19.CART infusion, ranging from 12.5% (17) to 57% (12), according to the doses of the lymphodepleting drugs fludarabine and CTX. Our patients received no or limited lymphodepleting chemotherapy (12.5 mg/kg CTX) that produced a maximum 30% reduction in lymphocyte counts: indeed, the 2 patients who had a CR received no pretreatment lymphodepletion. More profound lymphodepletion immediately before κ.CART infusion, by depleting Tregs, downregulating immunoinhibitory pathways such indoleamine 2,3-dioxygenase (IDO) (18) in the tumor environment, and creating a favorable homeostatic cytokine milieu for T cell expansion, will likely increase the therapeutic activity of κ.CARTs.
Our clinical study also enrolled patients with MM. There are few reported clinical data for MM patients treated with CARTs. A recent report from China describes the outcomes of 5 MM patients treated with CD138.CARTs, 4 of whom achieved up to 7 months of SD (19). We observed modest antimyeloma effects, documented by a reduction of the paraprotein, a selective reduction of free κ light chains, and an improvement of anemia. These effects were sustained for up to 24 months.
In preclinical studies, we did not observe deleterious effects of physiological levels of soluble Igs on the function of κ.CARTs (15). Indeed, soluble Igs did not impair the capacity of κ.CARTs to eliminate κ+ tumor cells and appeared to sustain the expansion of κ.CARTs, which may partially explain the observed clinical responses to κ.CARTs in patients with NHL. However, we also cannot exclude the possibility that higher levels of free κ light chains, such as those seen in κ-restricted MM, cause adverse effects on κ.CARTs, either by blocking the recognition of κ light chains on the surface of targeted cancer cells or by causing excessive T cell stimulation and exhaustion, thus limiting the therapeutic effect. On the other hand, plasma cells, including malignant counterparts, have minimal (if any) surface expression of light chains, and that can explain, to some extent, why the clinical responses were less striking in patients with MM. It would have been interesting to measure the distribution and persistence of infused κ.CARTs in the bone marrow of patients with MM, but the IRB-approved protocol did not mandate bone marrow studies as long as there was measurable disease by paraproteinemia, which was present in all of these patients.
How can we improve upon these outcomes? Clinical data in patients with ALL infused with CD19.CARTs suggest that the nature of the costimulation provided within the CD19.CAR may affect CART in vivo persistence. Indeed, while CD28 and 4-1BB costimulation of CD19.CARTs are equally effective in inducing clinical responses in patients with relapsed ALL, 4-1BB costimulation may ensure a longer persistence of CARTs and potentially more durable remissions (5). In addition, early in vitro studies and more recent animal experiments suggest that CARs that include long spacers derived from the constant regions of human Igs may cause activation of macrophages expressing the Fc receptor and compromise the antitumor activity of CARTs (20–22). Modifications of our current κ.CAR cassette to include the 4-1BB motif and remove the long spacer may enhance in vivo CART persistence and, therefore, the therapeutic effects of our proposed κ.CART therapy. Finally, if there is evidence that free κ light chains impair the function of the CAR, Ab libraries can be generated and screened to identify the scFv that has superior affinity for the membrane-bound form of the light chain versus the soluble form.
Hence, our study using autologous κ.CARTs shows that this strategy is safe and can produce antitumor activity. We believe a similar strategy applied to target the λ light chain will make our proposed approach broadly applicable to all mature B cell malignancies.
Methods
Clinical study.
We conducted a phase 1 study (ClinicalTrials.gov NCT00881920) designed to assess the feasibility and safety of infusing escalating doses of autologous, polyclonally activated peripheral blood T cells that were genetically modified to express a κ light chain–specific CAR (κ.CAR) in patients with relapsed or refractory κ-restricted lymphoproliferative disorders (NHL, CLL, and MM). All patients had to have measurable disease at the time of infusion (Figure 1). Patients with CLL or an ALC above 500/μl and who had not received any chemotherapy within the previous 4 weeks were given low-dose (12.5 mg/kg) CTX as a single infusion 4 days before administration of the first dose of κ.CARTs. Patients on chemotherapy at the time of treatment could receive κ.CARTs no sooner than 4 days after finishing their last cycle of chemotherapy. Otherwise, no chemotherapy was given to patients before or after κ.CART infusion. We administered 3 dose levels of κ.CARTs: 2 × 107 (dose level 1); 1 × 108 (dose level 2); and 2 × 108 (dose level 3) cells/m2. These doses were based on total cell numbers, regardless of the percentage of CAR+ cells. We used an interpatient dose escalation protocol that followed a continual reassessment method, which required safety to be demonstrated 6 weeks after infusion in 2 patients at each dose level before escalation. Additional infusions of the same dose of κ.CARTs were allowed as long as there was evidence of clinical benefit, defined as at least SD. Additional infusions were administered at least 6 weeks apart, and no chemotherapy was given before κ.CART infusion. Clinical and laboratory evaluations were performed at weeks 1, 2, 4, and 6 and months 3, 6, 9, and 12 after CART infusion, and yearly thereafter. Adverse events during and after T cell infusions were graded according to the NIH Common Terminology Criteria for Adverse Events (CTCAE), version 4 (http://ctep.cancer.gov). Responses were assessed by imaging or laboratory studies, as applicable, at week 6 after CART infusion and were defined as CR, PR, SD, or PD, according to the NCI’s Response Evaluation Criteria in Solid Tumors (RECIST) (23), with modifications based on the International Working Group criteria for NHL (24); the National Cancer Institute Working Group for CLL (25); or the modified (International Myeloma Working Group [IMWG]) uniform response criteria for MM (26).
Generation of retroviral constructs.
We cloned the Ab targeting the κ light chain of human Ig produced by the CRL-1758 mouse hybridoma (ATCC) as a single chain (scFv) and generated a second-generation CAR construct containing both CD28 and ζ endodomains (15). A spacer region derived from the human IgG1-CH2CH3 domains was cloned in-frame between the scFv and the signaling domains to facilitate detection by phenotypic analysis of the transgenic product. This cassette was then cloned into the SFG retroviral backbone. We generated the clinical-grade packaging cell line using PG13 cells (gibbon ape leukemia virus pseudotyping packaging cell line, CRL-10686; ATCC) (27). We used the highest-titer clone to establish a master cell bank and released the clone for clinical use only after safety testing and vector sequencing. We stored the final viral supernatant at –80°C and tested it before clinical release.
Generation and transduction of κ.CARTs.
To generate κ.CARTs, we transduced PBMCs obtained by Ficoll density centrifugation of peripheral blood obtained by phlebotomy 1–3 months before CART administration. We activated PBMCs with OKT3 Ab (Janssen Biotech) or CD3 and CD28 Abs (Miltenyi Biotec) and recombinant human IL-2 (IL-2, 100 U/ml; Prometheus) or IL-7 (10 ng/ml; R&D Systems) and IL-15 (5 ng/ml; R&D Systems) in 24-well plates precoated with a recombinant fibronectin fragment (FN CH-296, Retronectin Takara; Clontech). After transduction, T cells were expanded ex vivo in the presence of recombinant human IL-2 (rhIL-2) (100 U/ml) or IL-7 (10 ng/ml) and IL-15 (5 ng/ml), added twice a week. A change to the process of manufacture took place during the trial, when IL-7 and IL-15 replaced IL-2 because of our own and others’ observation that the combination of IL-7 and IL-15 better preserves the CD8+CD45RA+CCR7+ subset in culture, which is associated with superior expansion and persistence of CARTs in vivo (28). The characteristics of the T cell products are summarized in Supplemental Figure 1 and Supplemental Table 1. The generation of κ.CARTs required a median of 17 days (range 10–27) of culture in the 14 T cell products generated using IL-2 and 16 days (range 12–17 days) in the 13 T cell products expanded in the presence of IL-7 and IL-15. The expansion of T cells grown in IL-7 and IL-15 was overall better than that of T cells grown in IL-2 (Supplemental Figure 1A).
Immunophenotyping.
We used phycoerythrin (PE), FITC, peridinin chlorophyll protein (PerCP) or allophycocyanin-conjugated (APC-conjugated) CD3 (clone SK7); CD4 (SK3); CD8 (SK1); CD56 (B159); CD19 (SJ25C1); TCR-αβ (T10B9); TCR-γδ (B1); CD62L (DREG-56); CD27 (M‑T271); CD28 (CD28.2); CD45RA (HI100); CD45RO (UCHL1); CCR2 (48607); CCR4 (1G1); CCR5 (2D7/CCR5); CCR7 (3D12); CXCR3 (1C6/CXCR3); CXCR4 (12G5); CD162 (KPL-1); CD54 (HA58); CD38 (HB7); CD106 (51-10C9); CD11a (HI111); CD11c (B-ly6); CD18 (6.7); and κ (TB28-2) and λ (1-155-2) light chain Abs (all from BD Pharmingen) to stain the T cell products or tumor cells. We included control samples labeled with the appropriate isotype-matched Abs in each experiment. The κ.CAR was detected with an Fc-specific Alexa Fluor 647–conjugated goat polyclonal F(ab′)2 fragment anti-human IgG (catalog 109-606-088; Jackson ImmunoResearch), which recognizes the IgG1-CH2CH3 component of the CAR. Cells were analyzed by a FACScan (BD) equipped with a filter set for 4 fluorescence signals, using CellQuest software. Data analysis was performed using FlowJo Software (Tree Star). In selected experiments, frozen PBMCs from pre- and post-CART infusion time points were thawed and the number of CD19+ cells and of κ+ cells enumerated by FACS using CountBright beads (Invitrogen).
Cytotoxicity assays.
The cytotoxic specificity of each T cell line was measured in a standard 4-hour 51Cr release assay using effector-to-target (E:T) ratios of 40:1, 20:1, 10:1, and 5:1. Daudi (κ+ tumor cells), HDLM-2 (κ– tumor cells), and K562 (NK-sensitive cells) were used as target cells. The target cells were labeled simultaneously for 1 hour with 51Cr. We calculated the percentage of specific lysis as follows: [(experimental release – spontaneous release)/(maximum release – spontaneous release)] × 100.
Real-time quantitative PCR of the κ.CAR transgene.
We quantified the integrated genome of the retrovirus encoding the κ.CAR by real-time quantitative PCR (qPCR) (13). After extracting DNA from peripheral blood samples with the QIAamp DNA Blood Mini Kit (QIAGEN) following the manufacturer’s instructions, the DNA was amplified with primers and probes (Applied Biosystems) complementary to specific sequences within the retroviral vector (15). The standard curve was established using serial dilutions of the plasmid encoding the transgene. Amplifications were performed using the ABI 7900HT Fast Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific) according to the manufacturer’s instructions.
Cytometric bead array and multiplex assays.
We analyzed plasma or serum samples collected before and after κ.CART infusion using a BD cytokine cytometric bead array (CBA) kit (BD Pharmingen) or a Milliplex Kit (EMD Millipore), following the manufacturers’ instructions. In parallel with the samples, we used the human cytokine standards provided with the kits to prepare standard curves. The CBA assays were run using the BD FACSCalibur, and the data were analyzed from the Milliplex kits using the Luminex 200 System and Milliplex Analyst Software (EMD Millipore).
ELISpot assays.
IFN-γ enzyme-linked immunospot (ELISpot) assays were performed to identify precursor T cells specific for known TAAs. PBMCs were plated at 2 × 105 to 4 × 105 per well in triplicate in PI-Multiscreen plates (EMD Millipore), and IFN-γ production was measured in response to stimulation with peptide mixtures (pepmixes) consisting of 15-mer peptides overlapping by 11 aa and spanning the entire protein of interest for the following antigens: pp65, NY-ESO-1, MAGEA4, PRAME, and P53 (0.6 nmol of each peptide) (JPT Technologies). We included both negative (no pepmix) and positive (staphylococcal enterotoxin B) controls in each experiment. After 16 to 20 hours of incubation at 37°C in 5% CO2, the plates were developed for IFN-γ+ spots (29), dried overnight at room temperature in the dark, and sent to ZellNet Consulting (New York, New York, USA) for quantification. We expressed the frequency of T cells specific to each antigen as specific spot-forming cells (SFCs) per input cell number.
Statistics.
Descriptive statistics (means, median, ranges, and standard deviation or SEM) were used to summarize the data. The AUC was calculated using the trapezoidal rule. A 2-tailed Student’s t test or nonparametric Wilcoxon rank-sum test for between-groups comparisons was performed, while the nonparametric Wilcoxon signed-rank test was applied for within-subjects comparisons. A P value of less than 0.05 was considered statistically significant.
Study approval.
This study was approved by the US FDA, the Recombinant DNA Advisory Committee, and the IRB of the BCM and was conducted in accordance with Declaration of Helsinki principles. All participants provided written informed consent upon enrollment.
Author contributions
CAR, BS, BG, CMR, MKB, HEH, and GD conceptualized the overall strategy for the study and developed its clinical translation and implementation. The clinical protocol was written by CAR and MKB. CAR was the principal investigator of the protocol, and BG, MKB, and HEH were the investigational new drug (IND) sponsors. T cell manufacture, flow cytometry, and qPCR acquisition of clinical samples were performed by BB, HZ, OD, and EL, supervised by BS, and directed by APG and ZM. The manuscript was written by CAR, BS, MKB, and GD, and all authors discussed and interpreted the results. MHW and HL performed statistical analyses. CAR, GC, and RTK enrolled patients in the study and/or managed the patients, and VT was the study research nurse, assisting with enrollment, sample acquisition, and data safety monitoring of patients.
Supplementary Material
Acknowledgments
We thank the NCI, NIH (grant 3P50CA126752) and the LLS (SCOR grant 7018) for financial support. The clinical trial was also supported by grants from The Institute for Clinical and Translational Research of the BCM, shared resources of the Dan L. Duncan Cancer Center support (grant 5P30CA125123), and the National Gene Vector Biorepository.
Footnotes
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Conflict of interest: While this clinical trial was being conducted, the Center for Cell and Gene Therapy at BCM had a Collaborative Research Agreement with Celgene Corporation and Bluebird Bio.
Reference information:J Clin Invest. 2016;126(7):2588–2596. doi:10.1172/JCI86000.
Contributor Information
Barbara Savoldo, Email: barbar@email.unc.edu.
Vicky Torrano, Email: vxtorran@txch.org.
Brandon Ballard, Email: bballard@bellicum.com.
Huimin Zhang, Email: hxzhang@texaschildrens.org.
Olga Dakhova, Email: odakhova@bcm.edu.
Enli Liu, Email: eliu@mdanderson.org.
George Carrum, Email: gcarrum@bcm.edu.
Zhuyong Mei, Email: zmei@bcm.edu.
Meng-Fen Wu, Email: mengfenw@bcm.edu.
Hao Liu, Email: haol@bcm.edu.
Bambi Grilley, Email: grilley@bcm.edu.
Gianpietro Dotti, Email: GDotti@med.unc.edu.
References
- 1.Ramos CA, Dotti G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin Biol Ther. 2011;11(7):855–873. doi: 10.1517/14712598.2011.573476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brentjens RJ, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177): doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davila ML, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224): doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grupp SA, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee DW, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brentjens RJ, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalos M, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95): doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kochenderfer JN, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122(25):4129–4139. doi: 10.1182/blood-2013-08-519413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kochenderfer JN, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kochenderfer JN, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Savoldo B, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsui W, et al. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103(6):2332–2336. doi: 10.1182/blood-2003-09-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vera J, et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108(12):3890–3897. doi: 10.1182/blood-2006-04-017061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porter DL, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303): doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kochenderfer JN, et al. Anti-CD19 CAR T Cells Administered after Low-Dose Chemotherapy Can Induce Remissions of Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma. Presented at: 56th ASH Annual Meeting and Exposition; December 6–9, 2014; San Francisco, California, USA. [April 28, 2016];Abstract 550. https://ash.confex.com/ash/2014/webprogram/Paper72899.html
- 18.Ninomiya S, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. 2015;125(25):3905–3916. doi: 10.1182/blood-2015-01-621474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo B, et al. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. J Cell Immunother. 2016;2:28–35. doi: 10.1016/j.jocit.2014.11.001. [DOI] [Google Scholar]
- 20.Haso W, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121(7):1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudecek M, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3(2):125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hombach A, Hombach AA, Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010;17(10):1206–1213. doi: 10.1038/gt.2010.91. [DOI] [PubMed] [Google Scholar]
- 23.Therasse P, et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst. 2000;92(3):205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 24.Cheson BD, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579–586. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 25.Cheson BD, et al. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood. 1996;87(12):4990–4997. [PubMed] [Google Scholar]
- 26.Rajkumar SV, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011;117(18):4691–4695. doi: 10.1182/blood-2010-10-299487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pule MA, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14(11):1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Y, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR. Blood. 2014;123(24):3750–3759. doi: 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerdemann U, et al. Nucleofection of DCs to generate Multivirus-specific T cells for prevention or treatment of viral infections in the immunocompromised host. Mol Ther. 2009;17(9):1616–1625. doi: 10.1038/mt.2009.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.