

Abstract
The molecular mechanisms that underlie spleen development and congenital asplenia, a condition linked to increased risk of overwhelming infections, remain largely unknown. The transcription factor TLX1 controls cell fate specification and organ expansion during spleen development, and Tlx1 deletion causes asplenia in mice. Deregulation of TLX1 expression has recently been proposed in the pathogenesis of congenital asplenia in patients carrying mutations of the gene-encoding transcription factor SF-1. Herein, we have shown that TLX1-dependent regulation of retinoic acid (RA) metabolism is critical for spleen organogenesis. In a murine model, loss of Tlx1 during formation of the splenic anlage increased RA signaling by regulating several genes involved in RA metabolism. Uncontrolled RA activity resulted in premature differentiation of mesenchymal cells and reduced vasculogenesis of the splenic primordium. Pharmacological inhibition of RA signaling in Tlx1-deficient animals partially rescued the spleen defect. Finally, spleen growth was impaired in mice lacking either cytochrome P450 26B1 (Cyp26b1), which results in excess RA, or retinol dehydrogenase 10 (Rdh10), which results in RA deficiency. Together, these findings establish TLX1 as a critical regulator of RA metabolism and provide mechanistic insights into the molecular determinants of human congenital asplenia.
Introduction
The mammalian spleen is a secondary lymphoid organ that plays a central role in host defense. As a result, asplenia or hyposplenia and postsplenectomy patients often have an increased risk of overwhelming infections, particularly by encapsulated bacteria (1–5). Spleen development involves coordination of cell fate specification, migration, and proliferation to form a vascularized splenic primordium (6–9). In mice, these processes are coordinated by a limited set of transcription factors (10). Among these, T cell leukemia homeobox 1 (TLX1, also known as HOX11) (6, 11) acts downstream of the genetic cascade governing spleen development by promoting cell fate specification and organ expansion. At present, however, the precise downstream transcriptional networks and signaling pathways controlled by TLX1 remain unknown.
Tlx1-deficient mice are asplenic without any other abnormalities (6, 11), a phenotype resembling human isolated congenital asplenia (OMIM 271400), a condition in which the lack of the spleen exists as a sole organ defect (9, 12). Currently, only the ribosomal protein SA (RPSA) gene has been found mutated in isolated congenital asplenia patients (13); however, the precise role of RPSA during spleen development remains unknown. Congenital asplenia can be also associated with other abnormalities, such as laterality defects as observed in patients with Ivemark syndrome (OMIM 208530), patients with cardiac defects and transposition of great arteries (14), or patients with disorders of sexual development (OMIM 612965) carrying mutations in the gene encoding for steroidogenic factor 1 (SF-1/NR5a1) (15). Interestingly, a mutant form of SF-1 was recently shown to be defective in activating TLX1 transcription in patients with disorders of sexual development and asplenia, thus providing the first evidence that perturbation of TLX1 expression may be implicated in human congenital asplenia (16).
TLX1 regulates cellular proliferation and differentiation in different cellular systems (6, 8, 17–22). During spleen development, loss of Tlx1 causes reduced proliferation of the splenic mesenchyme (SPM) and growth arrest (8, 23). Conversely, ectopic expression of Tlx1 in thymocytes blocks differentiation and promotes leukemogenesis by altering the expression of genes involved in cell cycle regulation and thymocyte development (18, 19, 21, 24). At the molecular level, TLX1 can act as both an activator and a repressor of gene transcription depending on the cellular context and its interaction with transcriptional cofactors (25). For example, retinaldehyde dehydrogenase 1 (Raldh1, also known as Aldh1a1) is a gene that encodes a retinoic acid–synthesizing (RA-synthesizing) enzyme (26), and in mouse embryonic fibroblasts, TLX1 activates Aldh1a1 expression (24, 25, 27). In contrast, in the developing mouse spleen, TLX1 represses Aldh1a1 expression (25). At present, however, it remains unknown whether TLX1 plays a role in regulating retinoid signaling during spleen development, and whether deregulation in this pathway affects spleen organogenesis.
RA, the active metabolite of vitamin A, is an essential molecule required for vertebrate patterning and embryogenesis (15, 26, 28–31). RA binds to nuclear receptors (RARs) and regulates critical developmental pathways governing cellular proliferation, differentiation, organogenesis, and tissue homeostasis (32, 33). In the developing embryo, the activities of RA-synthesizing (RDHs, ALDHs) and degrading enzymes of cytochrome P450 family 26 (CYP26) regulate RA metabolism (31). Notably, elevated RA signaling in Cyp26b1–/– mutants causes aberrant cellular proliferation and differentiation, leading to several organ abnormalities including lymphatic vascular defects and altered germ cell development (33–36). Notably, RA controls the fate of germ cells in mice while SF-1 regulates RA metabolism during germ cell development (15, 37). Furthermore, elevated RA signaling in the form of teratogenic doses of RA in mice, rats, and nonhuman primates has also been associated with organ growth abnormalities (38–43).
Herein, we set out to uncover the molecular mechanism by which TLX1 regulates spleen development. Using gene expression profile analysis, we found that loss of Tlx1 in the SPM causes upregulation of several genes involved in RA metabolism. Conversely, the expression of Cyp26b1, which encodes an enzyme involved in RA degradation, is markedly reduced in the embryonic splenic anlage of Tlx1 mutant mice. Analysis of Cyp26b1 or retinol dehydrogenase 10 (Rdh10) mutants, which respectively exhibit an excess or deficiency in RA signaling, revealed severe spleen hypoplasia or agenesis, demonstrating the importance of finely regulating RA metabolism to ensure proper spleen development. Interestingly, loss of Sf-1 during spleen development also reduced Tlx1 and Cyp26b1 expression. Genome-wide analysis indicated that TLX1 binds the regulatory regions of RA-associated genes through the AP-1 site and cooperates with the AP-1 family of transcription factors to regulate gene expression. Importantly, pharmacological inhibition of RA signaling partially rescued the spleen phenotype of Tlx1 mutants. Collectively, our findings unveil molecular interactions critical for spleen development and shed light onto the pathogenesis of congenital asplenia.
Results
Loss of Tlx1 deregulates the RA signaling pathway.
We previously showed that loss of Tlx1 causes defects in specification and proliferation of spleen mesenchymal progenitors (8). However, the mechanisms by which TLX1 coordinates the initiation and expansion of the splenic anlage remain unknown. To identify deregulated genes and signaling pathways associated with loss of Tlx1, we performed gene expression analysis using mRNA obtained from Tlx1+/– heterozygous and Tlx1–/– homozygous embryonic spleens at E13.5 (Figure 1A). This time point was chosen because it coincides with the appearance of the spleen defect in Tlx1–/– homozygous embryos. Gene ontology analysis revealed statistically significant differences in the expression of genes related to developmental processes including spleen organogenesis (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI82956DS1). To identify deregulated pathways resulting from Tlx1 loss, we took advantage of the Gene Set Enrichment Analysis (GSEA) tool, a computational method that detects modest but coordinated changes in the expression of groups of functionally related genes (44). Remarkably, we found a highly significant deregulation of the RA pathway (FDR = 0.006) (Figure 1A). Quantitative PCR (qPCR) validation analysis revealed that genes encoding for the RA-synthesizing enzymes (i.e., Aldh1a1), RA nuclear receptors (i.e., Rara, Rarb), and vitamin A/RA transporters (i.e., Stra6/Crabp2) were all upregulated as a result of Tlx1 ablation (Figure 1A). Conversely, the expression of the RA-degrading enzyme Cyp26b1 was significantly reduced in the absence of Tlx1 (Figure 1A).
Figure 1. TLX1 controls RA signaling pathway.

(A) GSEA enrichment plots and heat maps of differentially expressed genes belonging to the RA pathway associated with loss of Tlx1. The bar-code plot indicates the position of the genes on the expression data rank-sorted by its association with Tlx1 mutants, with red and blue colors indicating over- and underexpression in the mRNA. Validation of RA-associated genes by qPCR was performed on Tlx1+/– and Tlx1–/– embryonic spleens. The means of triplicates ± SD are shown, *P < 0.05, **P < 0.01 (2-tailed Student’s t test). Data are representative of 1 of 2 different validation experiments with 8–10 pooled spleens for each genotype. (B) TLX1 peak annotation relative to the indicated genomic feature. TLX1 ChIP-seq was performed in the eSMC line, and peak call was generated by comparison with an unrelated ChIP-seq experiment using rabbit IgG as control. MEME motif prediction of DNA sequences enriched in TLX1 ChIP-seq. ChIP-qPCR analysis of TLX1 binding in eSMC line. Positive (R1, R3, and R5) and negative (R2, R4, and R6) binding regions are indicated relative to transcription start site (TSS). Data are normalized to amplification of the input chromatin. Data are representative of 1 of 3 independent experiments.
TLX1 binds genes associated with the RA pathway.
To identify whether TLX1 binds RA-associated genes, we performed ChIP sequencing (ChIP-seq) in the embryonic spleen mesenchymal cell (eSMC) line. Analysis of the profile distribution revealed preferential binding of TLX1 to intergenic (44%) and intronic (38%) regions and, to a lesser extent, to promoter (14%) regions (Figure 1B). We then searched for putative TLX1-binding sites by motif discovery analysis using the multiple EM for motif elicitation (MEME) algorithm and found enrichment for an AP-1 site consensus element (Figure 1B), suggesting control of genes bearing the AP-1 motif (45). Confirming this finding, ChIP-qPCR analysis showed binding of TLX1 to Cyp26b1, Rara, and Rxra genomic regions carrying the AP-1 sequence (Figure 1B and Supplemental Sequences 1–5). Moreover, we found that the AP-1 sequence was present in several RA-associated genes deregulated in our GSEA analysis (Figure 1A and Supplemental Table 1). Consistent with TLX1 acting as either a cotranscriptional repressor or activator of gene transcription, cotransfection of Tlx1 with Jdp2 or c-Jun — 2 AP-1 family members with opposite transcriptional functions — synergistically modulates the transactivation of an AP-1 luciferase reporter system (Supplemental Figure 2A). Expression of Jdp2 was strongly reduced in Tlx1 mutant as compared with control embryonic spleens (Supplemental Figure 2B), and, in agreement with these data, ChIP-seq analysis followed by ChIP-qPCR revealed direct binding of TLX1 to the Jdp2 locus (Supplemental Figure 2B). These findings raised the possibility that TLX1 and JDP2 may act in concert to regulate transcription. We therefore examined a possible physical association of TLX1 and JDP2, and found that these transcription factors interact in reciprocal coimmunoprecipitation assays (Supplemental Figure 2C), thus suggesting transcriptional coregulation of target genes, including RA-associated genes. Collectively, these findings demonstrate a direct transcriptional control of TLX1 on RA signaling during spleen organogenesis.
RA signaling and Tlx1 expression are mutually exclusive during spleen development.
To determine whether and where RA signaling is active during normal spleen development, we analyzed WT RARE-LacZ transgenic embryos during the initial formation of the splenic anlage (Figure 2). In this mouse model, β-gal expression is under the control of an RA-responsive element (RARE), and, as a result, LacZ staining reveals the domains of active RA signaling (46). We found that at E11.5 RA activity is absent in the newly formed splenic anlage (Supplemental Figure 3). In contrast, RA activity is detectable at E13.5 and is confined to the outer mesothelium layer, whereas the inner SPM remains devoid of RA signaling (Figure 2A). Interestingly, staining of Tlx1LacZ/+ heterozygous embryos, in which LacZ marks the domain of Tlx1 expression, revealed that during normal spleen development, Tlx1 is expressed only in the SPM (Figure 2B). In contrast, TLX1 is excluded from the outer mesothelial region exhibiting RA signaling (Figure 2A), thus indicating that TLX1 and RA are present in mutually exclusive domains during spleen development. Altogether, these results indicate that TLX1 may repress RA signaling in the SPM, possibly through the control of Cyp26b1 expression and RA degradation.
Figure 2. Tlx1 expression is excluded from the domains of RA signaling.

Transverse sections of RARE-LacZ embryos at E13.5 stained for LacZ to show RA signaling (A) and confocal images of E13.5 Tlx1+/– spleen sections to show TLX1 (β-GAL) (B). Scale bars: 50 μm and 25 μm. st, stomach; spm, splenic mesenchyme; m, mesothelium. Data are representative of 1 embryo of 5 embryos analyzed.
Uncontrolled RA signaling causes spleen growth defects.
Previous work has shown that Cyp26b1 deficiency causes developmental defects consistent with excessive RA activity (34, 35, 39), demonstrating the critical role of this enzyme in regulating RA homeostasis during embryonic development. The finding that Cyp26b1 is expressed during spleen development (47) and that it is strongly reduced in Tlx1 mutant embryonic spleens (Figure 1A) prompted us to test the hypothesis that impaired CYP26B1 function causes spleen growth defects. Remarkably, analysis of Cyp26b1 mutant embryos at E16.5 (Figure 3A) revealed the presence of only remnant splenic tissue when compared with littermate controls.
Figure 3. Spatiotemporal requirement of RA signaling during spleen development.
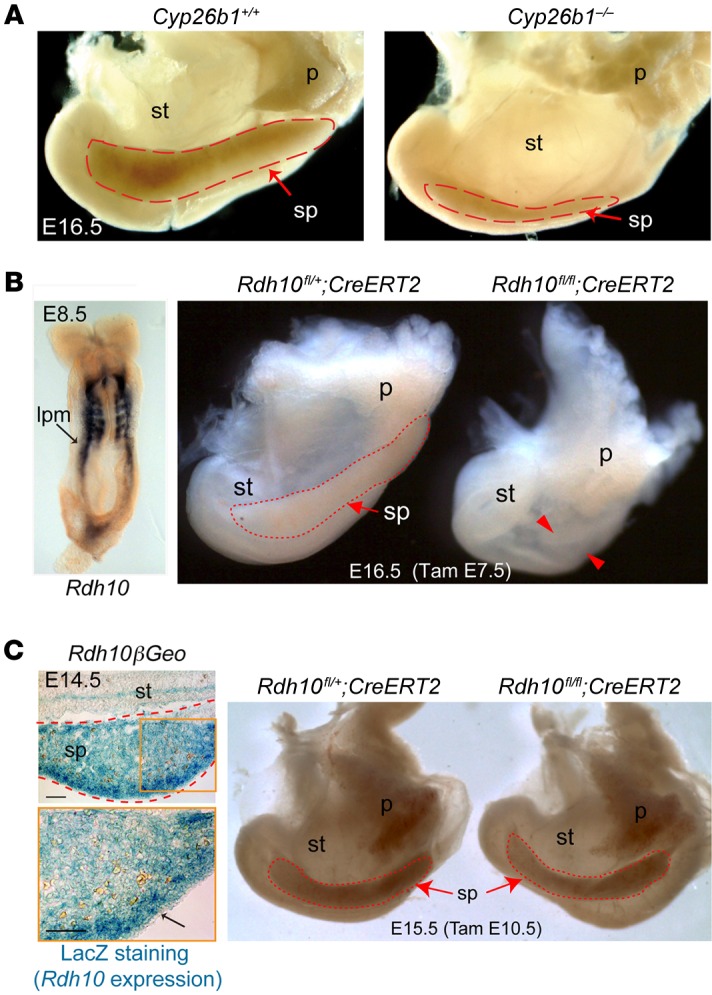
(A) Gross morphology of abdominal organs from Cyp26b1–/– and Cyp26b1+/+ embryos at E16.5. Data are representative of 1 embryo of 10 mutant and 8 control embryos analyzed from 4 different litters. (B) In situ hybridization for Rdh10 at E8.5 (left) and gross morphology of E16.5 abdominal organs from Rdh10fl/+;CreERT2 embryos injected with tamoxifen at E7.5 (right). Data are representative of 1 embryo of 6 mutant and 6 control embryos analyzed from different litters and time points for each genotype. (C) Expression of Rdh10 in the developing spleen as shown by LacZ staining of E14.5 spleens from Rdh10βGeo mice (left). Gross morphology of E15.5 abdominal organs from Rdh10fl/+;CreERT2 embryos injected with tamoxifen at E10.5 (right). Scale bars: 50 μm. Data are representative of 1 embryo of 15 mutant and 8 control embryos analyzed from 3 different litters. Dashed red lines indicate the developing spleen; red arrows indicate spleen normal situs. st, stomach; sp, spleen; p, pancreas; lpm, lateral plate mesoderm.
We then hypothesized that excessive RA signaling due to loss of Cyp26b1 expression inhibits spleen development, and therefore sought to assess whether physiological levels of RA are required for the proper patterning of spleen descendants in the lateral plate mesoderm (LPM), from which the spleen is thought to arise. We first performed lineage-tracing experiments to unequivocally demonstrate the contribution of the LPM to the SPM by crossing R26YFP reporter mice to Hoxb6-CreER mice, in which tamoxifen-dependent Cre expression is directed to the LPM (48). Injection of Hoxb6-CreER R26RYFP pregnant females at E8.5 with a single dose of tamoxifen followed by immunofluorescence analysis revealed a large contribution of the YFP+ LPM mesodermal cells to the developing SPM (Supplemental Figure 3).
Next, we crossed Rdh10fl/fl mice to the Cre-ERT2 tamoxifen-inducible transgenic line to conditionally ablate Rdh10, a critical enzyme involved in the first step of RA synthesis in the embryo (49, 50). Consistent with its expression in the LPM (Figure 3B), ablation of Rdh10 by a single dose of tamoxifen at E7.5 caused spleen agenesis in mutant embryos compared with littermate controls (Figure 3B). Conversely, despite expression of Rdh10 in developing spleen (Figure 3C), ablation of Rdh10 by administration of tamoxifen at E10.5 during initial formation of the spleen primordium did not affect organogenesis (Figure 3C). Collectively, these findings demonstrate a differential and precise spatiotemporal requirement for RA during spleen development.
CYP26B1 in spleen mesenchymal cells is required to prevent aberrant RA signaling.
To formally demonstrate that increased RA activity in the Tlx1 mutant SPM is caused by reduced Cyp26b1 expression, we exploited an in vitro system by coculturing RA reporter F9-LacZ cells (51) with an eSMC line that has been shown to mimic the native embryonic mesenchyme (9). Notably, silencing Tlx1 in eSMCs via shRNA recapitulates our in vivo data by causing deregulated expression of several RA-associated genes, including Cyp26b1 (Figure 4A and Supplemental Figure 4).
Figure 4. TLX1 controls RA activity.

(A) Western blot analysis of TLX1 protein in eSMCs upon shRNA silencing of Tlx1 (left). Anti-vinculin (Vcl) antibody is used as a loading control. Expression of Tlx1 and Cyp26b1 in sh-Ctrl and sh-Tlx1 eSMCs (right). Data are representative of 3 independent experiments. (B) Scheme of the coculture experiments with RARE-LacZ F9 reporter cells and sh-Ctrl or sh-Tlx1 eSMCs. Bright-field images of LacZ staining of F9-RARE-LacZ reporter cells cocultured for 48 hours with sh-Ctrl eSMCs (control), sh-Tlx1 eSMCs (silenced), or sh-Tlx1 eSMCs re-expressing Cyp26b1. Scale bars: 50 μm. Differences were measured by counting of the number of LacZ+ cells (in blue) over total cells. Data are representative of 1 of 3 independent experiments. (C) RARE-F9 cells transiently expressing Tlx1 or control vector were treated with RA and expression of indicated genes analyzed by qPCR 30 hours later. Data are representative of 1 of 3 independent experiments. (A–C)The means of triplicates ± SD are shown, *P < 0.05 (B and C), **P < 0.01 (A and C) (2-tailed Student’s t test).
To assess the functional consequences of Tlx1 and Cyp26b1 deficiency for RA signaling, we cocultured RA reporter F9-LacZ cells with shRNA-Tlx1 or shRNA-Ctrl control eSMCs, and performed LacZ staining to measure RA activity (Figure 4B). In the absence of TLX1, the number of LacZ+ cells was significantly higher than among reporter cells cultured with shRNA-Ctrl control eSMCs (Figure 4B). Interestingly, rescue of Cyp26b1 expression reduced the number of blue cells seen among shRNA-Tlx1 eSMCs to none or a few, similar to what is seen in shRNA-Ctrl control eSMCs (Figure 4B). These findings demonstrate that loss of CYP26B1 function in Tlx1 mutants causes increased RA signaling in the SPM.
To corroborate the role of CYP26B1 in limiting RA content and signaling in the SPM, we measured the expression of RA target genes under condition of CYP26B1 inhibition. RA-treated eSMCs cultured in the presence of the CYP26B1 inhibitor R116010 (52) displayed increased expression of RA-responsive genes (Supplemental Figure 3), thus demonstrating the critical role of CYP26B1 in restricting RA signaling in spleen mesenchymal cells.
TLX1 blocks RA-induced differentiation.
Since our data demonstrated a role for TLX1 as a repressor of RA signaling, we next evaluated the effect of RA on the differentiation of F9 cells, a cellular system previously used to study RA-induced differentiation (53, 54). We found that the expression levels of Lamb1 and Col4a1, 2 genes associated with RA-induced endodermal differentiation (53, 55), were significantly reduced in Tlx1-overexpressing F9 cells as compared with control cells (Figure 4C). Similarly, the expression of Rarb, a gene positively regulated by RA, was 50% lower in Tlx1-overexpressing cells than in control cells (Figure 4C), thus demonstrating that TLX1 counteracts RA-induced differentiation.
RA induces spleen growth arrest.
GSEA analysis revealed enrichment for pathways associated with cell cycle control. Notably, the genes included in this signature were downregulated in Tlx1 mutants (Supplemental Figure 5A). Consistent with this, we found that treatment of E13.5 primary spleen mesenchymal cells with RA significantly suppressed cellular proliferation compared with vehicle-treated cultures (Figure 5A). In line with this, RA treatment induced an upregulation of the cell cycle inhibitor Cdkn2b/p15. Notably, we found a significant upregulation of Cdkn2b/p15 mRNA levels in Tlx1 mutants as compared with control embryonic spleens (Figure 5A).
Figure 5. RA induces the expression of cell cycle inhibitors and growth arrest.

(A) Growth curve analysis of primary E13.5 spleen mesenchymal cells treated with RA or control vehicle (left). The means of triplicates ± SD are shown, *P < 0.05 (2-way ANOVA). Data are representative of 1 of 3 independent experiments. Expression of Cdkn2b/p15 in primary E13.5 spleen mesenchymal cells treated for 3 or 5 days with RA or control vehicle (middle). The means of triplicates ± SD are shown, **P < 0.01, ***P < 0.001 (2-tailed Student’s t test). Data are representative of 1 of 2 independent experiments. Heat map and validation of Cdkn2b/p15 expression by qPCR in E13.5 Tlx1+/– and Tlx1–/– spleens (right). The means of triplicates ± SD are shown, **P < 0.01 (2-tailed Student’s t test). Data are representative of 1 of 2 independent validation experiments. (B) Bright-field images of E13.5 explanted spleens cultured in the presence of RA or vehicle (DMSO). Arrows indicate mesenchymal cell sprouting. Quantification of the spleen size area (%) was calculated using ImageJ, as the ratio of spleen area at 24 hours versus 0 hours. The means of triplicates ± SD are shown, **P < 0.01 (2-tailed Student’s t test). Data are representative of 1 of 3 independent experiments with 8 explanted spleens for each condition.
To assess the functional effects of RA exposure on spleen growth, we performed organotypic cultures. E13.5 WT littermate spleens were divided into 2 groups and cultured, on a collagen layer, in the presence of RA or vehicle. Quantitative analysis of spleen size revealed that RA exposure significantly inhibits growth, which is accompanied by reduced sprouting of mesenchymal cells into the collagen layer (Figure 5B, arrows). In line with these data, silencing of Tlx1 causes G1 cell cycle arrest and deregulation of the RA signaling pathway (Supplemental Figure 4A and Supplemental Figure 5B). Altogether, these findings demonstrate that excessive RA signaling inhibits spleen organ expansion by reducing spleen mesenchymal cell proliferation.
Increased RA signaling induces premature differentiation and reduced vasculogenesis.
Retinoid excess in humans and mice causes a wide spectrum of malformations associated with patterning defects, premature differentiation, and organ growth arrest (29, 35, 36, 38–40, 56). Consistent with this phenomenon, our GSEA revealed enrichment of signatures associated with markers of lymphoid stromal cell maturation and extracellular matrix. Particularly, genes that belong to these pathways were induced in Tlx1 mutant as compared with control splenic anlage (Supplemental Figure 6). To test the possibility that increased RA activity in the Tlx1 mutant SPM results in premature cellular differentiation, we evaluated the expression of Desmin, a marker associated with maturation of spleen mesenchymal cells (57–59). Desmin was strongly upregulated at both the mRNA and the protein level in E13.5 Tlx1 mutants as compared with control embryonic spleens (Figure 6A). Consistent with this observation, treatment of E14.5 primary spleen mesenchymal cells with RA induced a significant upregulation of Desmin expression (Figure 6A). In addition, we assessed the expression of ER-TR7, an antigen expressed by differentiating spleen and lymph node mesenchymal cells (60). Importantly, immunofluorescence analysis showed that ER-TR7 expression starts only postnatally in WT mice (Supplemental Figure 6B). However, ER-TR7 is prematurely expressed in Tlx1 mutants as compared with controls (Supplemental Figure 6C). Finally, GSEA analysis revealed a significant increase in the expression of Hox genes that are known to be under the control of retinoid signaling during embryonic development (32) (Supplemental Figure 6D). Altogether, these findings indicate that the premature differentiation occurring in Tlx1 mutants is caused, at least in part, by deregulation of RA signaling.
Figure 6. Excessive RA due to loss of Tlx1 causes premature differentiation and reduced vasculogenesis.

(A) Heat map and validation of Desmin expression by qPCR in E13.5 Tlx1+/– and Tlx1–/– spleens (left). Confocal images of E13.5 spleen sagittal sections stained with anti-desmin antibody (green) (middle). Nuclei are visualized by DAPI staining (blue). Dashed red lines indicate the developing spleen. Data are representative of 1 embryo of 5 embryos analyzed for each genotype. Desmin expression by qPCR in primary E13.5 spleen mesenchymal cells after 48 hours of RA treatment (right). Data are representative of 1 of 2 independent experiments. (B) Expression of Vegf-a in primary E13.5 spleen mesenchymal cells treated for 48 hours with RA or control vehicle. Heat map and validation of Vegf-a expression by qPRCs in E13.5 Tlx1+/– and Tlx1–/– spleens. Data are representative of 1 of 2 independent experiments. IHC analysis on E13.5 Tlx1+/– and Tlx1–/– sagittal sections stained with anti–PECAM-1 antibody to reveal vascular networks and counterstained with hematoxylin to show nuclei. Microvessel density was calculated by counting of the number of PECAM-1+ vessels/μm2 in Tlx1+/– and Tlx1–/– embryonic spleen. Data are representative of 1 embryo of 5 embryos analyzed for each genotype. (C) Representative confocal images of E13.5 Tlx1+/– and Tlx1–/– spleen sagittal sections stained with anti–PECAM-1 antibody (red) to reveal endothelial cells and anti–β-gal antibody (green) to show TLX1-expressing cells in Tlx1-LacZ knock-in embryos. Nuclei are visualized by DAPI staining (blue). Data are representative of 1 embryo of 5 embryos analyzed for each genotype. (A and B) The means of triplicates ± SD are shown, *P < 0.05 (B), **P < 0.01 (A and B) (2-tailed Student’s t test). (A–C) Scale bars: 50 μm. st, stomach; sp, spleen.
In addition to mesenchymal cells, the splenic anlage comprises endothelial cells that form an expanding vascular network. RA was shown to regulate the expression of Vegf-a (61), the major mitogen for endothelial cell proliferation and vessel network formation. Consistent with this principle, treatment of E14.5 primary spleen mesenchymal cells with RA reduced Vegf-a expression as compared with vehicle-treated cells (Figure 6B). Also, Vegf-a expression was strongly reduced in E13.5 Tlx1 mutant as compared with controls, and this was accompanied by decreased microvessel density (Figure 6B). Moreover, TLX1 expression as indicated by β-gal staining of Tlx1LacZ/+ knock-in embryos was not expressed in PECAM-1+ endothelial cells (Figure 6C), thus indicating a non–cell-autonomous role of TLX1 during vasculogenesis. These results are consistent with the idea that RA affects vasculogenesis in a paracrine fashion.
Inhibition of RA signaling partially rescues the spleen phenotype.
We hypothesized that inhibition of RA signaling could potentially ameliorate the asplenic phenotype of Tlx1 mutants. To test this idea, we crossed Tlx1+/– mice and treated pregnant females with BMS493, a pan-RAR antagonist (62, 63), during the initiation of spleen development at E10.5. We took advantage of Tlx1 mutant mice expressing LacZ under the Tlx1 endogenous promoter, in which LacZ staining marks the distribution of TLX1+ spleen mesenchymal cells within the nascent splenic anlage. We observed that at E14.5, BMS493-treated Tlx1 mutant spleens were mostly formed by 1 single splenic anlage and were more compact compared with untreated controls that exhibited multiple unjoined splenules/accessory spleens (Figure 7A). However, an evaluation of spleen size, as assessed by measurement of the surface of the LacZ+ anlagen, did not reveal significant differences between homozygous mutant spleens from BMS493- and vehicle-treated mice (Figure 7A). Furthermore, qPCR analysis revealed that BMS493 treatment induced a significant rescue in the expression of Vegf-a, whereas Desmin expression was not rescued (Figure 7B). Importantly, the expression of 2 RA target genes — Col4a1 and Lamb1 — which are negatively regulated by RA in the SPM of Tlx1 mutants, was completely recovered by RA signaling inhibition (Figure 7B). Similarly, the expression of Rarb, a gene positively regulated by RA, was also completely rescued by BMS493 treatment, thus demonstrating the presence of increased RA signaling in the absence of Tlx1. Altogether, these findings indicate that repression of RA signaling is required for condensation of spleen mesenchymal cells and organ morphogenesis.
Figure 7. Inhibition of RA signaling partially rescues the spleen phenotype.

(A) Partial rescue of spleen morphogenesis in E14.5 Tlx1–/– embryonic spleens treated with BMS493 or control vehicle at E10.5. Number of unjoined splenules (outlined in white) and surface area measurements were normalized to littermate controls. Statistical significance in the number of unjoined splenules was calculated using χ2, *P < 0.05. For the surface area measurement, the means of triplicates ± SD are shown. NS, not significant (2-tailed Student’s t test). Data are from 15 (BMS493) and 14 (DMSO) treated embryonic spleens from 3 different litters. HOM, homozygous; HET, heterozygous. (B) Validation of Vegf-a, collagen 4a1 (Col4a1), laminin B1 (Lamb1), and Rarb expression by qPRCs in E14.5 Tlx1+/– and Tlx1–/– embryonic spleens treated with BMS493 or control vehicle. The means of triplicates ± SD are shown, *P < 0.05, **P < 0.01, ***P < 0.001 (2-tailed Student’s t test). Data are representative of a pool of 12–16 embryonic spleens for each group from 4 different experiments.
SF-1 controls Tlx1 and RA metabolism in the SPM.
Recent work by Zangen and collaborators reported that SF-1, which is required for human spleen development, transactivates the TLX1 promoter (16). In addition, loss of Sf-1 in mice causes a severe spleen hypoplasia defect (64). To test the hypothesis that Sf-1 is genetically upstream of Tlx1, we performed gene expression analysis using mRNA obtained from Sf1–/– homozygous or control splenic anlagen at E14.5. Consistent with the hypothesis, we found a significant reduction of Tlx1 mRNA levels in Sf1–/– mutants as compared with controls (Figure 8A). The expression of the RA-degrading enzyme Cyp26b1 was also severely reduced in Sf1–/– mutants as compared with controls (Figure 8A). In agreement with this observation, we found that SF-1 transactivates the Cyp26b1 promoter in a dose-dependent manner (Figure 8B). We then tested the possibility that, similar to TLX1, SF-1 may counteract the differentiation effects of RA. To this end, we treated F9 cells, transiently transfected with Sf-1, with RA and evaluated the expression of markers associated with cellular differentiation. Similarly to what we observed for Tlx1, overexpression of Sf-1 significantly inhibited cellular differentiation as demonstrated by reduction of the expression of Lamb1 and Col4a1, 2 genes associated with RA-induced endodermal differentiation (53, 55) (Figure 8C). Moreover, the expression of Rarb, a gene positively regulated by RA, was also significantly lower in Sf-1–expressing cells as compared with control cells (Figure 8C). Previous work showed that loss of Sf-1 during spleen development causes reduced vasculogenesis (64). Consistent with this finding, we observed a significant reduction in Vegf-a mRNA levels in Sf-1 mutants as compared with control embryonic splenic anlagen (Figure 8). Altogether these findings support a model in which SF-1 regulates Tlx1 expression and RA metabolism to ensure spleen development.
Figure 8. SF-1 controls Tlx1 and RA metabolism.

(A) Expression of Tlx1, Cyp26b1, and Vegf-a in E14.5 Sf-1+/+ and Sf-1–/– embryonic spleens. Data are representative of 1 of 2 independent experiments with 4 pooled spleens for each genotype. (B) Luciferase activity on a Cyp26b1 promoter reporter system was assessed at 48 hours in HEK293 cells transiently transfected with increasing concentrations of Sf-1 expression vector. Data are representative of 1 of 3 different experiments. (C) RARE-F9 cells transiently expressing Sf-1 or control vector were treated with RA and expression of indicated genes analyzed by qPCR 8 hours later. Data are representative of 1 of 3 independent experiments. (A–C) The means of triplicates ± SD are shown, *P < 0.05 (A and C), **P < 0.01 (A–C), ***P < 0.001 (B and C) (2-tailed Student’s t test).
Discussion
The lack of a spleen is often associated with deadly infections in humans. At present, the molecular mechanisms underlying asplenia remain mostly elusive. In the absence of Tlx1, spleen mesenchymal cells fail to proliferate and to form a discrete splenic primordium after E13.5 (6, 8, 11, 65). Our findings demonstrate that expression of Tlx1 in the SPM is required to control excessive RA signaling in order to assure the proliferation of mesenchymal precursors and the formation of the splenic anlage. In support of this conclusion, we showed that loss of Tlx1 leads to an increase in RA activity. This effect is due to reduced RA degradation caused by deregulated Cyp26b1 expression and to increased nuclear receptor expression and activation. RA may diffuse or be transported within the nascent SPM, causing aberrant morphogenesis, premature differentiation, and vascular abnormalities. Furthermore, the finding that Cyp26b1 mutant embryos exhibit only a remnant of splenic tissue clearly demonstrates the importance of restricting RA activity within the SPM to ensure proper spleen development. In support of this notion, re-expression of Cyp26b1 in Tlx1-deficient mesenchymal cells restored RA signaling to control levels, indicating that uncontrolled RA signaling in Tlx1 mutants is due to a deficit in CYP26B1 activity. While excessive RA signaling in Cyp26b1 mutants is detrimental during the formation of the splenic primordium, loss of RA synthesis at this stage did not affect spleen development. Nevertheless, ablation of retinoid synthesis in the LPM from which spleen mesenchymal precursors originate caused spleen agenesis, indicating the presence of a spatiotemporal window in which retinoids are required during spleen organogenesis.
Excess of retinoids has been shown to cause several developmental anomalies, including abnormal morphogenesis and organ growth inhibition (38–40, 43, 66). Consistent with these observations, we showed that Tlx1 mutant spleens exhibit reduced vasculogenesis and premature differentiation of mesenchymal progenitors. RA mediates these effects, since addition of RA to spleen organotypic cultures reduces Vegf-a expression while inducing Desmin and other markers normally expressed by mature lymphoid stromal cells (59). Also, Tlx1 is not expressed in endothelial cells of the SPM, indicating a non–cell-autonomous role in the vasculogenesis defect. Thus, reduced CYP26B1-degrading activity within the SPM causes increased levels of RA signaling that affects endothelial cell proliferation in a paracrine fashion.
We have previously shown that loss of Tlx1 leads to reduced proliferation of the SPM (8, 18). Consistent with this observation, we found that silencing Tlx1 in spleen mesenchymal cells increases RA signaling and reduces proliferation by promoting G1 cell cycle arrest. The notion that RA-induced growth inhibition is associated with G1 cell cycle arrest in different cellular systems (67) suggests that RA may contribute to the proliferation defect observed in Tlx1 mutant spleen. Supporting this hypothesis is the finding that Cdkn2b/p15, a gene that we showed to be induced by RA treatment in primary eSMCs, is significantly increased in Tlx1-deficient splenic anlage. However, the spleen phenotype of Tlx1 mutants is only partially rescued upon inhibition of RA signaling in vivo. One explanation for this discrepancy may be the inefficient activity of the BMS493 inhibitor on RA signaling in vivo. In addition, TLX1 may control gene transcription and cellular proliferation independently of RA as previously demonstrated (17, 18). Indeed, while expression of Col4a1, Lamb1, and Rarb was significantly rescued upon RA signaling inhibition, Vegf-a transcription was only modestly reactivated. In addition, the expression of Desmin and Cdkn2b/p15 (not shown) was not rescued upon RA inhibition.
At the molecular level, our ChIP-seq analysis revealed that TLX1 binds DNA preferentially through the AP-1 motif, a sequence found in the Vegf-a gene (61) and in several RA-associated genes, including Cyp26b1, Rara, and Rxra, which we found bound by TLX1. Among the TLX1 target genes, we also identified Jdp2, an AP-1 family transcription factor deregulated in the absence of Tlx1 and known to repress RA-induced differentiation (68). As with JDP2, we showed that TLX1 also represses RA-induced differentiation and that TLX1 and JDP2 physically interact. These findings support a scenario in which reduced expression of these transcription factors within the SMP causes deregulation of genetic programs controlled by RA. Thus, we propose that, in addition to controlling RA metabolism in the SPM, TLX1 directly regulates RA target genes containing the AP-1 sequence. Accordingly, our microarray and ChIP-seq data indicate that TLX1 represses the activation of nuclear RARs by binding to their regulatory regions. In the absence of TLX1, the transcription of RARs would be derepressed, while uncontrolled RA in the SPM would favor the conversion of RARs from corepressors into coactivators and the subsequent induction of c-Jun and other RA-regulated genes. In support of this idea, c-Jun, which is induced by RA in an AP-1–dependent manner, is strongly upregulated in Tlx1 mutant spleens compared with controls (not shown). In a similar manner, expression of Desmin, a marker associated with cellular differentiation positively regulated by RA and AP-1, is significantly increased in Tlx1 mutant spleens compared with controls.
In this scenario, our data further support the previous hypothesis by Zangen and collaborators (16) and implicate SF-1 as an upstream regulator of Tlx1 during spleen development. Our findings also indicate that SF-1 controls Cyp26b1 expression independently of TLX1. Thus, we propose a model in which SF-1 is required to activate Tlx1 transcription and both TLX1 and SF-1 independently control Cyp26b1 expression and RA metabolism (Figure 9). Supporting a role for SF-1 in the control of RA activity is the demonstration that SF-1 regulates Cyp26b1 expression during germ cell development (15). We showed that, similarly to what occurs with Tlx1 mutants, loss of Sf-1 causes reduced Vegf-a expression. Given that RA affects Vegf-a expression in spleen mesenchymal cells, it is likely that the severe vasculogenesis defect observed in Sf-1–deficient mice is caused, at least in part, by aberrant RA signaling (64).
Figure 9. Proposed model for SF-1 and TLX1 function during spleen development.

RA signaling starts at E13.5 and is confined in the outer mesothelial layer of the developing spleen, a domain negative for TLX1. On the contrary, SF-1 and TLX1 restrict RA signaling by promoting Cyp26b1 expression and RA degradation, and repression of RA nuclear receptors in the inner mesenchyme. In the absence of Sf-1 or Tlx1 the expression of Cyp26b1 is markedly reduced, thus causing increased RA content and activity. As a result, RA binds to nuclear receptors that activate RA-induced transcriptional programs. Under these conditions, RA signals in an autocrine (dashed arrows) and paracrine (solid arrows) fashion within the SPM, thus causing growth arrest due to premature cellular differentiation and reduced vasculogenesis. Thus, expression of SF-1 and TLX1 in the SPM is required to control RA signaling and ensure spleen development.
Interestingly, although mice deficient for Cyp26b1 have severe spleen hypoplasia, it has recently been reported that 2 siblings homozygous for a null allele of CYP26B1 have normal spleen size (39) (S. Robertson, unpublished observations). At present, the reason for this discrepancy remains unclear, though it is possible that the human CYP26B1-null allele is associated with incomplete clinical penetrance or that other CYP family members compensate for CYP26B1 deficiency in humans. Nevertheless, it is tentative to speculate that perturbations of Tlx1 expression, both in mice and in humans, may deregulate critical downstream pathways, including retinoid signaling that affects spleen development. Interestingly, an excess of retinoids in animal models causes heterotaxy, a syndrome associated with congenital asplenia and vascular defects in humans (39, 40, 61). In conclusion, our findings reveal the important role of TLX1 in controlling RA signaling during spleen organogenesis and provide novel mechanistic insights underlying the pathogenesis of congenital asplenia.
Methods
Mice.
Tlx1LacZ/+ (6), Cyp26b1+/– (33), Rdh10fl/fl (50), RARE-hsp68-LacZ (46), Hoxb6-CreER (48), Rdh10βGeo (50), Sf-1+/– (69–71), and Cre-ERT2 mice have been previously described (72). R26RYFP mice (73) were used as a reporter strain of Cre activity. C57BL/6N and CD1 mice were purchased from Charles River, Italy. Mice were bred and maintained at San Raffaele Scientific Institute in pathogen-free rooms under barrier conditions, with constant temperature, food and water ad libitum, and light/dark cycles of 12 hours. Tamoxifen (5 mg) and progesterone (1 mg) were dissolved in corn oil and administered via oral gavage to pregnant mice at the desired embryonic stage of development.
Pregnant Tlx1+/– or Tlx1–/– mice were treated with RAR antagonist BMS493 (Tocris Bioscience) or vehicle (DMSO; Sigma-Aldrich) at the dose of 5 mg/kg in corn oil. Mice were mated overnight, and the day of vaginal plug detection was marked as E0.5. Treatment was given by oral gavage twice daily in 10- to 12-hour intervals from E10.5 until E13.5.
RNA isolation, qPCR, and microarray analysis.
For gene expression profile analysis, total RNA was extracted from embryonic spleens harvested at E13.5 and E14.5, primary spleen mesenchymal cells (E14.5), and the established eSMC line with RNeasy Mini isolation kit (Qiagen). Reverse transcription of 1 μg of total RNA was performed using the ImProm II Reverse Transcription System kit with random primers (Promega Corp.). qPCRs were performed using the Universal Probe Library system on a LightCycler 480 (Roche). The Ct of Rpl13a (housekeeping) was subtracted from the Ct of the target gene, and the relative expression was calculated as 2–ΔCt. qPCRs were performed in triplicate and mean ± SD represented as relative expression. Primer sequences are described in Supplemental Table 2. Microarray analysis was performed using GeneChip Mouse Gene 1.0 ST Array from Affymetrix (GSE68519). Data were processed and normalized (GC robust multi-array average [GCRMA]) using GenePattern (74).
Chromatin immunoprecipitation.
ChIP experiments were performed using the eSMC line according to a protocol described previously (8). In brief, 1 × 108 cells were cross-linked with 1% formaldehyde at room temperature followed by addition of glycine at a final concentration of 0.125 M. Chromatin was fragmented by sonication with Sonicator Ultrasonic Processor XL (Misonix Inc.) with a 3/16-in. (4.8-mm) microtip (419A Tapered Microtip; Gilson Inc.) at 30% of amplitude for 30 seconds and for 35 times with a 1-minute interval. Samples were immunoprecipitated with 4 μg of the antibodies anti-Tlx1 (rabbit polyclonal, SC-880X; Santa Cruz Biotechnology Inc.) and the normal rabbit serum IgG (ChromoPure 011-000-003; Jackson ImmunoResearch Laboratories Inc.). Immunoprecipitated DNA was purified using Qiaquick PCR Purification kit (Qiagen), suspended in 45 μl of Tris-HCl 10 mM, and amplified by qPCR on a LightCycler 480 using TaqMan chemistries (Roche). Regions within Cyp26b1 (R1, R2), Rxra (R3, R4), Rara (R5, R6), and Jdp2 (R7, R8, R9) were tested using primers designed using the UPL Assay Design Center (Roche) and are listed in Supplemental Table 3. For ChIP sequencing (ChIP-seq) analysis the purified DNA was quantified using Quant-iT PicoGreen dsDNA kit (Invitrogen). For each ChIP-seq assay 40 ng of DNA was used directly for cluster generation and sequencing analysis with the HiSeq 2000 following the protocol of the manufacturer (Illumina).
Sequencing and data analysis.
Sequencing data (GSE81661) generated from the Illumina platforms were aligned to mouse reference genome (mm9) using Bowtie version 0.12.7. Only reads with unique alignment were retained for downstream analysis. Peak calling and bigWig files were generated using Model-Based Analysis for ChIP-seq (MACS) version 1.4. Only peaks with 10× –log P value ≥ 70 are considered for further processing. bigWig files were visualized using the University of California, Santa Cruz (UCSC) browser (http://genome.ucsc.edu). The list of mm9 annotated RefSeq genes used for the different analyses was downloaded from the UCSC database, and the data were processed by the MEME tool.
Immunofluorescence staining.
E13.5 and E16.5 embryos and P1 spleens were harvested and fixed 30 minutes to 1 hour at 4°C with 4% (wt/vol) PFA (Sigma-Aldrich), then washed in PBS 1× and dehydrated overnight in 30% sucrose (Sigma-Aldrich) at 4°C. Samples were embedded in Tissue-Tek OCT compound (Bio-Optica) and frozen in an ethanol dry-ice bath. Eight- to ten-micrometer-thick sections were placed onto glass slides (Bio-Optica), fixed in cold acetone for 5 minutes, dried, and kept at –80°C until used. Slices were incubated 30 minutes with a blocking solution of PBS at 0.5% FBS and 0.5% Tween (PBS-T 0.05%), followed by anti-desmin (mouse IgG1; D1033, clone DE-U-10; 1:200 stock 8.8 μg/μl; Sigma-Aldrich), anti–β-gal (mouse IgG1; G8021, clone GAL-13; 1:100 stock 5 μg/μl; Sigma-Aldrich), anti–CD31/PECAM-1 (PE rat IgG2a; 553373, clone MEC 13.3; 1:100 stock 0.2 μg/μl; BD), and anti–ER-TR7 (rat IgG2a; BM4018, clone ER-TR7; 1:200 stock 0.4 mg/ml; Acris) specific antibodies. Secondary anti-mouse Alexa Fluor 488 (1:500 stock 2 μg/μl; A10684; Invitrogen) or anti-rat Alexa Fluor 488 (1:500 stock 2 μg/μl; A21208; Invitrogen) antibody was diluted in PBS-T 0.05% blocking solution and incubated for 30 minutes. Nuclei were visualized with DAPI (Fluka), and mounting was performed with Mowiol (Calbiochem). Images were acquired using an Ultraview Leica TCS SP2 laser confocal microscope. Digital images were recorded in separately scanned channels with no overlap in detection of emission from the respective fluorochromes. Final image processing was performed with Adobe Photoshop and Illustrator.
IHC staining.
E13.5 embryos were harvested and fixed overnight at 4°C with 4% (wt/vol) PFA and processed into paraffin through graded alcohol series. Five- to eight-micrometer-thick sections were placed onto glass slides, dried overnight at 40°C–42°C, and kept at room temperature until used. Slices were deparaffinized, quenched for 5 minutes in 3% (vol/vol) hydrogen peroxide, and incubated with blocking solution of 3% of BSA (Sigma-Aldrich) in TBS (VWR) for 10 minutes at room temperature. Sections were stained with anti–CD31/PECAM-1 (rabbit polyclonal; 1:200 stock 200 μg/ml; RB-10333; Thermo Scientific) antibody overnight at 4°C. Immunolabeling was performed using Vectastain Elite ABC kit (Vector Laboratories) according to the manufacturer’s instructions. Slides were counterstained in Gill’s Hematoxylin (Sigma-Aldrich) and next mounted with xylene (Carlo Erba). Images were acquired using Aperio Scan Scope slide scanner (Leica Biosystems), and quantification of the microvessel density was performed with ImageJ software. Final image processing was performed with Adobe Photoshop and Illustrator.
Cell cultures, silencing, and transfections.
The embryonic spleen mesenchymal cell (eSMC) line, the F9-RARE-LacZ reporter cell line, and the embryonic endothelial cell line were previously described (9, 51, 75). Primary embryonic spleen mesenchymal cells were generated from E13.5 embryonic spleens. Cells were grown at 37°C, 5% CO2 in DMEM (Gibco) supplemented with 10% heat-inactivated FBS (Euroclone), 2 mM l-glutamine (l-Glu; Gibco), and 100 U/ml penicillin and 100 μg/ml streptomycin (Pen/Strep; Gibco). 293T (9) and NIH3T3 (8) cells were grown in Iscove’s modified Dulbecco’s medium (IMDM; Gibco) or in DMEM (Gibco), respectively, supplemented with 10% heat-inactivated FBS, l-Glu, and Pen/Strep. Embryonic endothelial cells were grown in 0.1% gelatin (Sigma-Aldrich) precoated dishes in DMEM supplemented with 20% FBS, l-Glu, Pen/Strep, 1 mM Na pyruvate (Gibco; Thermo Fisher Scientific), 100 μg/ml heparin (Sigma-Aldrich), and 5 μg/ml endothelial cell growth supplement (ECGS; Sigma-Aldrich). For silencing experiments, lentiviral particles were produced and used following the manufacturer’s instructions using the lentiviral plasmid pLKO.1-puro containing 2 different shRNA target sequences against murine Tlx1 or the scrambled sequence (Sigma-Aldrich). The shRNA-Tlx1 sequence was CCGGCGGTATCGACCAGATCCTCAACTCGAGTTGAGGATCTGGTCGATACCGTTTTTG; the control shRNA-scrambled sequence was a non-target shRNA. Selection of infected cells was performed in puromycin (Sigma-Aldrich), and cells were kept under culture conditions described above during all experiments.
For transfection experiments, cells were transfected using Lipofectamine 2000 (Invitrogen) or Amaxa MEF2 Nucleofector Kit (Lonza) with the following vectors: pCMV-Cyp26b1 (76), pcDNA3-Tlx1 (8), pcDNA3-Jun (77), pcDNA3-SF-1 (78), pcDNA4-Jdp2, and pAP1-Luc (53, 55). The Cyp26b1promo-Luc vector was generated by cloning of 4,000 bp of the mouse Cyp26b1 proximal promoter isolated by PCR from genomic DNA into a pGL3 luciferase reporter vector (Promega Corp.). Sequencing was performed to verify accuracy of the insert. Luciferase activity was assayed with a dual luciferase assay system (Promega Corp.), and activities were standardized against the internal control, Renilla luciferase or β-gal.
In situ hybridization.
Whole-mount in situ hybridization for Rdh10 was performed as previously described (49, 79) using digoxigenin-labeled probe with hybridization detected via NBT-BCIP or BM-purple (Roche).
Western blot analysis and immunoprecipitation assay.
Cells were lysed in RIPA buffer (5 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% NaDOC) containing protease inhibitor cocktail (Roche). Protein concentration was determined by the Bradford assay following the manufacturer’s instructions (Bio-Rad). Proteins were transferred onto a PVDF membrane (Immobilon-O; Millipore) and blocked in 5% skim milk, and immunoblotting was performed with anti–Hox11/Tlx1 (1:500 stock 200 μg/ml; SC-880; Santa Cruz Biotechnology Inc.) and anti-vinculin (1:10,000; V4505, clone VIN-11-5; Sigma-Aldrich) primary antibodies. Binding of HRP-labeled anti-rabbit or -mouse secondary antibodies (NA934 or NA931, respectively; GE Healthcare) was detected with the SuperSignal West Pico Chemiluminescent kit (Thermo Scientific). Immunoprecipitation was performed as previously described (80, 81). In brief, cells were harvested using PRO-PREP Protein Extraction Solution (iNtRON Biotechnology). The preparation of cell lysates, SDS-PAGE (8% or 10% gel), and Western blotting were performed as described elsewhere (80). In the case of sequential immunoprecipitation and Western blot analysis, 293T cells were transfected with pcDNA4-JDP2 or pcDNA3-TLX1 by Lipofectamine 2000 (Invitrogen), and after 48 hours, the cell lysates of each transformant were prepared for the sequential immunoprecipitation and Western blotting as described elsewhere (80). Each supernatant was precleared with protein A/G beads (Millipore), and incubated with antibodies specific for JDP2 (2) or TLX1 (1:550 stock 200 μl/ml; SC-880; Santa Cruz Biotechnology Inc.) at 4°C for 16 hours. The beads were preblocked with 1% BSA before being added to samples and then incubated at 4°C for 4 hours with rotation. The beads were pelleted, washed by chilled PBS, and loaded to Western blotting.
Proliferation assay.
For assaying proliferation of primary and immortal eSMCs, 5 × 103 cells per well were seeded in a 96-well plate. Primary spleen mesenchymal cells were obtained by trypsin digestion of pooled E14.5 spleens. Sixteen to eighteen hours after plating, cells were treated with 1 μM RA (in DMSO; Sigma-Aldrich) and 1 μM R116010 (in ethanol [EtOH]; ref. 52; provided by Miguel Torres, Centro Nacional de Investigaciones Cardiovasculares, Madrid, Spain). In controls and cultures treated for 24, 48, 72, or 96 hours, cells were fixed with 100 μl of Diff-Quick Fix solution (Medion Diagnostics) for 3 minutes at room temperature and then stained with 100 μl of Crystal Violet (Dade Behring) for 5 minutes at room temperature. Cells were washed with tap water and allowed to dry, and then 50 μl/well of Dissolving Solution (0.1% SDS, 50% EtOH, 0.25 M Tris-HCl pH 7.7, in 10 ml of ddH2O) was added. Optical absorbance was read at 570 nm with Microplate Reader Model 680 (Bio-Rad), and the data were analyzed with Microsoft Excel and GraphPad Prism 5.0c (GraphPad Software).
Coculture and organotypic experiments.
SMC shRNA-Scramble, shRNA-Tlx1, or shRNA-Tlx1/rescue-Cyp26b1 cells were seeded in a poly-l-Lysine–coated 12-well plate (Costar) with F9-RARE-LacZ cell lines in a 1:1 ratio (105 cells per well for each cell line). Coculture experiments were conducted in triplicate, and cells were maintained in complete DMEM with 5% CO2 at 37°C for 48 hours. Then LacZ staining or β-gal activity assays on the cocultured cells were performed. For organotypic experiments, embryonic spleens were microdissected from E13.5 CD1 embryos and embedded in 600 μl of 1.6% type I rat tail collagen solution (SERVA) in MEM and 16 mM sodium bicarbonate, using 12-well plates. Littermate spleens were divided into 2 groups, and after collagen polymerization, 1 ml of complete DMEM with 1 μM RA or vehicle (DMSO) was added to each well. All cultures were incubated at 5% CO2 at 37°C for 24 hours. Images were taken using Nikon Digital Camera DXM1200 mounted on a Nikon Eclipse TE200 microscope using the Nikon ACT-1 program. To further quantify the spleen areas, the images were analyzed with ImageJ software.
LacZ staining.
RARE-LacZ embryos were collected at E11.5 and E13.5 and fixed for 1 hour at 4°C in PBS containing 4% formaldehyde, 0.8% glutaraldehyde, 0.02% NP-40, and 1 mM MgCl2. After brief washing in PBS, embryos were stained overnight at 37°C in PBS 1× supplemented with 5-bromo-4-chloro-3-indolyl-d-galactopiranoside (X-Gal; 400 mg/ml) in 0.1 mM MgCl2, 20 mM potassium ferrocyanide, and 20 mM potassium ferricyanide. After the staining, embryos were incubated overnight at 4°C in PBS 1× containing 30% sucrose, and the day after, they were equilibrated for 20–30 minutes at room temperature in OCT-Killik (Bio-Optica) and then embedded in OCT-Killik. For the LacZ staining of the cocultured experiments, cells were fixed with LacZ fixing solution (0.5% glutaraldehyde, 0.1 M MgCl2, 5 mM EGTA pH 7.5), washed with LacZ washing buffer (0.2% Nonidet-P40, 2 mM MgCl2, 0.01% NaDOC in PBS 1× without MgCl2 and CaCl2), and then incubated overnight at 37°C in the dark with LacZ Staining solution (1 mg/ml X-Gal, 5 mM potassium ferrocyanide, and 5 mM potassium ferricyanide in LacZ washing buffer solution). Images were captured as previously described, and to further quantify the number of LacZ+ cells, the count of resulting blue cells was done with ImageJ software. To perform the β-gal activity assay the cocultured cells were lysed with Lysis Buffers (Promega Corp.) for 20 minutes at room temperature in agitation. After, 20 μl/well of cell lysates were transferred into a 96-well plate (Costar), and 140 μl/well of ONPG-MIX was added. The ONPG-MIX contained 1.4 μl/well of Mg2+ 100× (0.1 M MgCl2, 4.5 M b-MeEtOH), 104 μl/well of 0.1 M sodium phosphate solution pH 7.5 (0.06 M Na2HPO4 eptahydrate, 0.04 M NaH2PO4), and 34.6 μl/well of ONPG solution (3.6 mg/ml in 0.1 M sodium phosphate solution pH 7.5). The absorbance was measured with the Microplate Reader Model 680 (Bio-Rad) at 415 nm, and the data were analyzed using Microsoft Excel.
Statistics.
Statistical analysis using a 2-tailed unpaired Student’s t test or 2-way ANOVA or χ2 test, as indicated, was performed with GraphPad Prism and values expressed as mean ± SD. Differences were considered statistically significant at P less than 0.05.
Study approval.
All animal experiments were conducted in accordance with protocols approved by the IACUC of San Raffaele Scientific Institute.
Author contributions
EL, DF, LC, F Bernassola, and AB designed and performed experiments. KW and KKY performed immunoprecipitation experiments. LLS, NEBT, and PAT performed Rdh10 experiments. D Penkov, GL, D Pasini, and GT analyzed microarray and ChIP-seq data. NC and AD performed cloning experiments. KN, MW, F Blasi, KK, GM, and PAT provided reagents. EL and AB analyzed data, prepared figures, and wrote the manuscript with the contribution of PAT, and AB directed the study.
Supplementary Material
Acknowledgments
The authors are grateful to laboratory members for technical assistance and helpful suggestions. We are grateful to J. Rossant for providing the RARE-LacZ transgenic mice, Hiroshi Hamada for providing the Cyp26b1 mice, and Susan Mackem for providing the Hoxb6-CreERT mice. We are thankful to Daniela Talarico for comments on the manuscript. This work was supported by Associazione Italiana Ricerca sul Cancro Start-Up Grant 4780, grant IG 14511, and Special Program Molecular Clinical Oncology 5 per mille 9965, to A. Brendolan; Marie Curie Foundation IRG-2007 208932, to A. Brendolan; the Stowers Institute for Medical Research, to P.A. Trainor; the Taiwan government MOST-104-2320-B-037-033-My2, MOST-104-2314-B-037-002, NHRI-EX104-10416SI, and KMU-TP104A04, E24, G03, G04, to K.K. Yokoyama; Slovenian Research Agency project J7-7226, to G. Majdic; and the Russian Science Foundation (14-35-00026), to D. Penkov for ChIP-seq.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(7):2452–2464. doi:10.1172/JCI82956.
Contributor Information
Elisa Lenti, Email: lenti.elisa@hsr.it.
Diego Farinello, Email: farinello.diego@hsr.it.
Kazunari K. Yokoyama, Email: kazu@kmu.edu.tw.
Dmitry Penkov, Email: dmitry.penkov@ifom.eu.
Laura Castagnaro, Email: castagnaro.laura@hsr.it.
Giovanni Lavorgna, Email: giovanni.lavorgna@hsr.it.
Kenly Wuputra, Email: kenlywu@hotmail.com.
Lisa L. Sandell, Email: Lisa.Sandell@Louisville.edu.
Naomi E. Butler Tjaden, Email: naomitjaden@gmail.com.
Francesca Bernassola, Email: bernasso@uniroma2.it.
Nicoletta Caridi, Email: caridi.nicoletta@hsr.it.
Anna De Antoni, Email: anna.deantoni@ifom.eu.
Michael Wagner, Email: michael.wagner@downstate.edu.
Katja Kozinc, Email: Katja.Kozinc@vf.uni-lj.si.
Karen Niederreither, Email: niederre@igbmc.fr.
Francesco Blasi, Email: francesco.blasi@ifom.eu.
Diego Pasini, Email: diego.pasini@ieo.eu.
Gregor Majdic, Email: Gregor.Majdic@vf.uni-lj.si.
Giovanni Tonon, Email: tonon.giovanni@hsr.it.
Andrea Brendolan, Email: brendolan.andrea@hsr.it.
References
- 1.Ejstrud P, Kristensen B, Hansen JB, Madsen KM, Schonheyder HC, Sorensen HT. Risk and patterns of bacteraemia after splenectomy: a population-based study. Scand J Infect Dis. 2000;32(5):521–525. doi: 10.1080/003655400458811. [DOI] [PubMed] [Google Scholar]
- 2.Bisharat N, Omari H, Lavi I, Raz R. Risk of infection and death among post-splenectomy patients. J Infect. 2001;43(3):182–186. doi: 10.1053/jinf.2001.0904. [DOI] [PubMed] [Google Scholar]
- 3.Thomsen RW, et al. Risk for hospital contact with infection in patients with splenectomy: a population-based cohort study. Ann Intern Med. 2009;151(8):546–555. doi: 10.7326/0003-4819-151-8-200910200-00008. [DOI] [PubMed] [Google Scholar]
- 4.Ram S, Lewis LA, Rice PA. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin Microbiol Rev. 2010;23(4):740–780. doi: 10.1128/CMR.00048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kristinsson SY, Gridley G, Hoover RN, Check D, Landgren O. Long-term risks after splenectomy among 8,149 cancer-free American veterans: a cohort study with up to 27 years follow-up. Haematologica. 2014;99(2):392–398. doi: 10.3324/haematol.2013.092460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dear TN, et al. The Hox11 gene is essential for cell survival during spleen development. Development. 1995;121(9):2909–2915. doi: 10.1242/dev.121.9.2909. [DOI] [PubMed] [Google Scholar]
- 7.Burn SF, et al. The dynamics of spleen morphogenesis. Dev Biol. 2008;318(2):303–311. doi: 10.1016/j.ydbio.2008.03.031. [DOI] [PubMed] [Google Scholar]
- 8.Brendolan A, et al. A Pbx1-dependent genetic and transcriptional network regulates spleen ontogeny. Development. 2005;132(13):3113–3126. doi: 10.1242/dev.01884. [DOI] [PubMed] [Google Scholar]
- 9.Koss M, et al. Congenital asplenia in mice and humans with mutations in a Pbx/Nkx2-5/p15 module. Dev Cell. 2012;22(5):913–926. doi: 10.1016/j.devcel.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brendolan A, Rosado MM, Carsetti R, Selleri L, Dear TN. Development and function of the mammalian spleen. Bioessays. 2007;29(2):166–177. doi: 10.1002/bies.20528. [DOI] [PubMed] [Google Scholar]
- 11.Roberts CW, Shutter JR, Korsmeyer SJ. Hox11 controls the genesis of the spleen. Nature. 1994;368(6473):747–749. doi: 10.1038/368747a0. [DOI] [PubMed] [Google Scholar]
- 12.Mahlaoui N, et al. Isolated congenital asplenia: a French nationwide retrospective survey of 20 cases. J Pediatr. 2011;158(1):142–148. doi: 10.1016/j.jpeds.2010.07.027. [DOI] [PubMed] [Google Scholar]
- 13.Bolze A, et al. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science. 2013;340(6135):976–978. doi: 10.1126/science.1234864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Unolt M, et al. Transposition of great arteries: new insights into the pathogenesis. Front Pediatr. 2013;1: doi: 10.3389/fped.2013.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kashimada K, et al. Antagonistic regulation of Cyp26b1 by transcription factors SOX9/SF1 and FOXL2 during gonadal development in mice. FASEB J. 2011;25(10):3561–3569. doi: 10.1096/fj.11-184333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zangen D, et al. Testicular differentiation factor SF-1 is required for human spleen development. J Clin Invest. 2014;124(5):2071–2075. doi: 10.1172/JCI73186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawabe T, Muslin AJ, Korsmeyer SJ. HOX11 interacts with protein phosphatases PP2A and PP1 and disrupts a G2/M cell-cycle checkpoint. Nature. 1997;385(6615):454–458. doi: 10.1038/385454a0. [DOI] [PubMed] [Google Scholar]
- 18.Riz I, Hawley RG. G1/S transcriptional networks modulated by the HOX11/TLX1 oncogene of T-cell acute lymphoblastic leukemia. Oncogene. 2005;24(36):5561–5575. doi: 10.1038/sj.onc.1208727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Owens BM, Hawley TS, Spain LM, Kerkel KA, Hawley RG. TLX1/HOX11-mediated disruption of primary thymocyte differentiation prior to the CD4+CD8+ double-positive stage. Br J Haematol. 2006;132(2):216–229. doi: 10.1111/j.1365-2141.2005.05850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riz I, et al. TLX1/HOX11-induced hematopoietic differentiation blockade. Oncogene. 2007;26(28):4115–4123. doi: 10.1038/sj.onc.1210185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Keersmaecker K, et al. The TLX1 oncogene drives aneuploidy in T cell transformation. Nat Med. 2010;16(11):1321–1327. doi: 10.1038/nm.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dadi S, et al. TLX homeodomain oncogenes mediate T cell maturation arrest in T-ALL via interaction with ETS1 and suppression of TCRalpha gene expression. Cancer Cell. 2012;21(4):563–576. doi: 10.1016/j.ccr.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Kanzler B, Dear TN. Hox11 acts cell autonomously in spleen development and its absence results in altered cell fate of mesenchymal spleen precursors. Dev Biol. 2001;234(1):231–243. doi: 10.1006/dbio.2001.0239. [DOI] [PubMed] [Google Scholar]
- 24.Riz I, Hawley TS, Johnston H, Hawley RG. Role of TLX1 in T-cell acute lymphoblastic leukaemia pathogenesis. Br J Haematol. 2009;145(1):140–143. doi: 10.1111/j.1365-2141.2008.07556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rice KL, Izon DJ, Ford J, Boodhoo A, Kees UR, Greene WK. Overexpression of stem cell associated ALDH1A1, a target of the leukemogenic transcription factor TLX1/HOX11, inhibits lymphopoiesis and promotes myelopoiesis in murine hematopoietic progenitors. Leuk Res. 2008;32(6):873–883. doi: 10.1016/j.leukres.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Niederreither K, Dolle P. Retinoic acid in development: towards an integrated view. Genetics. 2008;9(7):541–553. doi: 10.1038/nrg2340. [DOI] [PubMed] [Google Scholar]
- 27.Greene WK, Bahn S, Masson N, Rabbitts TH. The T-cell oncogenic protein HOX11 activates Aldh1 expression in NIH 3T3 cells but represses its expression in mouse spleen development. Mol Cell Biol. 1998;18(12):7030–7037. doi: 10.1128/MCB.18.12.7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell. 2008;134(6):921–931. doi: 10.1016/j.cell.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niederreither K, Vermot J, Messaddeq N, Schuhbaur B, Chambon P, Dolle P. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development. 2001;128(7):1019–1031. doi: 10.1242/dev.128.7.1019. [DOI] [PubMed] [Google Scholar]
- 30.Vermot J, Gallego Llamas J, Fraulob V, Niederreither K, Chambon P, Dolle P. Retinoic acid controls the bilateral symmetry of somite formation in the mouse embryo. Science. 2005;308(5721):563–566. doi: 10.1126/science.1108363. [DOI] [PubMed] [Google Scholar]
- 31.Rhinn M, Dolle P. Retinoic acid signalling during development. Development. 2012;139(5):843–858. doi: 10.1242/dev.065938. [DOI] [PubMed] [Google Scholar]
- 32.Cunningham TJ, Duester G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Mol Cell Biol. 2015;16(2):110–123. doi: 10.1038/nrm3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yashiro K, et al. Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing mouse limb. Dev Cell. 2004;6(3):411–422. doi: 10.1016/S1534-5807(04)00062-0. [DOI] [PubMed] [Google Scholar]
- 34.Sakai Y, et al. The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 2001;15(2):213–225. doi: 10.1101/gad.851501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uehara M, Yashiro K, Takaoka K, Yamamoto M, Hamada H. Removal of maternal retinoic acid by embryonic CYP26 is required for correct Nodal expression during early embryonic patterning. Genes Dev. 2009;23(14):1689–1698. doi: 10.1101/gad.1776209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowles J, et al. Control of retinoid levels by CYP26B1 is important for lymphatic vascular development in the mouse embryo. Dev Biol. 2014;386(1):25–33. doi: 10.1016/j.ydbio.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 37.Bowles J, et al. Retinoid signaling determines germ cell fate in mice. Science. 2006;312(5773):596–600. doi: 10.1126/science.1125691. [DOI] [PubMed] [Google Scholar]
- 38.Makori N, Peterson PE, Lantz K, Hendrickx AG. Exposure of cynomolgus monkey embryos to retinoic acid causes thymic defects: effects on peripheral lymphoid organ development. J Med Primatol. 2002;31(2):91–97. doi: 10.1034/j.1600-0684.2002.01013.x. [DOI] [PubMed] [Google Scholar]
- 39.Laue K, et al. Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am J Hum Genet. 2011;89(5):595–606. doi: 10.1016/j.ajhg.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee LM, et al. A paradoxical teratogenic mechanism for retinoic acid. Proc Natl Acad Sci U S A. 2012;109(34):13668–13673. doi: 10.1073/pnas.1200872109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garnaas MK, et al. Rargb regulates organ laterality in a zebrafish model of right atrial isomerism. Dev Biol. 2012;372(2):178–189. doi: 10.1016/j.ydbio.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oh SP, Li E. The signaling pathway mediated by the type IIB activin receptor controls axial patterning and lateral asymmetry in the mouse. Genes Dev. 1997;11(14):1812–1826. doi: 10.1101/gad.11.14.1812. [DOI] [PubMed] [Google Scholar]
- 43.Wasiak S, Lohnes D. Retinoic acid affects left-right patterning. Dev Biol. 1999;215(2):332–342. doi: 10.1006/dbio.1999.9465. [DOI] [PubMed] [Google Scholar]
- 44.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Cancer. 2003;3(11):859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 46.Rossant J, Zirngibl R, Cado D, Shago M, Giguere V. Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 1991;5(8):1333–1344. doi: 10.1101/gad.5.8.1333. [DOI] [PubMed] [Google Scholar]
- 47.Abu-Abed S, MacLean G, Fraulob V, Chambon P, Petkovich M, Dolle P. Differential expression of the retinoic acid-metabolizing enzymes CYP26A1 and CYP26B1 during murine organogenesis. Mech Dev. 2002;110(1–2):173–177. doi: 10.1016/s0925-4773(01)00572-x. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen MT, Zhu J, Nakamura E, Bao X, Mackem S. Tamoxifen-dependent, inducible Hoxb6CreERT recombinase function in lateral plate and limb mesoderm, CNS isthmic organizer, posterior trunk neural crest, hindgut, and tailbud. Dev Dyn. 2009;238(2):467–474. doi: 10.1002/dvdy.21846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sandell LL, et al. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007;21(9):1113–1124. doi: 10.1101/gad.1533407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sandell LL, Lynn ML, Inman KE, McDowell W, Trainor PA. RDH10 oxidation of Vitamin A is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PLoS One. 2012;7(2): doi: 10.1371/journal.pone.0030698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wagner M, Han B, Jessell TM. Regional differences in retinoid release from embryonic neural tissue detected by an in vitro reporter assay. Development. 1992;116(1):55–66. doi: 10.1242/dev.116.1.55. [DOI] [PubMed] [Google Scholar]
- 52.Van Heusden J, et al. Inhibition of all-TRANS-retinoic acid metabolism by R116010 induces antitumour activity. Br J Cancer. 2002;86(4):605–611. doi: 10.1038/sj.bjc.6600056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin C, et al. JDP2, a repressor of AP-1, recruits a histone deacetylase 3 complex to inhibit the retinoic acid-induced differentiation of F9 cells. Mol Cell Biol. 2002;22(13):4815–4826. doi: 10.1128/MCB.22.13.4815-4826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Su D, Gudas LJ. Gene expression profiling elucidates a specific role for RARgamma in the retinoic acid-induced differentiation of F9 teratocarcinoma stem cells. Biochem Pharmacol. 2008;75(5):1129–1160. doi: 10.1016/j.bcp.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jin C, Li H, Ugai H, Murata T, Yokoyama KK. Transcriptional regulation of the c-jun gene by AP-1 repressor protein JDP2 during the differentiation of F9 cells. Nucleic Acids Res Suppl. 2002;(2):97–98. doi: 10.1093/nass/2.1.97. [DOI] [PubMed] [Google Scholar]
- 56.Gutierrez-Mazariegos J, Theodosiou M, Campo-Paysaa F, Schubert M. Vitamin A: a multifunctional tool for development. Semin Cell Dev Biol. 2011;22(6):603–610. doi: 10.1016/j.semcdb.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 57.Bajenoff M, et al. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25(6):989–1001. doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Immunology. 2009;9(9):618–629. doi: 10.1038/nri2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Castagnaro L, et al. Nkx2-5(+)islet1(+) mesenchymal precursors generate distinct spleen stromal cell subsets and participate in restoring stromal network integrity. Immunity. 2013;38(4):782–791. doi: 10.1016/j.immuni.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Katakai T, Hara T, Sugai M, Gonda H, Shimizu A. Lymph node fibroblastic reticular cells construct the stromal reticulum via contact with lymphocytes. J Exp Med. 2004;200(6):783–795. doi: 10.1084/jem.20040254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lai L, Bohnsack BL, Niederreither K, Hirschi KK. Retinoic acid regulates endothelial cell proliferation during vasculogenesis. Development. 2003;130(26):6465–6474. doi: 10.1242/dev.00887. [DOI] [PubMed] [Google Scholar]
- 62.Chen F, Cao Y, Qian J, Shao F, Niederreither K, Cardoso WV. A retinoic acid-dependent network in the foregut controls formation of the mouse lung primordium. J Clin Invest. 2010;120(6):2040–2048. doi: 10.1172/JCI40253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van de Pavert SA, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature. 2014;508(7494):123–127. doi: 10.1038/nature13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morohashi K, et al. Structural and functional abnormalities in the spleen of an mFtz-F1 gene-disrupted mouse. Blood. 1999;93(5):1586–1594. [PubMed] [Google Scholar]
- 65.Roberts CW, Sonder AM, Lumsden A, Korsmeyer SJ. Development expression of Hox11 and specification of splenic cell fate. Am J Pathol. 1995;146(5):1089–1101. [PMC free article] [PubMed] [Google Scholar]
- 66.Chazaud C, Chambon P, Dolle P. Retinoic acid is required in the mouse embryo for left-right asymmetry determination and heart morphogenesis. Development. 1999;126(12):2589–2596. doi: 10.1242/dev.126.12.2589. [DOI] [PubMed] [Google Scholar]
- 67.Wang L, Mear JP, Kuan CY, Colbert MC. Retinoic acid induces CDK inhibitors and growth arrest specific (Gas) genes in neural crest cells. Dev Growth Differ. 2005;47(3):119–130. doi: 10.1111/j.1440-169X.2005.00788.x. [DOI] [PubMed] [Google Scholar]
- 68.Jin C, et al. Regulation of histone acetylation and nucleosome assembly by transcription factor JDP2. Nat Struct Mol Biol. 2006;13(4):331–338. doi: 10.1038/nsmb1063. [DOI] [PubMed] [Google Scholar]
- 69.Budefeld T, Grgurevic N, Tobet SA, Majdic G. Sex differences in brain developing in the presence or absence of gonads. Dev Neurobiol. 2008;68(7):981–995. doi: 10.1002/dneu.20638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Majdic G, et al. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology. 2002;143(2):607–614. doi: 10.1210/endo.143.2.8652. [DOI] [PubMed] [Google Scholar]
- 71.Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77(4):481–490. doi: 10.1016/0092-8674(94)90211-9. [DOI] [PubMed] [Google Scholar]
- 72.Ruzankina Y, et al. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1(1):113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 74.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2. Nat Genet. 2006;38(5):500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 75.Balconi G, Spagnuolo R, Dejana E. Development of endothelial cell lines from embryonic stem cells: A tool for studying genetically manipulated endothelial cells in vitro. Arterioscler Thromb Vasc Biol. 2000;20(6):1443–1451. doi: 10.1161/01.ATV.20.6.1443. [DOI] [PubMed] [Google Scholar]
- 76.Takeuchi H, Yokota A, Ohoka Y, Iwata M. Cyp26b1 regulates retinoic acid-dependent signals in T cells and its expression is inhibited by transforming growth factor-β. PLoS One. 2011;6(1): doi: 10.1371/journal.pone.0016089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bakiri L, Matsuo K, Wisniewska M, Wagner EF, Yaniv M. Promoter specificity and biological activity of tethered AP-1 dimers. Mol Cell Biol. 2002;22(13):4952–4964. doi: 10.1128/MCB.22.13.4952-4964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schepers G, Wilson M, Wilhelm D, Koopman P. SOX8 is expressed during testis differentiation in mice and synergizes with SF1 to activate the Amh promoter in vitro. J Biol Chem. 2003;278(30):28101–28108. doi: 10.1074/jbc.M304067200. [DOI] [PubMed] [Google Scholar]
- 79.Nagy A, Gertsenstein M, Vintersten K, Behringer R. In situ hybridization of mouse embryo and tissue sections with radiolabeled RNA probes. CSH Protoc. 2007;2007: doi: 10.1101/pdb.prot4821. [DOI] [PubMed] [Google Scholar]
- 80.Pan J, et al. Suppression of cell-cycle progression by Jun dimerization protein-2 (JDP2) involves downregulation of cyclin-A2. Oncogene. 2010;29(47):6245–6256. doi: 10.1038/onc.2010.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aronheim A, Zandi E, Hennemann H, Elledge SJ, Karin M. Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol Cell Biol. 1997;17(6):3094–3102. doi: 10.1128/MCB.17.6.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.