

Abstract
High levels of arginine metabolizing enzymes, including inducible nitric oxide synthase (iNOS) and arginase (ARG), are typical in asthmatic airway epithelium; however, little is known about the metabolic effects of enhanced arginine flux in asthma. Here, we demonstrated that increased metabolism sustains arginine availability in asthmatic airway epithelium with consequences for bioenergetics and inflammation. Expression of iNOS, ARG2, arginine synthetic enzymes, and mitochondrial respiratory complexes III and IV was elevated in asthmatic lung samples compared with healthy controls. ARG2 overexpression in a human bronchial epithelial cell line accelerated oxidative bioenergetic pathways and suppressed hypoxia-inducible factors (HIFs) and phosphorylation of the signal transducer for atopic Th2 inflammation STAT6 (pSTAT6), both of which are implicated in asthma etiology. Arg2-deficient mice had lower mitochondrial membrane potential and greater HIF-2α than WT animals. In an allergen-induced asthma model, mice lacking Arg2 had greater Th2 inflammation than WT mice, as indicated by higher levels of pSTAT6, IL-13, IL-17, eotaxin, and eosinophils and more mucus metaplasia. Bone marrow transplants from Arg2-deficient mice did not affect airway inflammation in recipient mice, supporting resident lung cells as the drivers of elevated Th2 inflammation. These data demonstrate that arginine flux preserves cellular respiration and suppresses pathological signaling events that promote inflammation in asthma.
Introduction
One of the most common chronic diseases in the United States, asthma is a syndrome of airway inflammation and airflow obstruction. The airway inflammation of asthma is characterized by high fractional exhaled nitric oxide (FENO) generated by inducible NO synthase (iNOS; Enzyme Commission [EC] no. 1.14.13.39) expressed in the airway epithelium, where it catalyzes the conversion of arginine to NO and citrulline (refs. 1–3 and Figure 1A). Although iNOS expression and FENO are increased in asthma and promote inflammatory injury via nitration (2, 4), airway hyper-responsiveness is not diminished in mice with genetic deletion of iNOS (5), and arginine-analog inhibitors of iNOS pathways have poor efficacy in asthma (6). This caused us to consider that metabolism of arginine may mechanistically contribute to asthma through pathways other than generation of NO. In fact, despite higher levels of iNOS and FENO, intracellular arginine levels in the epithelium of asthmatic individuals are 3-fold higher than in healthy controls (7). Although arginine may be taken into cells by cationic amino acid transporter (CAT) proteins, cells producing NO over prolonged periods synthesize arginine at levels sufficient to sustain NO production (8–10). Cells synthesize arginine endogenously (11) via argininosuccinate synthetase (ASS; EC 6.3.4.5), which uses citrulline and aspartate as substrates to form argininosuccinate that is subsequently cleaved by argininosuccinate lyase (ASL; EC 4.3.2.1) to generate arginine, such that the N for NO is actually derived from aspartate, which in turn receives the N from glutamate via transamination of oxaloacetate (OAA), a TCA cycle intermediate (Figure 1A). Arginine regeneration by ASS is rate-limiting for iNOS activity in most systems (9, 12, 13). All this suggests that the airway epithelium in asthma manifests an arginine metabolic endotype.
Figure 1. Arginine metabolism and bioenergetics in asthma.

(A) The citrulline-NO and tricarboxylic acid (TCA) cycles. iNOS converts arginine to citrulline and NO. ARG2 converts arginine to ornithine and urea. Ornithine aminotransferase (OAT) converts ornithine to glutamate, which gives rise to αKG to enter the TCA cycle producing reducing equivalents for electron transport chain (ETC) to generate membrane potential for ATP. Aspartate transaminase (AST) uses glutamate to transaminate TCA cycle intermediate oxaloacetate and produces aspartate. Argininosuccinate synthetase (ASS) uses citrulline and aspartate as substrates to form argininosuccinate, which is cleaved by argininosuccinate lyase (ASL) to form arginine and fumarate, linking arginase and iNOS pathways via the citrulline-NO and TCA cycles. (B) Expression in bronchial epithelial cell lysates by Western blot. Cytokeratin confirms epithelial cells obtained by airway brushing. Replicate samples run on parallel gels are presented. (C) Relative units (means ± SEM) in human bronchial epithelial cells (control n ≥ 4, asthma n ≥ 10); 2-tailed t test except 1-tailed t test for complex III-1 and complex IV-4. (D–G) IHC of ARG2 and ASS in endobronchial biopsies of controls and asthmatics. ARG2 is more prominent in asthmatic bronchial epithelium (F) than control (D). Images representative of multiple sections from 15 asthmatics and 7 controls. Bronchial epithelial cells of asthma (G) show stronger cytoplasmic positivity for ASS than control (E). Images representative of multiple sections from 5 asthmatics and 5 controls. Scale bars: 40 μm. (H–K) Immunogold electron microscopy analyses of ARG2 and ASS in epithelium from endobronchial biopsies. I, higher magnification of H, and K of J. White arrowhead shows gold particles. Images representative of 5 individuals. M, mitochondrion. Scale bars: 250 nm.
In addition to higher iNOS and arginine levels, arginase activity (ARG; EC 3.5.3.1), which catabolizes arginine to ornithine and urea, is also increased in asthma (Figure 1A and refs. 6, 14–19). ARG1 is present exclusively in the cytosol of hepatic cells as part of the urea cycle, but ARG2 is found in mitochondria of many tissues without a functioning complete urea cycle, including the lung (18). Intriguingly, ARG2 gene variants, which lie within an asthma linkage region on chromosome 14q24, are some of the earliest and most consistent SNPs discovered in GWAS of asthma, and are strongly linked to origins and severity of asthma (20–22). ARG2 is composed of 354 amino acid residues, including an NH2-terminal presequence for mitochondrial targeting and import (23). The function of mitochondrial ARG2 is unknown and unexplored in healthy or asthmatic airways. In the mitochondria, ornithine generated by ARG2 will give rise to glutamate via ornithine aminotransferase (OAT). Glutamate participates in several transamination reactions, including forming α-ketoglutarate (αKG) that may enter the TCA cycle and increase cycle intermediates and flux (Figure 1A). Transamination of glutamate with OAA forms aspartate, the source of N for NO production via ASS and ASL reactions (Figure 1A). Thus, arginine metabolism via ARG2 in the mitochondria may support the bioenergetic state of the cell, and simultaneously provide endogenous arginine for high NO synthesis in asthma.
Mitochondrial provision of intermediates by arginine/ornithine for oxidative metabolism may have important consequences for signal transducers of airway inflammation. Adaptive transcriptional responses to TCA cycle activity and oxygen levels are mediated by hypoxia-inducible factors (HIFs). Prolyl hydroxylases utilize oxygen and αKG in catalytic hydroxylation of HIFs, targeting them for degradation (ref. 24 and Figure 1A). HIF-1α is expressed ubiquitously, but HIF-2α expression is restricted to certain tissues, including the lung (25). Emerging evidence identifies HIFs in the mechanistic pathway of allergic airway inflammation, in part by regulation of IL-13 production (26–28). IL-13 is sufficient to induce typical features of asthma, including induction of mucin production and secretion, and recruitment of eosinophils to the lungs (29–31). The majority of these downstream allergic inflammatory effects of IL-13 are mediated through signal transducer and activator of transcription 6 (STAT6). Taking all this into consideration in asthma, we hypothesized that greater arginine metabolism through ARG2 increases TCA cycle flux and oxidative metabolism and dampens signal transduction via HIF and STAT6, thereby providing beneficial effects to suppress airway inflammation and asthma severity.
Results
Clinical population.
The clinical characteristics of the study subjects are displayed in Supplemental Table 1 (supplemental material available online with this article; doi:10.1172/JCI82925DS1). Not all subjects participated in all experimental studies; the numbers of subjects assessed for each experiment are listed with each result.
Increased whole-body metabolism of arginine in asthmatic individuals.
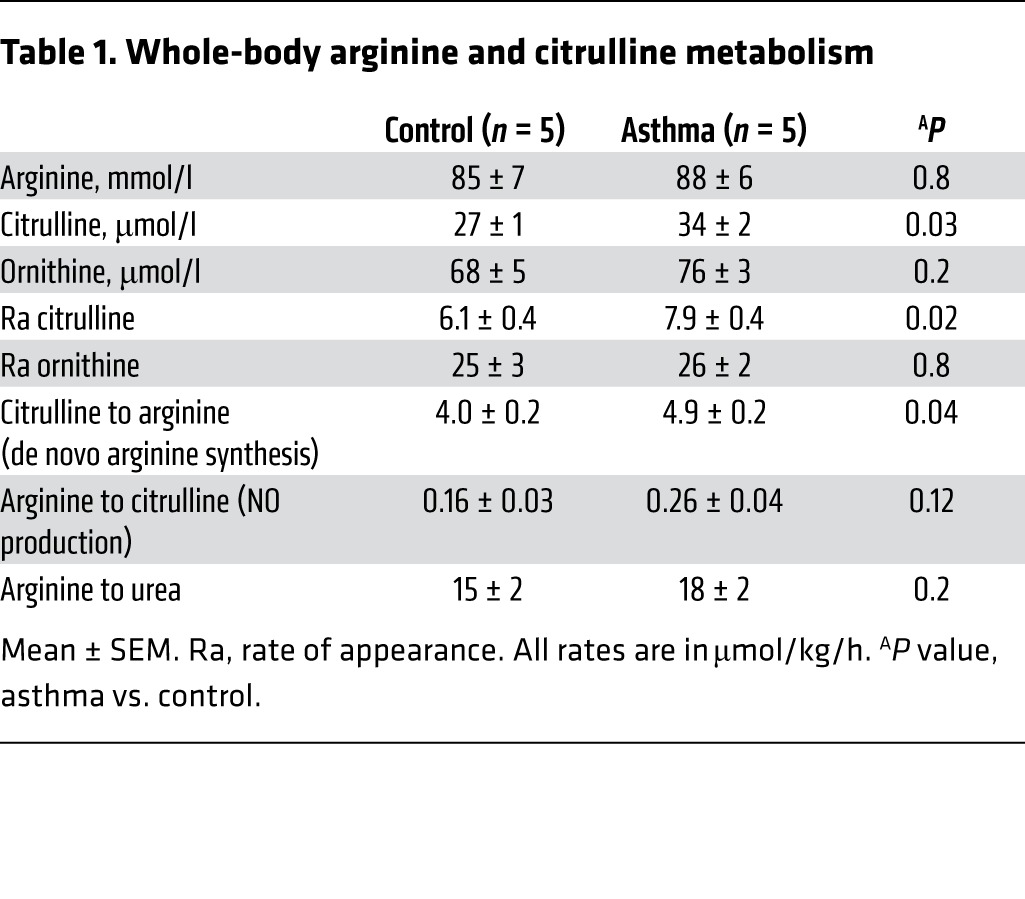
We (1, 2, 4, 19) and others (6, 14–18) have shown that arginine metabolic pathways, including iNOS and ARG, are increased in the airway epithelium in asthma. To address whether de novo arginine synthesis was increased, individuals with asthma and healthy controls were studied using stable isotope–labeled arginine and citrulline tracers to determine the whole-body rate of metabolism of arginine and its de novo synthesis from citrulline (32, 33). Arginine and other urea cycle amino acids are highly compartmentalized in various tissues, and their intracellular pools do not equilibrate rapidly with extracellular pools. Thus, studies of whole-body kinetics as measured by tracers in the plasma are likely to be underestimates of total flux, but still useful to assess differences in relative flux between groups. As shown in Table 1, the plasma concentration of citrulline, its rate of appearance, and the contribution of citrulline-to-arginine (de novo arginine synthesis) were significantly higher in asthma (n = 5) as compared with controls (n = 5) (Table 1).
Table 1. Whole-body arginine and citrulline metabolism.
Higher ARG2 and arginine biosynthesis in asthmatic airway.
To identify specific arginine metabolic pathways in the human asthmatic airway, expression and localization of ARG1, ARG2, and iNOS were evaluated in freshly obtained airway epithelium, bronchoalveolar lavage (BAL) cells (Supplemental Table 2), and lung biopsies of asthmatics and healthy controls. ARG2 was expressed at higher levels in the human airway epithelium obtained at bronchoscopy of asthmatics (n = 10) compared with healthy controls (n = 6, P = 0.01) (Figure 1, B and C). IHC analyses confirmed that ARG2 in asthmatic bronchial epithelial cells was consistently greater than in controls (control n = 7, asthma n = 15, Figure 1, D and F), while ARG1 was undetectable by Western analyses or by IHC (Figure 1B). Immunogold electron microscopy analyses demonstrated that ARG2 was located in the mitochondria of airway epithelium (Figure 1, H and I). ARG2 was undetectable in mononuclear cells from freshly obtained BAL from asthmatics or healthy controls by Western blot analysis (Supplemental Figure 1A). ARG2 was highly expressed in the bronchial epithelium but not in airway smooth muscles in asthma airways (n = 4, Supplemental Figure 1, B and C) or control airways (n = 2) (data not shown). This identified that bronchial epithelium was the primary cellular site of ARG activity in the airways (34). As previously reported (7), iNOS was abundantly expressed in the bronchial epithelial cells in asthma (Figure 1B). The epithelial localization of iNOS and ARG2 directed our studies to focus on arginine metabolism and its consequences in the airway epithelium. As in prior studies (7), iNOS expression was significantly correlated with FENO levels (R2 = 0.55, P = 0.001). Interestingly, ARG2 expression was positively correlated with iNOS expression (R2 = 0.26, P = 0.05), and tended to be positively correlated with FENO (R2 = 0.18, P = 0.1). These findings suggest that arginine metabolic pathways (iNOS and ARG2) are coordinately induced in asthma and not in competition for arginine substrate.
De novo arginine synthesis is the major intracellular source for arginine (11). In support of greater arginine synthesis in asthma, ASS and ASL proteins were present at higher levels in freshly obtained asthmatic airway epithelial cells (n = 10) as compared with control (n = 4, ASS, P = 0.03; ASL, P = 0.04; Figure 1, B and C). Immunostaining identified that ASS expression was also consistently more abundant in asthmatic bronchial epithelial cells (n = 5, Figure 1G) as compared with control (n = 5) (Figure 1E). In contrast to ARG2, immunogold labeling showed ASS was expressed in the cytosol (Figure 1, J and K). Previous studies have shown that CAT2 is increased in a murine experimental asthma model and that CAT2-deficient mice have spontaneous eosinophilic lung inflammation (18, 35). However, expression of CAT2 and CAT3 was similar in bronchial epithelial cells from asthma and control, while CAT1 was undetectable in all samples (n ≥ 3) (data not shown). Altogether, the findings suggest that there is a higher capacity for intracellular de novo arginine synthesis in the asthmatic airway epithelium to support the greater arginine metabolism via iNOS and ARG2, which is consistent with the tracer-measured increase in whole-body de novo arginine synthesis in asthma.
Increased mitochondrial electron transport chain complex proteins and ATP in asthma.
ARG2 is located in the mitochondria, and its higher expression and production of ornithine in asthma may impact cellular bioenergetics. Complex I, II, III, and IV proteins were examined in the freshly obtained human airway epithelial cells from asthma and controls (Figure 1, B and C). The expressions of complex III-1, complex III-2, and complex IV-4 proteins were all significantly higher in airway epithelial cells from asthma (n = 10) as compared with control (n = 5) (Figure 1, B and C). Complex I and II expressions were similar between asthma and control (data not shown). Indicators of energy production by cellular respiration, such as ATP, pyruvate, and lactate, were measured in lysates of freshly obtained bronchial epithelial cells. Asthma (n = 16) and controls (n = 10) had similar intracellular levels of pyruvate and lactate (pyruvate pmol/μg protein, control 3.5 ± 1.2, asthma 2.4 ± 0.6, P = 0.4; lactate pmol/μg protein, control 6 ± 2, asthma 5 ± 2, P = 0.8). Intracellular concentration of ATP was higher in asthma (ATP pmol/μg protein, control 0.17 ± 0.05, asthma 0.33 ± 0.08, P = 0.06), resulting in higher ATP/pyruvate ratio (control 0.05 ± 0.02, asthma 0.35 ± 0.13, P = 0.04) in asthma as compared with control.
Mitochondrial ultrastructure was evaluated by electron microscopic images (Figure 2, A−F) of human airway endobronchial biopsies. Images revealed mitochondria, some with tightly coiled inner membrane (Figure 2F) and some with greater electron density (Figure 2F), in airway cells of asthma as compared with those of control (Figure 2C). Visual counts of mitochondria numbers in electron microscopy images suggested more mitochondria in asthmatic bronchial epithelial cells (mitochondrial number per cell, control 61 ± 5, n = 6 subjects, asthma 91 ± 11, n = 4 subjects, P = 0.03). Stereologic quantitation of mitochondria in electron microscopy images performed in anonymized fashion also suggested a greater mitochondria volume in asthmatic bronchial epithelial cells (volume of mitochondria/volume of cell, control 0.10 ± 0.01, n = 5, asthma 0.13 ± 0.01, n = 4 subjects, P = 0.05, 1-tailed t test), i.e., asthmatic epithelium may have larger and/or more mitochondria. These findings of alterations in mitochondrial ultrastructure and/or numbers are consistent with prior observations in airways of an allergen–murine asthma model (36, 37).
Figure 2. Ultrastructure of mitochondria in asthma airway epithelium.

Ultrastructural analyses of bronchial epithelium from control (A–C) and asthma (D–F). C, close-up view of A; B and F, close-up of D and E. Cilia are visible on cells (A and D). Mitochondria are concentrated in apical regions. High power reveals hyperdense mitochondria with tightly coiled inner membrane (F, white arrowhead) and lesser electron-dense mitochondria (F, black arrowhead) in asthma and control (C, arrow). Images representative of 4 asthmatics and 6 controls. N, nuclear. All scale bars: 1 μm.
ARG2 overexpression shifts metabolism to oxidative pathways and dampens the activation of proinflammatory signal transducers, HIFs, and STAT6.
Although iNOS/NO, its nitrosylation and nitration products, and, recently, nitrite and nitrate command a multitude of diverse metabolic functions (3, 38), the effects of arginine flux through ARG2 to produce ornithine in the mitochondria have not been studied. Here, ARG2 effects were evaluated using human bronchial epithelial cells (BET1A) in vitro. Compared with control vector transfection, transfection of ARG2 increased ARG activity (P = 0.04), measured as the rate of release of tracer urea in the medium following incubation with labeled arginine; the specific ARG inhibitor S-(2-boronoethyl)-l-cysteine (BEC) returned ARG activity to low levels (P = 0.04) (Figure 3A). BET1A cells were particularly useful in these experiments because they have very low to undetectable endogenous expression of ARG2 or ARG1, or when transfected with empty expression vector, but expressed high levels of ARG2 when transfected with the mitochondrial-targeted ARG2 expression vector (Figure 3, B and C). ARG2 localization in mitochondria was confirmed by subcellular fractionation of the overexpressing cells (Figure 3B). A very faint band for ARG2 was noted in the cytosolic fraction by Western analysis, likely related to protein synthesis in the cytosol and trafficking into the mitochondria. There was not a concomitant increase in iNOS expression in ARG2-overexpressing cells (Figure 3C). However, compared with control empty vector, ARG2 transfection led to an approximately 1.7-fold increase in OAT expression that was abrogated by inhibition of ARG activity with BEC (P = 0.003, ANOVA) (Figure 3, C and D). This suggests that ARG2 overexpression induces OAT, which metabolizes ornithine to glutamate. Interestingly, CAT proteins also increased with ARG2 overexpression, and the effect was blunted by BEC (Figure 3, C and D). Ultrastructural analyses (n ≥ 3 replicate experiments) revealed similar mitochondrial numbers between groups over the relatively short time of ARG2 temporary transfection, but the mitochondria in BET1A cells overexpressing ARG2 (Figure 3, G and H) were more electron dense than those in cells transfected with empty vector (Figure 3, E and F), or other control transfections with other mitochondrial localized proteins, such as mitochondrial GFP (Figure 3, I and J, and Supplemental Figure 2) or manganese superoxide dismutase (Figure 3, K and L, and Supplemental Figure 2). Overall, the findings indicate that transient ARG2 overexpression in bronchial epithelial cells in vitro can cause changes in arginine metabolism–related pathways and mitochondrial ultrastructure.
Figure 3. Protein expression and functional activity of bronchial epithelial cells with ARG2 overexpression.

(A) Arginase activity in bronchial epithelial cells (BET1A) transfected with ARG2 is greater than control vector (n ≥ 2 replicate experiments). Arginase inhibitor S-(2-boronoethyl)-l-cysteine (BEC) returns urea levels to baseline, confirming ARG activity. Two-tailed t test. (B) Mitochondrial (Mito) and cytosolic (Cyto) fractions for ARG2, voltage-dependent anion channel (VDAC), and enolase in BET1A cells transfected with control vector or ARG2 (n = 3). (C) Protein expression in BET1A cells transfected with control or ARG2 vector in the presence and absence of BEC for the doses indicated (n ≥ 3 replicate experiments). Enolase as a loading control. Replicate samples run on parallel gels are presented. (D) Quantification of relative expressions in BET1A cells transfected with control or ARG2 vector in the presence and absence of BEC (n ≥ 3 replicate experiments, ANOVA). #P < 0.05, 2-tailed t test, ARG2-expressing vs. control-transfected cells. *P < 0.05, 2-tailed t test, ARG2-expressing cells without BEC vs. with BEC. (E–L) Ultrastructure of mitochondria in BET1A cells transfected with ARG2 vector (G and H), control vector (E and F), mitochondrial GFP (MtGFP) (I and J), or mitochondrial localized manganese superoxide dismutase (MnSOD) (K and L) (n ≥ 3 replicate experiments). Mitochondria in BET1A cells overexpressing ARG2 have greater electron density than cells with empty vector or overexpressing other mitochondrial-localized proteins. F, H, J, and L are close-up views of boxed areas in E, G, I, and K, respectively. N, nuclear. All scale bars: 1 μm. White arrows indicate mitochondrion.
The effect of ARG2 on bioenergetics of BET1A cells was assessed by the measurement of the oxygen consumption rate (OCR) and the extracellular acidification rate (ECAR), the latter a measure of proton production and lactate, using the Seahorse XF24 analyzer (ref. 39 and Figure 4, A and B). The OCR of ARG2-transfected cells was higher than those with vector alone, suggesting higher oxidative metabolism. Addition of BEC to the incubation medium of ARG2-transfected cells resulted in suppression of the OCR to levels similar to those of vector-alone cells (n = 3) (Figure 4A). The coupling efficiency was similar among groups (P > 0.05). In all cells, uncoupling of oxidative phosphorylation with carbonyl cyanide-ρ-trifluoromethoxyphenylhydrazone (FCCP) increased the rate of oxygen consumption, indicating that ATP synthesis is a checkpoint for oxygen consumption in epithelial cells (Figure 4A). Treatment with rotenone (inhibitor of complex I) and antimycin A (inhibitor of complex III) resulted in a decrease in oxygen consumption to negligible levels, indicating that the OCR was attributable to mitochondrial sources (Figure 4A). Consistent with the greater oxidative metabolism, BET1A cells transfected with ARG2 had lower ECAR than vector-transfected cells, indicating a lower rate of release of lactate into the medium. Inhibition of ARG2 with BEC caused ECAR to return to basal levels (n = 3) (Figure 4B). Blocking complex V (ATP synthesis) with oligomycin caused ECAR to increase in all cells; however, the ARG2-transfected cells were less sensitive to oligomycin and maintained a lower ECAR (Figure 4B). Addition of 2-deoxyglucose, a pharmacologic inhibitor of glycolysis, decreased ECAR to near zero, indicating that the measure was related to glycolysis (Figure 4B).
Figure 4. Bioenergetics of bronchial epithelial cells with ARG2 overexpression.

(A) The oxygen consumption rate (OCR) of cells (n = 3 replicate experiments). Inhibitors assess mitochondrial function. The ATPase inhibitor oligomycin, the mitochondrial uncoupler FCCP, and rotenone (ETC complex I inhibitor) and antimycin A (ETC complex III inhibitor) were injected sequentially after measurement of basal rates. OCR relative to vector: the OCR of ARG2-transfected cells normalized to basal OCR of BET1A cells transfected with control vector. (B) Extracellular acidification rate (ECAR) of cells (n = 3 replicate experiments). Glucose, oligomycin, and the glycolysis inhibitor 2-deoxyglucose (2-DG) were injected sequentially. ECAR relative to vector: ECAR of ARG2-transfected cells normalized to basal OCR of BET1A cells transfected with control vector. mpH, pmoles H+/min. (C) Radioisotope studies of glucose metabolism in BET1A cells transfected with ARG2 or control vector (n = 3 replicate experiments). (D) Radioisotope studies of glycine cleavage in BET1A cells transfected with ARG2 vector (n = 3 replicate experiments). All 2-tailed t tests except B, Van der Waerden test, and lactate production in C, median test.
Parallel experiments were performed in order to quantify the rate of oxidation of glucose and production of lactate using radioactive glucose tracer (Figure 4C). As shown, in accordance with the OCR data (Figure 4A), the rate of oxidation of glucose (production of [14C]-O2 from [14C]-glucose) by ARG2-transfected cells was significantly higher than the rate seen with vector alone (n = 3, P = 0.003). In addition, the rate of production of [14C]-lactate from [14C]-glucose was significantly less by the ARG2-transfected cells, suggesting a higher rate of pyruvate oxidation (n = 3, P = 0.02) and thus a significantly higher ratio of rate of oxidation of glucose relative to rate of production of lactate (n = 3, P = 0.02). Inhibition of ARG with BEC in ARG2-transfected cells decreased the rate of oxidation of glucose and increased the production of lactate, indicating that the effects of ARG2 were dependent on increase in ARG2 activity (Figure 4C). These results confirmed the greater oxidative metabolism (of glucose) in ARG2-transfected cells.
An increase in oxidative metabolism would be expected to impact the mitochondrial redox state. We measured the activity of the glycine cleavage system (GCS) in the mitochondria as an index of NAD+ availability, i.e., mitochondrial redox. The GCS is a mitochondrial enzyme composed of 4 loosely associated proteins bound to the mitochondrial inner membrane that catalyze oxidative cleavage of glycine to CO2, NH4+, and a methylene group, and requires NAD+ as cofactor (40). We quantified the activity of GCS by monitoring the conversion of [14C]-glycine to [14C]-O2. ARG2-transfected cells had significantly greater GCS activity (n = 3, P = 0.03) (Figure 4D). These data provide independent confirmation that ARG2 increases mitochondrial oxidative metabolism. Finally, ATP production was determined in BET1A cells transfected with ARG2 or with vector alone. Consistent with a higher rate of oxidative metabolism, ARG2-expressing cells had higher ATP production as compared with vector alone (pmol/μg protein, vector alone 42 ± 12, n = 4, ARG2 72 ± 1, n = 2, P = 0.04).
We speculated that the effects of ARG2 on bioenergetics were likely due to greater influx of ornithine into the mitochondria and consequently higher influx of αKG into the TCA cycle. Alterations in αKG and TCA cycle intermediates influence the activity of αKG-dependent dioxygenases, i.e., prolyl hydroxylases (PHDs), which regulate HIF expression (41). To test whether ARG2 affected HIF, a hypoxia responsive element–luciferase reporter construct (42) was cotransfected with the ARG2 vector. The luciferase activities in BET1A cells transfected with empty vector or ARG2 vector were low, and the ARG inhibitor BEC had no effect on luciferase activities (all P values NS; Figure 5A). Upon exposure to the PHD inhibitor dimethyloxalylglycine (DMOG), cells with vector alone had an increase in luciferase activity, indicating HIF expression and binding to hypoxia responsive element (HRE) and driving luciferase expression (n ≥ 8, P < 0.0001). The DMOG-exposed ARG2-expressing cells had less luciferase expression than vector-alone cells (n ≥ 8, P = 0.03). Exposure to the ARG inhibitor BEC further increased luciferase activity in the DMOG-exposed ARG2-expressing cells (n ≥ 8, P < 0.0001; Figure 5A); BEC did not significantly increase luciferase activity in the DMOG-exposed empty vector–transfected cells (P > 0.05) (Figure 5A). These data indicate that ornithine production by ARG2 may feed intermediates into the TCA cycle via generation of glutamate and αKG, which blunts HIF expression and activity (Figure 5A).
Figure 5. HIF luciferase reporter activity and STAT6 activation in bronchial epithelial cells with ARG2 overexpression.

(A) HRE-driven luciferase activity in BET1A cells transiently transfected with ARG2 vector or control vector and cotransfected with WT HRE-luciferase reporter and Renilla construct, treated with the prolyl hydroxylase (PHD) inhibitor dimethyloxalylglycine (DMOG) (which competes with αKG for PHD) and/or the ARG inhibitor BEC. Data are fold induction over untreated control vector (n ≥ 8 replicate experiments). #P = 0.03, DMOG-exposed ARG2-expressing cells vs. DMOG-exposed control vector–transfected cells, 2-tailed t test. (B and C) IL-4–induced STAT6 activation in BET1A cells transfected with ARG2 or control vector. After transfection, cells were cultured in the presence and absence of IL-4 (10 ng/ml) for the times indicated. (B) Cell nuclear extracts were collected and blotted for phospho-STAT6 (pSTAT6), with lamin B as loading control. (C) IL-4–induced phosphorylation of STAT6 as fold induction over cells transfected with control vector at time point 5 minutes (n ≥ 3, ANOVA).
STAT6 activation is the central signal transducer for Th2 events in the airway, and its activation is redox dependent (43). To determine whether ARG2 overexpression in the epithelial cells regulates STAT6 activation, activated phospho-STAT6 (pSTAT6) was analyzed in ARG2-transfected cells. ARG2-expressing cells had lesser levels of IL-4–induced phosphorylation of STAT6 (Figure 5, B and C), indicating that arginine metabolism blunts Th2 activation of STAT6 in the epithelium. These data indicate that ARG2 overexpression can suppress HIF and STAT6 activation, mechanistically linking mitochondrial arginine metabolism to signal transduction important to the origins of asthma inflammation.
Mitochondrial membrane potential and ROS in airway epithelium of arginase type 2–/–mice.
To determine whether ARG2 affects mitochondrial function in vivo, 6- to 8-week-old mice genetically deficient for arginase type 2 (Arg2 KO) were evaluated (44). Arg2 KO mice have metabolic abnormalities and hypertension (44–46). Here, the whole-body rate of oxygen consumption was lower in Arg2 KO mice as compared with the WT (data not shown). Western analyses of lungs, livers, and kidneys of mice with Arg2 KO or WT mice showed that, similar to the expression pattern in human tissues (18), ARG2 expression was prominent in lungs and kidneys in WT mice, while liver mainly had ARG1 (Figure 6A). ARG2 was not detectable in any organ from Arg2 KO mice. ARG1 expression was not increased in lung tissue of Arg2 KO mice (Figure 6A). Ultrastructural analyses of lung tissues revealed similar morphology and number of mitochondria between WT and Arg2 KO mice (not shown). Arg2 KO had higher levels of HIF-2α and the downstream expression of carbonic anhydrase IX (CAIX) protein in lungs by IHC and Western analyses (HIF-2α, WT 1 ± 0.1, n = 7, Arg2 KO 2.2 ± 0.4, n = 6, P = 0.03; CAIX, WT 1 ± 0.1, n = 4, Arg2 KO 3.4 ± 1.5, n = 4, P = 0.04) (Figure 6, B−D). HIF-1α protein expression was very low in the nonstimulated mouse lung and similar between the Arg2 KO and WT mice (Figure 6D).
Figure 6. ARG2 expression and metabolic rates in Arg2–/– (KO) mice.

(A) Western blot analyses of lungs, livers, and kidneys of mice with Arg2 KO or WT (n ≥ 4 replicate experiments). GAPDH as a loading control. (B and C) Increased expression of carbonic anhydrase IX (CAIX) in lungs of mice with Arg2 KO (C) compared with WT (B). Images representative of 2 Arg2 KO and 2 WT lungs. a, airways. Scale bars: 40 μm. (D) Western blot analyses of HIF-2α and downstream gene CAIX expression in lungs of mice with Arg2 KO (n = 6) compared with WT (n = 7). Lamin B as a loading control for nuclear protein. Enolase as a loading control for whole cell extract. (E–H) Mitochondrial membrane potential and mitochondrial superoxide in mouse airway epithelial cells. Cells stained with JC-1 (E) and MitoSOX (G), respectively. Contour plots show gating of JC-1 red+ or MitoSOX+ cells. Overlaying black contour graphs show fluorescence levels of unstained controls. Histogram overlays of JC-1 red+ (F) or MitoSOX+ cells (H) show a decrease in Arg2 KO mice (n = 10) compared with WT mice (n = 10). Bar graph quantification of mean fluorescence intensities in JC-1+ or MitoSOX + subsets. Two-tailed t test. (I and J) Mitochondrial (Mito) and cytosolic (Cyto) fractions analyzed via Western blot for peroxiredoxin-SO3 (PRX-SO3) and total PRX3 expression in lungs of mice with Arg2 KO (n = 7) compared with WT (n = 7). Two-tailed t test.
Mitochondria function in airway epithelial cells was analyzed by fluorescent probes in flow cytometric analysis of mitochondrial potential and superoxide (Figure 6, E−H). JC-1, a cationic carbocyanine dye that accumulates in mitochondria, was used as a marker for mitochondrial membrane potential. MitoSOX is oxidized by mitochondrial superoxide and is used as a quantitative measure of mitochondrial redox. Freshly obtained mouse airway epithelial cells from Arg2 KO mice (n = 10) had lower mitochondrial membrane potential as compared with WT (n = 10, P = 0.04) (Figure 6, E and F) and less ROS production (P = 0.02) (Figure 6, G and H). Mitochondrial redox was also assessed by cysteine oxidation of the mitochondrial-localized peroxiredoxin 3 (PRX3), a 2-Cys PRX family member that scavenges mitochondrial H2O2 (47, 48). In oxidative conditions, the PRX3 sulfenic intermediate (Cys51-SOH) is overoxidized to cysteine sulfinic acid (Cys51-SO2H) or cysteine sulfonic acid (Cys51-SO3H). Using a specific antibody that recognizes both sulfinic and sulfonic forms of PRX family members, mitochondrial fractions were evaluated for sulfinylation levels of PRX (PRX3-SO3) relative to total PRX3 expression. The mitochondria levels of PRX3-SO3 were significantly less in Arg2 KO mice lungs as compared with WT (Figure 6, I and J). Sulfonic forms of other PRXs were not detected in cytosolic fractions (Figure 6I). Thus, mice deficient in Arg2 (Arg2 KO) had lower mitochondrial membrane potential, less ROS production, and consequently less hyperoxidation of PRX3 (Prx3-SO3). Notably, CAT2 and CAT3 expression was similar in lung tissue of Arg2 KO and WT mice, and CAT1 was not detectable in lung tissues (data not shown). These data indicate that ARG2 is important for maintenance of mitochondrial membrane potential, redox state, and regulation of hypoxia sensing.
Greater inflammation and mucus metaplasia in Arg2 KO mice in an asthma model.
To determine whether ARG2 regulates severity of airway inflammation and remodeling, Arg2 KO mice were evaluated using the ovalbumin (OVA) allergen asthma model. The OVA-sensitized and PBS-challenged control mice (OVA/PBS) were compared with the OVA-sensitized and OVA-challenged (OVA/OVA) asthma model. The OVA/PBS Arg2 KO mice (n = 3) had visibly more HIF-2α than control OVA/PBS WT mice (n = 3), and higher levels of the HIF-regulated gene product CAIX (Figure 7A). HIF-1α expression was low in lungs of mice irrespective of genotype or the OVA asthma model (Figure 7A). Interestingly, the Arg2 KO OVA/PBS group had detectable expression of activated pSTAT6 in lungs, while the WT OVA/PBS mice did not. Arg2 KO mice had similar iNOS protein induction in the OVA/OVA asthma model as compared with WT (Figure 7A). Arg2 KO mice did not have a higher level of NO production as measured by nitrate in the BAL fluid. In fact, nitrate levels were significantly lower in Arg2 KO mice (nitrate μM, WT 6.1 ± 2.0, Arg2 KO 2.5 ± 0.4, P = 0.008), indicating that iNOS/NO did not account for the greater inflammation and remodeling in the Arg2 KO mice and underscoring the role of ARG2 in supporting the cell-autonomous production of NO by providing N via aspartate (Figure 1A). Airway hyper-responsiveness of Arg2 KO was similar to that of WT mice (data not shown), which is in line with prior studies that also show a disconnect between airway inflammation and airway hyper-responsiveness (49).
Figure 7. Arg2 genetic deletion in inflammation and goblet cell metaplasia in OVA model.

(A) Representative protein expressions in lungs of Arg2 KO challenged with aerosolized OVA or PBS. Lamin B as loading control for nuclear protein. Enolase as a loading control for whole cell extract. n ≥ 3 replicate experiments. (B) Flow cytometry of IL-13 in cytokeratin-positive cells from lungs of OVA/OVA–treated Arg2 KO and WT. IL-13 expression in cytokeratin-positive cells (cytokeratin+, epithelial marker) increases in WT and Arg2 KO in the OVA/OVA model, but IL-13 is higher in Arg2 KO compared with WT. n ≥ 3 replicate experiments. (C–F) IHC of IL-13. Positive staining in airway epithelium of Arg2 KO OVA/OVA and WT OVA/OVA. Arg2 KO OVA/OVA has greater IL-13 expression than WT OVA/OVA. n ≥ 3 replicate experiments. Scale bars: 40 μm. (G–K) Elevated total cells (G), eosinophils (H), eotaxin-1 (I), IL-13 (J), and IL-17 (K) in BAL of OVA/OVA–treated Arg2 KO. n ≥ 3 replicate experiments. K, 2-tailed t test; G, H, J, Wilcoxon, and I, median test. (L) Mucin-positive cells by PAS in airways of Arg2 KO compared with WT in OVA/OVA. n ≥ 4 replicate experiments. Two-tailed t test. (M–T) Airway goblet cell metaplasia (PAS staining) in Arg2 KO OVA/OVA mice. N, close-up of M, P of O, R of Q, and T of S. Goblet cells not observed in medium-sized or small airways in OVA/PBS–treated WT (M and N) or Arg2 KO (Q and R). Goblet cell metaplasia (red cells, black arrowheads) is more prominent in Arg2 KO OVA/OVA (S and T) than WT OVA/OVA (O and P). n ≥ 4 replicate experiments. Black arrows show inflammatory infiltrate; arrowheads, positive PAS staining. a, airways. Scale bars: 40 μm.
As compared with WT OVA/OVA mice, Arg2 KO OVA/OVA mice (n ≥ 3) had significantly higher IL-13 expression in cytokeratin-positive epithelial cells by flow cytometry (IL-13+cytokeratin+) (P = 0.001) (Figure 7B). IHC confirmed increased expression of IL-13 in airway epithelium of Arg2 KO OVA/OVA mice (Figure 7, C–F). IL-13 expression in CD45+ leukocytes (IL-13+CD45+) was similar between WT and Arg2 KO in the OVA/OVA model, indicating that IL-13 expression in leukocytes was not different between Arg2 KO and WT OVA/OVA mice (data not shown). Arg2 KO OVA/OVA mice (n ≥ 3) had greater Th2 inflammation including higher levels of the eosinophil chemoattractant eotaxin-1 (Figure 7I) and total cells and eosinophil numbers (Figure 7, G and H), as well as higher IL-13 (Figure 7J) and IL-17 levels (Figure 7K) in the BAL. Consistent with higher levels of IL-13, the Arg2 KO OVA/OVA mice (n ≥ 3) had greater downstream activation of pSTAT6 (WT 1 ± 0.07, Arg2 KO 2.02 ± 0.10, P = 0.002) (Figure 7A) and more airway goblet cell metaplasia (P = 0.004) (Figure 7, L–T) as compared with WT mice (n ≥ 3). Overall, the data identify higher levels of epithelial IL-13 in Arg2 KO animals in the OVA model, with adverse consequences of greater mucus production and inflammation. At baseline, Arg2 KO mice and WT mice had similarly low IL-13 expression in epithelium, indicating that OVA sensitization and OVA challenge are required for the appearance of exocrine IL-13 in the airways.
To further define the relative contributions of epithelial and/or leukocyte effects on the more severe airway inflammation in Arg2 KO mice, mice underwent bone marrow ablation and reconstitution prior to OVA sensitization and challenge (Figure 8A). Airway inflammation as determined by total number of cells (Figure 8B), eosinophils (Figure 8C), eotaxin-1 (Figure 8D), or IL-13 levels (Figure 8E) in BAL and mucus hypersecretion as determined by periodic acid–Schiff (PAS) staining (Figure 8, G–O) were unaffected in mice receiving bone marrow transplant from Arg2 KO (n ≥ 4) or from WT mice (n ≥ 4; all P values NS). These results suggest that the more severe airway inflammation in Arg2 KO mice was largely due to resident lung cells. In contrast, the BAL level of IL-17, which is produced primarily by leukocytes, was higher in Arg2 KO mice receiving bone marrow transplant from Arg2 KO but not from WT (P = 0.01) (Figure 8F). These results suggest that Arg2 deficiency impacts more than airway epithelium, and that interactions of resident lung cells and leukocytes of the Arg2 KO contribute to the greater inflammation in the OVA model.
Figure 8. Effects of bone marrow ablation and reconstitution between Arg2 KO and WT in the OVA/OVA asthma model.

(A) Arg2 KO and WT underwent bone marrow ablation and reconstitution, then OVA sensitization and challenge. (B–F) Total cells (B), eosinophils (C), eotaxin-1 (D), IL-13 (E), and IL-17 (F) in BAL of Arg2 KO and WT receiving bone marrow transplant from Arg2 KO or WT in the OVA/OVA asthma model. n ≥ 4 replicate experiments. #P = 0.01, Arg2 KO recipients receiving bone marrow from Arg2 KO vs. WT, 2-tailed t test. (G) Quantification of PAS staining in airways of Arg2 KO and WT mice receiving bone marrow from Arg2 KO or WT in the OVA/OVA model. n ≥ 4 replicate experiments. Two-tailed t test. (H−O) Airway goblet cell metaplasia (PAS staining) in airways of Arg2 KO and WT receiving bone marrow from Arg2 KO or WT in the OVA/OVA model. I, close-up view of H, K of J, M of L, and O of N. n ≥ 4 replicate experiments. Black arrows show inflammatory infiltrate; arrowheads, PAS+ staining. a, airways. Scale bars: 40 μm.
Discussion
Regulation of gene expression in response to nutritional substrates was a landmark in our understanding of how metabolites modulate cellular functions in unicellular organisms. Although amino acids and carbohydrates participate in gene regulation in humans (50, 51), there is little to no understanding of how metabolites contribute to disease pathophysiology. This study provides new knowledge on the mechanism(s) by which mitochondrial metabolism of arginine modulates transcription factors, gene expression, cell function, and inflammation. Greater arginine flux through ARG2 into the mitochondria increases oxidative metabolism, dampens proinflammatory signal transduction events that are central to asthma origins, and serves as a brake on Th2 inflammation.
This study and prior work define asthma as an arginine-driven metabolic disorder (1, 2, 16, 17, 19). Although arginine may be taken into cells by CAT proteins, in 1990, Vane’s group (8) identified that cells producing NO over prolonged periods, such as the asthmatic airway epithelium, synthesize arginine at levels sufficient to sustain NO production. Many reports now confirm the importance of the cell-autonomous citrulline-arginine-NO cycle (Figure 1A) for sustained NO production by NOS enzymes, including iNOS (8–10). Asthmatics in this study are found to have greater whole-body de novo synthesis of arginine, and have all the enzymes necessary in airway cells to synthesize arginine from citrulline for ARG and iNOS activity. It is important to note that although tracer studies provide comparable quantitative data, whole-body tracer measures in plasma may not accurately reflect the cellular/subcellular levels found at specific anatomical/cellular sites relevant to asthma because of the unique metabolism and compartmentalization of arginine, citrulline, and ornithine in organs. Arginine is rapidly taken up by the liver but not released because of its rapid conversion to ornithine by ARG1 in urea cycle. Citrulline is synthesized by the gut from glutamine, not taken up by liver, but rather delivered to the kidney, where it is converted into arginine and released into the circulation. Despite the unique metabolism of these amino acids, greater whole-body conversion of citrulline into arginine is identifiable in asthma, supporting the idea that asthma is characterized by excessive arginine metabolism.
Although higher-than-normal levels of arginine/NO have been long described in asthma, and FENO has been standardized for clinical assessment of inflammation in asthma, we (19) and others (6, 14–18) have found that ARG activity is increased in asthma, but little was known of the consequences of increased ARG activity for asthma pathobiology. This study reveals that the greater mitochondrial arginine metabolism is linked to changes in mitochondrial endotype and a shift toward oxidative bioenergetic pathways. The loss of mitochondrial arginine metabolism in mice genetically deficient in Arg2 results in lower production of NO, lower mitochondrial membrane potential, greater activation of hypoxia sensing and Th2 signaling pathways, and more severe allergen-induced asthma. Studies of pharmacologic inhibition of ARGs in murine models of airway inflammation have provided inconsistent results, i.e., airway inflammation and hyper-reactivity are diminished or worsened (14–16, 52, 53). The findings here contrast with prior ambiguous reports of broad-based pharmacologic inhibitors of ARG (14–16, 52, 53), which are not mitochondrial-targeted and may have off-target effects on other arginine pathways. Taken together, the findings in this study indicate that ARG2 is protective for the mitochondrial function, particularly in the intracellular environment of high-level NO synthesis. Prior studies have shown that NO has inhibitory effects on the respiratory chain (complex I and III) and consequently decreases oxygen consumption and cellular respiration (54). Inhibition of the respiratory chain by NO results in an increase in the rate of aerobic glycolysis (Warburg effect), and an increase in the conversion of pyruvate to lactate. The release of lactate during aerobic glycolysis is primarily to maintain cellular redox (NAD/NADH ratio) so that the various processes, primarily glycolysis, requiring NAD can be sustained. ARG2 counteracts these effects: it increases oxygen consumption, increases oxidation of glucose, decreases lactate release, sustains cellular redox balance (NAD/NADH), and accelerates the TCA cycle by providing NAD and carbon backbone (αKG and other TCA intermediates). Thus, cells with continuous high levels of NO production, such as in asthmatic airway epithelium, apparently benefit from ARG2 production of ornithine and subsequently glutamate in 2 ways. First, the greater ornithine to glutamate can supply TCA cycle intermediates, maintain redox balance, and fuel oxidative phosphorylation. Second, the greater ornithine to glutamate enables endogenous arginine synthesis via the citrulline-NO cycle to support NO production. Consistent with this concept, Arg2-deficient mice had less NO production, despite iNOS expression at high levels in the asthma model, supporting the importance of the intracellular citrulline-NO cycle for regeneration of arginine and maintenance of iNOS activity.
Prior studies have identified mitochondrial structural and functional changes in the allergen-induced murine model of asthma (36), which mechanistically linked mitochondrial function to asthmatic features such as inflammation; however, the underlying mechanisms of mitochondrial change in asthma were unknown (37). Likewise, prior study showed that platelets from asthmatic individuals have less reliance on glycolysis and greater TCA cycle turnover (55). Here, we show greater mitochondrial electron transport chain complex proteins, mitochondria numbers, and higher ATP/pyruvate ratio in human asthmatic epithelial cells as compared with control, and link the changes to ARG2 expression and activity. Metabolic measures indicate that ARG2 metabolism of arginine in the mitochondrion can generate αKG, which enters the TCA cycle for greater cycle activity (Figure 1A). The TCA cycle has a primary role in oxidation of substrate for energy metabolism, but also serves critical biosynthetic functions in which intermediates enter and leave the cycle to regulate cell metabolism and signal transduction (56). For example, glucose oxidative metabolism via the TCA cycle enhances PHD activity and suppresses HIF (57). HIFs are present at higher levels in murine asthma models and in epithelial cells of endobronchial biopsies of asthmatics after allergen challenge (28). Although airway epithelial cells are exposed directly to air (4), HIF is expressed in the normal and asthmatic airway epithelium (28), suggesting that HIF protein abundance in the airway may be adjusted in response to nonhypoxic stimuli (57). This study shows that mitochondrial arginine metabolism, independent of hypoxia, is a primary factor controlling HIF and downstream activation of Th2 genes in the airways. Asthma is typified by Th2 cytokines (30, 31) that lead to activation of STAT6 (1, 2), eotaxin-1 that attracts eosinophils into the airway (58), and airway goblet cell metaplasia with increase in mucin glycoproteins, i.e., MUC5AC (26). The promoter regions of IL-13, eotaxin, and MUC5AC genes all have HREs (26), supporting that metabolic changes that promote higher HIF levels would lead to greater expression of these genes and more severe asthma. Likewise, pharmacologic inhibition of HIF, as well as conditional deletion of HIF in mice, results in markedly less IL-13 and airway inflammation (28). Thus, HIF is a key mechanistic pathway in allergic airway inflammation, in part by regulation of IL-13 production (26–28). IL-13 is sufficient to induce typical features of asthma, including induction of mucin production and secretion and recruitment of eosinophils to the lungs (29). Airway epithelium has been identified as a major site of IL-13 expression, and the epithelial production of IL-13 induces many features of asthma primarily via its actions on resident epithelial and smooth muscle cells (29–31). Here, epithelial IL-13 levels were higher in Arg2-deficient animals, which was associated with greater mucus production and inflammation.
There are limitations to mechanistic studies using temporary transfections and animal models of disease. However, the multiple model systems used and the complementary findings in human asthma support that our findings are relevant for understanding arginine metabolism in the pathobiology of asthma. Altogether these studies show that mitochondrial ARG in the asthmatic airway epithelium, by transporting ornithine into the mitochondrion, contributes (a) nitrogen (via aspartate) for the cytosolic cell-autonomous de novo synthesis of arginine for NO production by iNOS, and (b) TCA cycle intermediates that increase oxidative metabolism, modulate redox, and suppress pathological HIF signaling events that lead to activation of IL-13 and STAT6, which are central to asthma. Understanding how metabolism regulates inflammation provides new potential targets to modify the severity of asthma and move us several steps forward in our ability to evaluate novel metabolic therapies for asthma.
Methods
Study population.
Asthma was verified based on American Thoracic Society guidelines, which include positive methacholine challenge and/or reversible airflow obstruction. Healthy controls had normal spirometry and negative methacholine challenge. All were recruited from Cleveland Clinic, and gave written informed consent by signing a consent document approved by the Cleveland Clinic Institutional Review Board. Exclusion for asthma and healthy controls included having any of the following: current smoking, smoking within the past year, former smoking with ≥5 pack-year total history, or pregnancy. Spirometry was performed with an automated spirometer, and FENO measured according to the standards of the American Thoracic Society. A subgroup of participants underwent bronchoscopy for endobronchial biopsy, BAL, and differential cell count (19).
Stable isotopic studies and de novo arginine synthesis.
Tracer/tracee ratios of plasma arginine, citrulline, and ornithine were measured by liquid chromatography–tandem mass spectrometry. Rate of appearance or total flux and endogenous flux of arginine, citrulline, ornithine, and urea, the rate of conversion of citrulline to arginine, and the rate of NO synthesis were calculated as previously described (33, 59). Details are provided in Supplemental Methods.
Western analyses.
Whole cell lysates and BAL cell protein were prepared as previously described (60). Nuclear extracts were prepared according to standardized protocol (Panomics Inc.). Mitochondrial and cytosolic fractions were prepared according to protocol provided by Thermo Scientific (Mitochondria Isolation Kit for Cultured Cells & Mitochondria Isolation Kit for Tissue). Total protein was measured using the Pierce Coomassie Plus Protein Assay (Thermo Scientific). Equal amounts of protein (whole cell lysates 80–100 μg/lane, nuclear extracts 40–60 μg/lane, and mitochondrial fraction 40 μg/lane) were loaded per lane for samples from BET1A cells or mouse tissues. For human bronchial epithelial cells, the volumes of whole cell lysates loaded per lane were adjusted to have similar cytokeratin expression after 5 μl lysates prerun to measure cytokeratin. Protein was separated by electrophoresis on a 4%–15% Tris-HCl or Any kD precast gel (Bio-Rad Laboratories), and transferred onto PVDF membranes (Millipore Corp.). Amersham ECL Western blot detection reagents, Amersham Hyperfilm ECL (GE Healthcare Life Sciences), and Kodak M35 X-OMAT Automatic Processors were used for detection of signals. By direct scanning of the Western blot films with an HP Scanjet 4890 and processing with ImageJ software (NIH), the bands were quantitated in densitometry units. Details of antibodies used are provided in Supplemental Methods.
Immunohistochemistry.
Paraffin-embedded formalin-fixed tissues were sectioned at 5 μm. For bronchial biopsies and open lung biopsies, rabbit anti-ARG2 and ASS polyclonal antibodies (Santa Cruz Biotechnology) were used for analyses after antigen retrieval with sodium citrate buffer (pH 6.0). ARG2 and ASS expressed in renal tubules of mouse kidney served as positive control, while negative control of secondary antibody alone was performed on each section of tissue studied. For analyses of mouse lung tissues, rabbit anti-CAIX antibody (Novus Biologicals) was used after antigen retrieval (42). For IL-13 staining, rabbit anti–IL-13 antibody (ab106732; Abcam) was used after antigen retrieval with a Tris/borate/EDTA buffer (pH 8.0–8.5) (Ventana Medical System Inc.). IHC staining was performed using the Discovery ULTRA automated stainer from Ventana. The antibody was visualized using the OmniMap anti-rabbit HRP secondary antibody (760-4311; Ventana) and the ChromoMap DAB detection kit (Ventana). The slides were counterstained with hematoxylin.
Ultrastructural analyses.
Ultrastructure of airway epithelial cells and BET1A cells was studied using an Tecnai G2 Spirit BioTWIN Transmission Electron Microscope (FEI Co.) and samples prepared as previously described (38) for stereologic quantitation of mitochondria. Details are provided in Supplemental Methods.
Immunogold electron microscopy.
Samples were fixed in PBS containing 0.05% glutaraldehyde and 4% paraformaldehyde, dehydrated in ascending ethanol up to 100%, embedded in LR white resin, and polymerized at 50°C for 4 days. Ultrathin sections cut with a diamond knife (85 nm) were mounted on nickel grids coated with formvar. The grids were rehydrated with PBS and incubated in PBS with 0.1% BSA to block nonspecific reactions for 15 minutes. The sections were incubated with rabbit anti-ARG2 or ASS polyclonal antibody (Santa Cruz Biotechnology) at 1:40 dilution in PBS with 0.1% BSA for 3 hours, washed with PBS, and then incubated for 1 hour with secondary antibodies (10 nm gold-conjugated goat anti-rabbit IgG from Ted Pella Inc.) at 1:10 dilution in PBS with 0.1% BSA. After PBS wash, samples were fixed with 1% glutaraldehyde in PBS for 7 minutes, stained briefly with uranyl acetate and lead citrate, washed with distilled water, and examined with a Tecnai G2 Spirit BioTWIN Transmission Electron Microscope (FEI Co.) at 60 kV.
ATP, pyruvate, and lactate.
ATP in cells was measured using a luciferase-based luminescence assay kit (PerkinElmer). Pyruvate and lactate concentrations in cell lysates were determined with a Pyruvate Assay Kit or Lactate Assay Kit (BioVision).
Cell culture.
BET1A cells, a human bronchial cell line transformed by SV40 T antigen, were cultured in serum-free LHC-8 (Biofluids Inc.) with 0.33 nM retinoic acid and 2.75 μM epinephrine on precoated plates. A549 cells, an epithelial cell line from lung adenocarcinoma, were cultured in MEM (Invitrogen Corp.) with 10% heat-inactivated FCS, or 24 hours before cytokine stimulation with 1% FCS. A549 was exposed to cytokines containing 10,000 U/ml IFN-γ, 0.5 ng/ml IL-1β, and 10 ng/ml TNF-α. Human IFN-γ was a gift from Genentech or purchased from R&D Systems Inc. Recombinant human IL-1β and TNF-α were purchased from Biosource.
Transient transfection and luciferase assay.
The 1.097-kb human ARG2 cDNA containing the ATG start codon, mitochondrial-targeting region, and TAG stop codon was inserted into expression vector that contained cytomegalovirus promoter, to create an ARG2 expression plasmid (ARG2, CCF88). WT HRE-luciferase reporter construct was a gift from M.C. Simon (University of Pennsylvania School of Medicine, Philadelphia, PA) (42). Using Lipofectamine 2000 Reagent (Invitrogen Corp.), BET1A cells were transiently transfected for 6 hours with ARG2, empty vector, or manganese superoxide dismutase (MnSOD) expression vector (MnSOD cDNA cloned into adenovirus shuttle vector pAVS6, CCF35), or cotransfected with HRE construct and Renilla luciferase construct for normalization of transfection efficiency. After transfection, cells were treated with 50 μM ARG inhibitor S-(2-boronoethyl)-l-cysteine (BEC) or doses as indicated, 300 μM nonselective PHD inhibitor DMOG, or 10 ng/ml IL-4 (Invitrogen Corp.) or left untreated. BET1A cells were labeled with mitochondrial GFP (Life Technologies). Forty-eight hours after transfection, cells were assayed by Dual-Luciferase Reporter Assay System (Promega Corp.). Relative luciferase activity in BET1A cells was the value of HRE luciferase activity normalized with dual (Renilla) luciferase assay and protein.
Arginase activity.
Arginase activity was measured in the intact cells by quantification of the rate of production of [15N2]-urea from [15N2]-arginine and expressed as nanomoles of urea produced per hour normalized to total protein. 1 × 106 to 2 × 106 BET1A cells were incubated in 9 ml LHC9 medium at 37°C for 40 hours. At time 0 h, the medium was enriched with [15N2]-arginine in order to achieve 15N2 enrichment in arginine of 30 moles % access. Unlabeled urea 1.75 mM was added to the incubation medium. Media samples for measurement of 15N2 in urea were obtained at 0, 2, 16, 24, and 40 hours. The 15N2 enrichment of urea was quantified using gas chromatography–mass spectrometry as described previously (61). Following addition of labeled arginine, likely because of increase in the amount of arginine in the media, there was an initial increase in the label in urea in all conditions. Therefore, we calculated ARG activity assuming the 2 hours sample as the initial sample.
Oxygen consumption rate and extracellular acidification rate.
The ECAR and OCR were measured using the Seahorse Extracellular Flux (XF24) Analyzer (Seahorse Bioscience Inc.). BET1A cells were detached, washed 3 times with the appropriate Seahorse assay medium, and resuspended in assay medium. Sixty thousand cells in 150 μl assay medium were added to the wells of a Seahorse cell plate and adhered using BD Cell-Tak (BD Biosciences) according to Seahorse protocol for nonadherent cells, with 2 or 4 wells per plate left empty for background correction. Seahorse assay media (DMEM without glucose, l-glutamine, phenol red, sodium pyruvate, and sodium bicarbonate [Sigma-Aldrich] supplemented with either 6 mM glucose, 1.85 g/l sodium chloride, 1 mM sodium pyruvate, and 15 mg/l phenol red [MitoStress Assay] or 1.85 g/l sodium chloride and 3 mg/l phenol red [GlycoStress Assay]) was supplemented with 2 mM l-glutamine and the pH adjusted to 7.35 with sodium hydroxide. The plate was incubated in a 37°C non-CO2 incubator for 25 minutes. Additional assay medium was added to bring the final per-well volume to 500 μl (MitoStress) or 720 μl (GlycoStress), and the plate incubated in a 37°C non-CO2 incubator for an additional 15 minutes. The plate was then transferred to the Seahorse XF24 Analyzer for analysis. Once in the XF24, BET1A cells underwent a MitoStress test (basal measurement of oxygen consumption followed by successive treatments with oligomycin A [0.5 μM], FCCP [carbonyl cyanide-ρ-trifluoromethoxyphenylhydrazone; 0.5 μM], and rotenone and antimycin A [1.5 μM]) or a GlycoStress test (basal measurement of extracellular acidification followed by successive treatments with glucose [10 mM], oligomycin [0.25 μM], and 2-deoxyglucose [100 mM]). All MitoStress OCR measures were done 3 times in a 3-2-3-minute mix-wait-measure cycle, while GlycoStress ECAR measures were done 3 times in a 3-3-minute mix-measure cycle. Measures were normalized with basal OCR or ECAR level of BET1A cells transfected with empty vector.
Radioisotope studies of glucose and glycine metabolism.
The rate of oxidation of glucose to CO2 was measured by incubation of 1 × 106 to 2 × 106 BET1A cells in LHC9 media containing 6 mM glucose with [14C]-glucose or [14C]-glycine in an atmosphere of 95% O2 and 5% CO2 in an airtight Erlenmeyer flask. Cells were incubated for 3 hours. The CO2 generated was flushed by addition of sodium bicarbonate and sulfuric acid into the medium. CO2 was trapped in hydroxide of hyamine (10 M) placed in a plastic cup suspended in the vessel. The radioactive CO2 trapped in hyamine was counted in a scintillation counter. The rate of oxidation of glucose or glycine was calculated using the precursor-product relationship. The rate of glycolysis (glucose to lactate) was measured as follows: Lactate was separated from the medium at the end of incubation using ion exchange chromatography. The radioactivity in the isolated lactate was measured using a scintillation counter. The rate of conversion of glucose to lactate was calculated using precursor-product relationship.
Animal experiments.
WT or Arg2–/– female C57BL/6 mice 6–8 weeks old were used. WT mice were purchased from The Jackson Laboratory, and Arg2–/– mice were donated by W.E. O’Brien (Baylor College of Medicine, Houston, TX) (44). Animals were immunized twice 7 days apart by i.p. injection with OVA (Sigma-Aldrich) [20 μg, adsorbed in Al(OH)3]. Seven days after the second i.p. injection, mice were challenged with aerosolized OVA in a chamber kept saturated with nebulized OVA solution (1% wt/vol in sterile PBS) or sham-challenged with PBS aerosol for 2 or 4 successive days. Upon 24 hours after the final challenge, BAL, airway epithelial cells, and lung tissues were collected for analysis. Eotaxin-1, IL-13, and IL-17 levels in BAL fluid were measured by Quantikine ELISA kits (R&D Systems Inc.). Transplantation of bone marrow was performed by whole-body sublethal irradiation (total irradiation dose of 1,000 cGy) of recipient mice followed by tail vein injection of 5 × 106 bone marrow cells obtained from donor mice. Bone marrow–reconstituted mice were used for experiments 4 weeks after the bone marrow transfusion. To quantify the mucus production in mouse airways, the area of PAS+ epithelial cells and the total area of epithelial cells per bronchiole were acquired using Image-Pro Plus 7.0 software (Media Cybernetics Inc.). The relative PAS staining values in airways (percent) were calculated as the area of PAS+ epithelial cells per bronchiole divided by the total area of epithelial cells.
Flow cytometry.
MitoProbe JC-1 and MitoSOX red were used (Life Technologies) to measure the mitochondrial potential and superoxide, respectively. Both probes are selectively retained in the mitochondria by the mitochondrial potential. Murine airway epithelial cells were harvested by instillation of 0.05% trypsin/EDTA into trachea of mice that were sacrificed and had previously had pulmonary vasculature perfused with PBS. Cells were filtered through a 40-μm filter to remove clumps, any rbc were lysed with ammonium chloride lysis buffer, and cells were purified by seeding on a 35-mm Primaria tissue culture dish for 1 hour in mouse tracheal epithelial cell (MTEC) Basic media (DMEM/F-12) containing 10% FBS, which allows fibroblast cells to attach while leaving epithelial cells in suspension. The suspended cells collected were epithelial cells as confirmed by positive staining with cytokeratin. The suspended cells were collected and stained with probe JC-1 or MitoSOX in Mg2+/Ca2+–free buffers. Unstained samples were used as controls. All samples were run on an LSRII flow cytometer (BD Biosciences). The blue 488 nm argon laser was used to excite JC-1. Green fluorescence signal was collected using long pass (LP) 505 nm dichroic and band pass (BP) 525/50 nm filter in photomultiplier tube (PMT) A. Red fluorescence signal was collected using LP 570 nm dichroic and BP 585/42 nm filter in PMT B. MitoSOX was excited using the green 532 nm laser, and signals were collected with the standard phycoerythrin (PE) detector. SPHERO Ultra Rainbow calibration beads (Spherotech) were used to standardize PMT sensitivity for comparison of measurements over time. A minimum of 1,000 events was collected. Data were analyzed using FlowJo Vx software (Tree Star). For JC-1 analysis, compensation was set using cells stained with JC-1, but mitochondrial potential collapsed with carbonyl cyanide m-chlorophenyl hydrazone (CCCP). Time and forward scatter (FSC)/side scatter (SSC) plots were used to select for true cellular events. Gating strategy for analysis of JC-1 and MitoSOX is shown in Figure 6.
IL-13 flow cytometry.
Lungs and tracheas were minced and digested to a single cell suspension (62). GolgiStop (BD Biosciences) was used in all buffers to retain cytokines in cells. DNase I (Sigma-Aldrich) was used to prevent cell clumping. Cells were bulk-stained with live/dead fixable blue cell stain (Thermo Scientific). Nonspecific Fc binding sites were blocked by preincubation of cells in Fc-block (eBioscience). Cell surface staining for anti-CD45–Alexa 700 (1:50 in 1% BSA in PBS, 100 μl/test, 30 minutes’ incubation at room temperature) was performed prior to fixation of the cells in 4% paraformaldehyde for 10 minutes at room temperature. Permeabilization and blocking of intracellular nonspecific binding was performed by incubation of the cells in 0.5% Tween-20, 2% FBS in PBS for 15–30 minutes, followed by anti-cytokeratin–FITC (1/10; Abcam) and anti–IL-13 (1/200; eBioscience). Both antibodies were diluted in 0.5% Tween-20, 2% FBS in PBS, and used at 100 μl/test, 30 minutes’ incubation time at room temperature. After incubation, cells were washed 2 times in 0.5% Tween-20, 2% FBS in PBS. Cells were analyzed on an LSRFortessa (BD Biosciences) flow cytometer equipped with 5 laser lines (355 nm, 407 nm, 488 nm, 561 nm, and 641 nm) with standard configuration. Data were analyzed using FlowJo 10 software (Tree Star). Aggregates were excluded on FSC-A/FSC-H dot plots, followed by dead cell exclusion. Cell debris was eliminated on an FSC/SSC plot. Expression of IL-13 in CD45+ cells and cytokeratin+ cells was analyzed. Unstained cells were used to define boundaries. Cells from control animals served as biological controls. All antibodies were titrated to determine optimal concentrations. AbC Total Antibody Compensation Bead Kit (Thermo Scientific) was used to prepare spectral overlap compensation controls for antibodies.
Statistics.
Data are shown as mean ± SEM. All statistical comparisons were performed using 2-tailed Student’s t test except when otherwise specified, nonparametric analyses, or ANOVA as appropriate. Bivariate analysis was used to study the relationship between 2 variables. The level of significance for P was chosen at 0.05. All data were analyzed with the statistical program JMP Pro 10 (SAS Institute).
Study approval.
The human study was approved by the Cleveland Clinic Institutional Review Board (Cleveland, Ohio, USA). Written informed consent was received from participants prior to inclusion in the study. Discarded residual lung tissue from explanted lungs not used for transplant was obtained under an exempt IRB. All animal experiments were approved by the Cleveland Clinic IACUC.
Author contributions
WX, KA, CCK, RMT, SCK, and SCE designed the research studies. WX, SG, KA, AJJ, DAM, CDB, LLG, BBG, KAQ, CCK, and RMT performed the experiments. SAAC, SHW, and RMT acquired data. WX, KA, AJJ, SCK, and SCE analyzed the data. JMP provided reagents. WX, SCK, and SCE wrote the manuscript.
Supplementary Material
Acknowledgments
Thanks to C. Farver, G. Cheng, M. Aronica, T. Okamoto, M. Yin, K. Simmerman, J. Peterson, and C. Croniger for help with the study. This work was supported by awards from the National Heart, Lung, and Blood Institute (HL081064, HL103453, and HL109250); this project was also supported in part by the National Center for Advancing Translational Sciences (ULTR000439). S.C. Erzurum is supported in part by the Alfred Lerner Chair for Biomedical Research; K. Asosingh is a scholar of the International Society for Advancement of Cytometry.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(7):2465–2481. doi:10.1172/JCI82925.
Contributor Information
Weiling Xu, Email: xuw@ccf.org.
Sudakshina Ghosh, Email: sudakshina_g@hotmail.com.
Kewal Asosingh, Email: asosink@ccf.org.
Brian B. Graham, Email: brian.graham@ucdenver.edu.
Samuel H. Wedes, Email: sam.wedes@gmail.com.
Rubin M. Tuder, Email: rubin.tuder@ucdenver.edu.
References
- 1.Comhair SA, Erzurum SC. Redox control of asthma: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2010;12(1):93–124. doi: 10.1089/ars.2008.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghosh S, et al. Nitrotyrosine proteome survey in asthma identifies oxidative mechanism of catalase inactivation. J Immunol. 2006;176(9):5587–5597. doi: 10.4049/jimmunol.176.9.5587. [DOI] [PubMed] [Google Scholar]
- 3.Larsen FJ, et al. Dietary inorganic nitrate improves mitochondrial efficiency in humans. Cell Metab. 2011;13(2):149–159. doi: 10.1016/j.cmet.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Dweik RA, et al. Nitric oxide synthesis in the lung. Regulation by oxygen through a kinetic mechanism. J Clin Invest. 1998;101(3):660–666. doi: 10.1172/JCI1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathrani VC, Kenyon NJ, Zeki A, Last JA. Mouse models of asthma: can they give us mechanistic insights into the role of nitric oxide? Curr Med Chem. 2007;14(20):2204–2213. doi: 10.2174/092986707781389628. [DOI] [PubMed] [Google Scholar]
- 6.Bratt JM, Zeki AA, Last JA, Kenyon NJ. Competitive metabolism of L-arginine arginase as a therapeutic target in asthma. J Biomed Res. 2011;25(5):299–308. doi: 10.1016/S1674-8301(11)60041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo FH, et al. Molecular mechanisms of increased nitric oxide (NO) in asthma: evidence for transcriptional and post-translational regulation of NO synthesis. J Immunol. 2000;164(11):5970–5980. doi: 10.4049/jimmunol.164.11.5970. [DOI] [PubMed] [Google Scholar]
- 8.Hecker M, Sessa WC, Harris HJ, Anggard EE, Vane JR. The metabolism of L-arginine and its significance for the biosynthesis of endothelium-derived relaxing factor: cultured endothelial cells recycle L-citrulline to L-arginine. Proc Natl Acad Sci U S A. 1990;87(21):8612–8616. doi: 10.1073/pnas.87.21.8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie L, Gross SS. Argininosuccinate synthetase overexpression in vascular smooth muscle cells potentiates immunostimulant-induced NO production. J Biol Chem. 1997;272(26):16624–16630. doi: 10.1074/jbc.272.26.16624. [DOI] [PubMed] [Google Scholar]
- 10.Nagasaki A, et al. Coinduction of nitric oxide synthase, argininosuccinate synthetase, and argininosuccinate lyase in lipopolysaccharide-treated rats. RNA blot, immunoblot, and immunohistochemical analyses. J Biol Chem. 1996;271(5):2658–2662. doi: 10.1074/jbc.271.5.2658. [DOI] [PubMed] [Google Scholar]
- 11.Morris SM., Jr Arginine: beyond protein. Am J Clin Nutr. 2006;83(2):508S–512S. doi: 10.1093/ajcn/83.2.508S. [DOI] [PubMed] [Google Scholar]
- 12.Erez A, et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat Med. 2011;17(12):1619–1626. doi: 10.1038/nm.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Husson A, Brasse-Lagnel C, Fairand A, Renouf S, Lavoinne A. Argininosuccinate synthetase from the urea cycle to the citrulline-NO cycle. Eur J Biochem. 2003;270(9):1887–1899. doi: 10.1046/j.1432-1033.2003.03559.x. [DOI] [PubMed] [Google Scholar]
- 14.Bratt JM, Franzi LM, Linderholm AL, Last MS, Kenyon NJ, Last JA. Arginase enzymes in isolated airways from normal and nitric oxide synthase 2-knockout mice exposed to ovalbumin. Toxicol Appl Pharmacol. 2009;234(3):273–280. doi: 10.1016/j.taap.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ckless K, et al. Inhibition of arginase activity enhances inflammation in mice with allergic airway disease, in association with increases in protein S-nitrosylation and tyrosine nitration. J Immunol. 2008;181(6):4255–4264. doi: 10.4049/jimmunol.181.6.4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maarsingh H, Zaagsma J, Meurs H. Arginase: a key enzyme in the pathophysiology of allergic asthma opening novel therapeutic perspectives. Br J Pharmacol. 2009;158(3):652–664. doi: 10.1111/j.1476-5381.2009.00374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris CR, et al. Decreased arginine bioavailability and increased serum arginase activity in asthma. Am J Respir Crit Care Med. 2004;170(2):148–153. doi: 10.1164/rccm.200309-1304OC. [DOI] [PubMed] [Google Scholar]
- 18.Zimmermann N, et al. Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest. 2003;111(12):1863–1874. doi: 10.1172/JCI17912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lara A, et al. Alterations of the arginine metabolome in asthma. Am J Respir Crit Care Med. 2008;178(7):673–681. doi: 10.1164/rccm.200710-1542OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, et al. Genetic polymorphisms in arginase I and II and childhood asthma and atopy. J Allergy Clin Immunol. 2006;117(1):119–126. doi: 10.1016/j.jaci.2005.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salam MT, Islam T, Gauderman WJ, Gilliland FD. Roles of arginase variants, atopy, and ozone in childhood asthma. J Allergy Clin Immunol. 2009;123(3):596–602. doi: 10.1016/j.jaci.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vonk JM, Postma DS, Maarsingh H, Bruinenberg M, Koppelman GH, Meurs H. Arginase 1 and arginase 2 variations associate with asthma, asthma severity and beta2 agonist and steroid response. Pharmacogenet Genomics. 2010;20(3):179–186. doi: 10.1097/FPC.0b013e328336c7fd. [DOI] [PubMed] [Google Scholar]
- 23.Mori M. Regulation of nitric oxide synthesis and apoptosis by arginase and arginine recycling. J Nutr. 2007;137(6 suppl 2):1616S–1620S. doi: 10.1093/jn/137.6.1616S. [DOI] [PubMed] [Google Scholar]
- 24.Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007;2007(407): doi: 10.1126/stke.4072007cm8. [DOI] [PubMed] [Google Scholar]
- 25.Wiesener MS, et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003;17(2):271–273. doi: 10.1096/fj.02-0445fje. [DOI] [PubMed] [Google Scholar]
- 26.Young HW, et al. Central role of Muc5ac expression in mucous metaplasia and its regulation by conserved 5′ elements. Am J Respir Cell Mol Biol. 2007;37(3):273–290. doi: 10.1165/rcmb.2005-0460OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi LZ, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huerta-Yepez S, et al. Hypoxia inducible factor promotes murine allergic airway inflammation and is increased in asthma and rhinitis. Allergy. 2011;66(7):909–918. doi: 10.1111/j.1398-9995.2011.02594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wills-Karp M. Interleukin-13 in asthma pathogenesis. Immunol Rev. 2004;202:175–190. doi: 10.1111/j.0105-2896.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- 30.Allahverdian S, Harada N, Singhera GK, Knight DA, Dorscheid DR. Secretion of IL-13 by airway epithelial cells enhances epithelial repair via HB-EGF. Am J Respir Cell Mol Biol. 2008;38(2):153–160. doi: 10.1165/rcmb.2007-0173OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Semlali A, Jacques E, Koussih L, Gounni AS, Chakir J. Thymic stromal lymphopoietin-induced human asthmatic airway epithelial cell proliferation through an IL-13-dependent pathway. J Allergy Clin Immunol. 2010;125(4):844–850. doi: 10.1016/j.jaci.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 32.Castillo L, Beaumier L, Ajami AM, Young VR. Whole body nitric oxide synthesis in healthy men determined from [15N] arginine-to-[15N]citrulline labeling. Proc Natl Acad Sci U S A. 1996;93(21):11460–11465. doi: 10.1073/pnas.93.21.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kao CC, Bandi V, Guntupalli KK, Wu M, Castillo L, Jahoor F. Arginine, citrulline and nitric oxide metabolism in sepsis. Clin Sci (Lond) 2009;117(1):23–30. doi: 10.1042/CS20080444. [DOI] [PubMed] [Google Scholar]
- 34.Xu W, et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004;18(14):1746–1748. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- 35.Rothenberg ME, et al. Cationic amino acid transporter 2 regulates inflammatory homeostasis in the lung. Proc Natl Acad Sci U S A. 2006;103(40):14895–14900. doi: 10.1073/pnas.0605478103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mabalirajan U, Dinda AK, Sharma SK, Ghosh B. Esculetin restores mitochondrial dysfunction and reduces allergic asthma features in experimental murine model. J Immunol. 2009;183(3):2059–2067. doi: 10.4049/jimmunol.0900342. [DOI] [PubMed] [Google Scholar]
- 37.Trian T, et al. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J Exp Med. 2007;204(13):3173–3181. doi: 10.1084/jem.20070956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu W, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A. 2007;104(4):1342–1347. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dranka BP, Hill BG, Darley-Usmar VM. Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radic Biol Med. 2010;48(7):905–914. doi: 10.1016/j.freeradbiomed.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hampson RK, Barron LL, Olson MS. Regulation of the glycine cleavage system in isolated rat liver mitochondria. J Biol Chem. 1983;258(5):2993–2999. [PubMed] [Google Scholar]
- 41.Sasaki M, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488(7413):656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fijalkowska I, et al. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol. 2010;176(3):1130–1138. doi: 10.2353/ajpath.2010.090832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma P, et al. Redox regulation of interleukin-4 signaling. Immunity. 2008;29(4):551–564. doi: 10.1016/j.immuni.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi O, Morris SM., Jr Zoghbi H, Porter CW, and O’Brien WE. Generation of a mouse model for arginase II deficiency by targeted disruption of the arginase II gene. Mol Cell Biol. 2001;21(3):811–813. doi: 10.1128/MCB.21.3.811-813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Navarro LA, et al. Arginase 2 deficiency results in spontaneous steatohepatitis: a novel link between innate immune activation and hepatic de novo lipogenesis. J Hepatol. 2015;62(2):412–420. doi: 10.1016/j.jhep.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huynh NN, et al. Arginase II knockout mouse displays a hypertensive phenotype despite a decreased vasoconstrictory profile. Hypertension. 2009;54(2):294–301. doi: 10.1161/HYPERTENSIONAHA.108.121731. [DOI] [PubMed] [Google Scholar]
- 47.Kil IS, et al. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol Cell. 2012;46(5):584–594. doi: 10.1016/j.molcel.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 48.Woo HA, et al. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300(5619):653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 49.De Sanctis GT, et al. Contribution of nitric oxide synthases 1, 2, and 3 to airway hyperresponsiveness and inflammation in a murine model of asthma. J Exp Med. 1999;189(10):1621–1630. doi: 10.1084/jem.189.10.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Towle HC. Metabolic regulation of gene transcription in mammals. J Biol Chem. 1995;270(40):23235–23238. doi: 10.1074/jbc.270.40.23235. [DOI] [PubMed] [Google Scholar]
- 51.Fafournoux P, Bruhat A, Jousse C. Amino acid regulation of gene expression. Biochem J. 2000;351(pt 1):1–12. doi: 10.1042/0264-6021:3510001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Niese KA, et al. Bone marrow cell derived arginase I is the major source of allergen-induced lung arginase but is not required for airway hyperresponsiveness, remodeling and lung inflammatory responses in mice. BMC Immunol. 2009;10: doi: 10.1186/1471-2172-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takahashi N, et al. Direct inhibition of arginase attenuated airway allergic reactions and inflammation in a Dermatophagoides farinae-induced NC/Nga mouse model. Am J Physiol Lung Cell Mol Physiol. 2010;299(1):L17–L24. doi: 10.1152/ajplung.00216.2009. [DOI] [PubMed] [Google Scholar]
- 54.Iglesias DE, Bombicino SS, Valdez LB, Boveris A. Nitric oxide interacts with mitochondrial complex III producing antimycin-like effects. Free Radic Biol Med. 2015;89:602–613. doi: 10.1016/j.freeradbiomed.2015.08.024. [DOI] [PubMed] [Google Scholar]
- 55.Xu W, Cardenes N, Corey C, Erzurum SC, Shiva S. Platelets from Asthmatic Individuals Show Less Reliance on Glycolysis. PLoS One. 2015;10(7): doi: 10.1371/journal.pone.0132007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. 2002;277(34):30409–30412. doi: 10.1074/jbc.R200006200. [DOI] [PubMed] [Google Scholar]
- 57.Fong GH, Takeda K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008;15(4):635–641. doi: 10.1038/cdd.2008.10. [DOI] [PubMed] [Google Scholar]
- 58.Asosingh K, Hanson JD, Cheng G, Aronica MA, Erzurum SC. Allergen-induced, eotaxin-rich, proangiogenic bone marrow progenitors: a blood-borne cellular envoy for lung eosinophilia. J Allergy Clin Immunol. 2010;125(4):918–925. doi: 10.1016/j.jaci.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Castillo L, et al. Plasma arginine and citrulline kinetics in adults given adequate and arginine-free diets. Proc Natl Acad Sci U S A. 1993;90(16):7749–7753. doi: 10.1073/pnas.90.16.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu W, et al. STAT-1 and c-Fos interaction in nitric oxide synthase-2 gene activation. Am J Physiol Lung Cell Mol Physiol. 2003;285(1):L137–L148. doi: 10.1152/ajplung.00441.2002. [DOI] [PubMed] [Google Scholar]
- 61.Tserng KY, Kalhan SC. Gas chromatography/mass spectrometric determination of [15N]urea in plasma and application to urea metabolism study. Anal Chem. 1982;54(3):489–491. doi: 10.1021/ac00240a031. [DOI] [PubMed] [Google Scholar]
- 62.Asosingh K, et al. Nascent endothelium initiates Th2 polarization of asthma. J Immunol. 2013;190(7):3458–3465. doi: 10.4049/jimmunol.1202095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.