

Abstract
The balance between protein folding and misfolding is a crucial determinant of amyloid assembly. Transient intermediates that are sparsely populated during protein folding have been identified as key players in amyloid aggregation. However, due to their ephemeral nature, structural characterization of these species remains challenging. Here, using the power of nonuniformly sampled NMR methods we investigate the folding pathway of amyloidogenic and nonamyloidogenic variants of β2-microglobulin (β2m) in atomic detail. Despite folding via common intermediate states, we show that the decreased population of the aggregation-prone ITrans state and population of a less stable, more dynamic species ablate amyloid formation by increasing the energy barrier for amyloid assembly. The results show that subtle changes in conformational dynamics can have a dramatic effect in determining whether a protein is amyloidogenic, without perturbation of the mechanism of protein folding.
Introduction
Protein unfolding is commonly associated with amyloid formation. This view is supported by the large number of intrinsically unfolded proteins that are the causative agents of amyloid diseases, such as α-synuclein, amyloid-β peptide (Aβ), and amylin.1−3 In terms of folded protein precursors, a link between local or global unfolding and the onset of aggregation has been documented.4 Indeed, decreased native state stability and a reduction in co-operativity have been linked with enhanced amyloidogenicity of several proteins, including human lysozyme,5 transthyretin,6 prion protein,7 superoxide dismutase (SOD),8 and β2-microglobulin (β2m).9,10 These findings have guided theoretical approaches to predict aggregation-prone regions of folded proteins by combining the intrinsic propensity of a protein sequence to self-assemble with its probability to become exposed (e.g., by increased predicted hydrogen-exchange rates).11,12 Along similar lines, strategies to stabilize native-like conformations by the use of small molecules,13,14 aptamers,15 molecular chaperones16,17 or via other protein–protein interactions18,19 all reduce amyloid formation. However, a quantitative link between the conformational properties and dynamics of individual partially folded species and amyloid propensity remains elusive, in part because of difficulties in identifying and characterizing amyloid intermediates in atomic detail. In addition, the complexity of the energy landscape of proteins, which involves many potentially amyloidogenic species, exacerbates the intricacy of the system.
In this study we investigate the structural, kinetic, and thermodynamic properties of sparsely populated intermediates of human and murine β2m (hβ2m, mβ2m, respectively) and link their properties to the known, very different amyloid propensities of these proteins.20 Hβ2m forms amyloid fibrils in the joints of patients undergoing hemodialysis, in a pathological condition known as dialysis-related amyloidosis.21,22 Previous studies have examined the link between the folding pathway of β2m and its aggregation propensity. These studies showed that hβ2m does not aggregate at neutral pH unless additives such as Cu2+,23 trifluoroethanol (TFE),24 or other cosolvents,25,26 which partially unfold the protein, are added. As hβ2m contains a thermodynamically unfavorable cis prolyl-peptide bond at position 32, protein folding involves a slow folding phase which is attributed to trans–cis proline isomerization of this bond.9,27 A native-like intermediate containing a trans-Pro 32 (IT) accumulates during folding of hβ2m, which can be trapped by removal of the N-terminal six residues of the protein, to create the variant ΔN6.28,29 At neutral pH, the concentration of IT correlates directly with the rate of amyloid formation of hβ2m,9 suggesting that formation of this on-pathway native-like folding intermediate is a key determinant of amyloid formation. This supposition is confirmed by the ability of ΔN6 to aggregate readily at pH 6.2.28 On the other hand, mβ2m, despite being 70% identical in sequence to hβ2m and also containing a cis-X Pro 32 bond, does not form amyloid fibrils at neutral pH, or even when unfolded under acidic conditions (unless high concentrations of salt are added) (Figure 1A and 1B).19,30 The basis of such dramatically different amyloid propensities despite the sequence and structural similarities of these two proteins remained unclear.
Figure 1.

Structure and amyloidogenicity of mβ2m. (A) The NMR structure of monomeric hβ2m (gray-2XKS28) overlaid with the crystal structure of mβ2m bound to the MHC-I complex (green-1LK238) or with the solution structure of ΔN6 (red-2XKU28). (B) Aggregation assay of 80 μM hβ2m (gray), mβ2m (green), or ΔN6 (red) in 10 mM sodium phosphate buffer, pH 6.2 (three replicates for each protein). Negative stain electron micrographs, color-coded with the same scheme, are shown on the right (scale bar = 500 nm). (C) The 1H–15N HSQC spectrum of mβ2m in the same buffer as (B). (D) Correlation between experimental RDCs measured for mβ2m in 10 mg/mL phage PF1 and those back-calculated from the crystal structure 1LK238 (R2 = 0.85).
Here, we set out to investigate the molecular origins of the reduced amyloidogenicity of mβ2m. We characterize the stability, structure, and dynamics of the native protein and show that despite its inability to form amyloid, mβ2m is kinetically and thermodynamically less stable than its human counterpart. The folding pathway of mβ2m is then explored using real-time NMR, taking advantage of the power of nonuniformly sampling (NUS) methods to reveal detailed information on the energy landscape of mβ2m folding. Combined with other biophysical methods, we show that while mβ2m also folds through an IT state, this species is relatively more flexible than its hβ2m counterpart and in conformational exchange with other, less-structured non-native states.
The molten globule-like characteristics of the mβ2m folding intermediate reduce the lifetime of the structured, well-folded IT state, which now represents only a minor substate in the structural ensemble. Our findings confirm the vital role of IT (and hence a highly structured, yet non-native species) in determining the aggregation of β2m. Moreover, the results highlight the importance of defining the energy landscape of amyloidogenic proteins in detail to allow prediction of their amyloid propensity. The findings presented also suggest that targeting a defined non-native species should be a successful means of controlling the fate of assembly of β2m and, in principle, that of other amyloidogenic proteins which aggregate via a specific, non-native precursor.
Methods
Protein Preparation
The pINK plasmid containing the hβ2m, mβ2m, or ΔN6 gene was transformed into E. coli cells of the BL21 DE3 plysS- strain. Starter cultures were generated by inoculating 100 mL of LB medium with cells containing the relevant gene and 50 μg/mL carbenicilin and 50 μg/mL chloramphenicol and incubating overnight at 37 °C, 200 rpm. 2 L flasks containing 1 L of LB or HDMI (1 g/L 15N-NH4Cl, 2 g/L 13C-glucose) medium were inoculated with 10 mL of starter culture. Cells were incubated at 37 °C, 200 rpm until they reached an OD600 of ∼0.6 and then the expression of β2m was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG-final concentration of 1 mM). Expression was allowed to continue overnight at 37 °C, and cells were harvested next morning using a Heraus continual action centrifuge performing at 15 000 rpm. The cell pellet containing β2m as inclusion bodies was chemically lysed by the addition of 50–100 mL of lysis buffer (100 μg/mL lysozyme, 50 μg/mL DNase I, 50 μg/mL phenylmethylesulfonyl fluoride (PMSF), 10 mM Tris-HCl pH 8.0). Further cell disruption was performed using a constant cell disrupter system (Constantsystems) at a high pressure of 20.0 kpsi. Inclusion bodies were separated using centrifugation (15 000 rpm using a Sorvall SS34 rotor) in a Beckman centrifuge for 40 min at 4 °C, and the inclusion body pellet was washed with 10 mM Tris-HCl pH 8.0 buffer four times. Finally, β2m was solubilized in 10–20 mM Tris-HCl pH 8.0 (hβ2m, ΔN6) or 10–20 mM Tris-HCl pH 8.5 (mβ2m) containing 8 M urea (MP biomedicals) and refolded by dialysis (3000 MW cutoff) against 2–5 L of the same buffer but lacking urea. The refolded protein was centrifuged for 30 min at 15000 rpm (Sorvall SS34 rotor) to pellet insoluble material, and the supernatant was loaded on a Q-Sepharose (GE Healthcare) column already equilibrated with 2 column volumes of 20 mM Tris-HCl pH 8.0 (hβ2m, ΔN6) or 20 mM Tris-HCl pH 8.5 (mβ2m) for anion exchange purification. Bound protein was eluted with a gradient of 0–400 mM NaCl (in the same buffer) over 800 mL and was freeze-dried after dialysis in dH2O or concentrated using 3000 MW cutoff centricons (Avanti LTD). Freeze-dried protein was resuspended in 10 mM sodium phosphate buffer pH 7.0 (hβ2m, ΔN6) and 10 mM sodium phosphate buffer pH 8.2 (mβ2m) filtered through 0.2 μm filters (Fisher Scientific) and gel-filtered using a HiLoad Superdex-75 Prep column (Amersham Biosciences), calibrated with a standard gel filtration calibration kit (GE Healthcare). The monomer peak was collected, concentrated, aliquoted, and stored at −80 °C or freeze-dried.
NMR Spectroscopy
Assignments of the backbone atoms of mβ2m were obtained using samples of 500 or 750 μM uniformly labeled (15N, 13C) protein in 10 mM sodium phosphate buffer pH 6.2, 83.3 mM NaCl, 0.02% (w/v) NaN3, and 10% (v/v) D2O. Three-dimensional (3D) NMR experiments were recorded at 25 °C using Varian Inova spectrometers (Agilent) operating at proton frequencies of 500 MHz (HNCA, HNCO, CBCA(CO)NH, HN(CA)CO) and 750 MHz (HNCACB), equipped with a room temperature or cryogenic probe, respectively. Samples for H/D exchange experiments were prepared in 10 mM sodium phosphate buffer pH 6.2 and then freeze-dried. Freeze-dried protein was dissolved in 100% (v/v) D2O containing 83.3 mM NaCl and placed into the NMR tube after manual mixing. The loss of intensity of amide proton resonances was then monitored by SOFAST 1H–15N HSQC31 spectra (5–10 min each) at 25 °C. Residual dipolar coupling (RDC) experiments (J modulated series) were carried out using a sample of 200 μM 15N-mβ2m in 10 mM sodium phosphate pH 8.2 and aligned in 11 mg/mL bacteriophage PF1 (Asla Scientific). RDC data were back-calculated from crystal structures using PALES.32 For relaxation experiments, a sample of 80 μM 15N-mβ2m was prepared in 10 mM sodium phosphate buffer at pH 6.2 with 10% (v/v) D2O, 0.02% (w/v) NaN3, 83.3 mM NaCl. T2 experiments were perfomed with 2048 and 128 complex points in the direct and indirect dimension using 18.8, 37.7, 56. 6, 75.5, 94.4, 113.2, 132,1, 151.0 ms as relaxation delays. The relaxation delays for the T1 experiments were 0.0.16, 0.32, 0.48, 0.64, 0.80, 0.96, 1.12, 1.44 s. For 1H–15N NOE cross-relaxation experiments saturation of amide protons was achieved with a train of 120° pulses for 3.5 s prior to the experiment. All relaxation and RDC experiments were performed at 25 °C, using a 600 MHz Varian Inova spectrometer equipped with a room tempreature probe. Direct carbon detection experiments were performed using a sample of 13C, 15N mβ2m at 1.3 mM on a 950 MHz Bruker spectrometer equipped with a cryogenic TCI probe (1024 and 160 complex points in the direct and indirect dimensions).
For real-time refolding experiments two refolding protocols were followed: (1) protein samples were made in 10 mM sodium phosphate buffer pH 6.2 and freeze-dried. Unfolding was performed by dissolving the freeze-dried protein (2–3 mg) in 30–60 μL of the same buffer containing 8 M urea at 37 °C for 1 h, and the protein was then refolded by rapid 10-fold dilution in 10 mM sodium phosphate pH 6.2, 10% (v/v) D2O, and 0.02% (w/v) NaN3; (2) protein samples were made in 250 μL of 10 mM sodium phosphate and 10% (v/v) D2O, and the pH was adjusted to 2.0 (or pH 3.6) using Tris-HCl. Refolding was then initiated by addition of 50 μL of 300 mM sodium phosphate buffer, pH 7.2 (final pH 6.1–6.3). Both refolding protocols were found to give rise to similar spectra of the intermediate species. This observation demonstrates that the increased flexibility of the murine intermediate (see Results) is not the result of the residual 0.8 M urea present at the end of the first refolding protocol. The refolding from IT to native hβ2m was monitored by a series of SOFAST 1H–15N HSQC spectra31,33 at 25 °C, with 80 increments in the indirect dimension, two scans per increment and 512 complex points, resulting in a total acquisition time of 45 s.
To assign the I1 state of mβ2m, refolding was monitored by continuous acquisition of NUS NMR spectra. 3D HNCO+ and HNCA+34 with a total acquisition time of ca. 17 h for each spectrum and 2D 1H–15N BEST-TROSY35 were collected on separate samples (800 MHz Bruker AVANCE III HD spectrometer with 3 mm TCI cryoprobe). 2D 1H–15N BEST-TROSY, having the highest sensitivity, was chosen as a reference spectrum to guide 3D spectra multidimensional decomposition (MDD) coprocessing with a sliding time frame window, resulting in a temporal resolution of a few minutes36 (see Supplementary Methods).
To aid assignment of the real-time spectra, two samples were prepared in which the early intermediate of mβ2m was highly populated. The first consisted of 600 μM of uniformly labeled 13C, 15N mβ2m in 10 mM sodium phosphate and 10 mM sodium acetate pH 3.6, and the second consisted of 250 μM of uniformly labeled 13C, 15N mβ2m in 10 mM sodium phosphate pH 6.2 with 1 M urea added. Both samples gave rise to HNCA spectra that closely resembled the real-time HNCA spectrum of the early intermediate of mβ2m. Additional 3D spectra were performed using these samples including HNCA, HNCO, and CBCACONH utilizing a 600 MHz Varian Inova spectrometer equipped with a room tempreature probe. TALOS+37 was used to predict the backbone order parameter (S2). TALOS+ uses H, NH, Co, Cα, and Cβ backbone chemical shifts to calculate the random coil chemical shift index which is then converted to backbone S2. Aggregation assays performed on these samples confirmed that the early folding intermediate of mβ2m is not aggregation-prone.
Aggregation Assays
Samples containing 60 μM protein in 10 mM sodium phosphate buffer, pH 6.2, or 10 mM sodium phosphate, with 10 mM sodium acetate pH 3.6, or in 10 mM sodium phosphate buffer, pH 6.2, with 1 M urea, with the appropriate amount of NaCl added to give a total ionic strength of 100 mM), 0.02% (w/v) NaN3 and 10 μM Thioflavin T (ThT) were incubated at 37 °C in sealed 96 well plates (Thermo Scientific) with agitation at 600 rpm. Fluorescence was monitored at 480 ± 10 nm after excitation at 440 ± 10 nm using a FLUOROstar Optima microplate reader (BMG Labtech).
Equilibrium Unfolding
Urea stock solutions containing 75 mM sodium phosphate buffer, pH 6.2, and either no urea or 10.5 M urea were made, and the exact concentration of urea was determined using the measured refractive index (Ceti refractometer). The stock solutions were used to make samples of protein containing 0–10 M urea in 0.2 M increments, with a final protein concentration of 0.2 mg/mL. Samples were incubated at 25 °C for 12 h before analysis using tryptophan fluorescence. Fluorescence was excited at 295 nm, and the emission was monitored at 340 nm using a Photon Technology International QM-1 spectrofluorimeter (PTI). The data were then globally fit to a two-state model:
| 1 |
where ΔGoUF (kJ mol–1) is the equilibrium stability, MUF is the equilibrium m-value, a and c represent the denaturant-dependence of the folded and unfolded signal intensities, respectively, and b and d are the signal intensities of the folded and unfolded states, respectively, in the absence of denaturant.
Electron Microscopy (EM)
Carbon coated copper grids were prepared by the application of a thin layer of Formvar with an overlay of carbon. Samples were centrifuged (14 000g, 10 min), and the pellets were resuspended in fresh 10 mM sodium phosphate buffer, pH 6.2, diluted to a final protein concentration of 12 μM with deionized water and then applied to the grid in a dropwise fashion. The grid was then carefully dried with filter paper before it was negatively stained by the addition of 18 μL of 2% (w/v) uranyl acetate. Micrographs were recorded on a Philips CM10 or a JEOL JEM-1400 electron microscope.
Stopped Flow Experiments
Experiments were performed using an Applied-Photophysics SX1.8MV stopped-flow fluorimeter. The temperature was held constant at 37 °C (±0.1 °C) using a Neslab circulating water bath system. Experiments were performed in buffered solutions containing 10 mM sodium phosphate (pH 6.2) and 83.3 mM sodium chloride with or without 0.4 M sodium sulfate. Refolding experiments were performed by 1:10 dilution of 80 μM protein in buffer containing 8 M urea, into buffered solutions with final urea concentrations in the range 0.75–8.0 M. The final urea concentration ranged from 3.0 to 8.0 M for unfolding experiments. To obtain refolding data at 0 M urea, a pH jump was performed by a 1:10 dilution of 80 μM protein in 10 mM phosphate (pH 2.5) into 80 mM sodium phosphate (pH 6.2). Data were normalized to the signal of the folded and unfolded protein in 0 and 8 M urea, respectively. At each urea concentration at least seven kinetic traces were obtained, averaged, and fitted to a single exponential function using the manufacturer’s software. ΔGoUI1 was determined by plotting the fluorescence at the end point of a 20 s kinetic trace of folding against urea concentration and by plotting the fluorescence of the unfolded state against urea concentration (in the latter case, the values at low urea concentration were obtained by linear extrapolation from the values at high urea concentration). The fluorescence of the IT state decreased with increasing urea concentration until it approached the fluorescence of the unfolded state. To estimate ΔGoUI1 at 0 M urea, data were also recorded in the presence of 0.4 M Na2SO4 and the data in 0 and 0.4 M Na2SO4 were fitted globally to eq 1.
Results
The native state of mβ2m is thermodynamically unstable
As well as being 70% identical in sequence, mβ2m and hβ2m/ΔN6 have similar structures (backbone RMSD ≈ 1.5 Å) (Figure 1A). However, only ΔN6 is able to aggregate into amyloid fibrils at pH 6.2 as monitored by the increase in ThT fluorescence and by negative stain EM (Figure 1B), in agreement with previous studies.19 To enable NMR studies of mβ2m, chemical shift assignments were obtained for the native monomeric protein using a range of 3D NMR experiments (BMRB 19772) (85% of backbone atoms were assigned; see Methods). The 1H–15N HSQC spectrum of native mβ2m at pH 6.2 shows a single set of well-dispersed intense peaks, characteristic of a folded protein in solution that undergoes limited chemical exchange on the ms time scale (Figure 1C).
The only available structure of mβ2m is the crystal structure of the protein bound to the heavy chain of the major histocompatibility complex (MHC-I).38 In the case of hβ2m, binding to the heavy chain causes conformational changes particularly in the AB loop and the D strand of the protein.39 To determine whether the crystal structure of mβ2m bound to the MHC-I complex constitutes a good representation for the structure of the monomeric protein in solution, residual dipolar couplings (RDC) were measured. Figure 1D shows that there is excellent agreement (R2 = 0.85) between the measured RDCs and those back-calculated from the crystal structure of MHC-I-bound mβ2m (Figure 1D), confirming the identity of the solution structure of monomeric mβ2m with that bound to MHC-I. Therefore, differences in the structures of the native proteins cannot explain the different amyloid potential of the two variants.
We next assessed whether differences in thermodynamic and/or kinetic stability between mβ2m and hβ2m/ΔN6 could rationalize their different amyloid propensity. Equilibrium denaturation experiments revealed that mβ2m is less stable than hβ2m (ΔΔG°un = −12.4 kJ/mol) (Figure 2A, 2B and Supplementary Table 1). Remarkably, mβ2m is less stable than the aggregation prone ΔN6 (Figure 2A, 2B) demonstrating that thermodynamic instability cannot explain the inability of mβ2m to form amyloid. Notably, ΔN6 shows a reduced m-value compared with hβ2m and mβ2m, consistent with exposure of hydrophobic residues that are normally buried in the core of hβ2m (10 of the 17 core residues become more exposed in ΔN628). Hydrogen exchange experiments monitored by 1H–15N HSQC spectra revealed that mβ2m is also kinetically less stable than hβ2m, with amide protons exchanging with the solvent more rapidly than hβ2m, while ΔN6 is the least kinetically stable of the three proteins studied here (Figure 2C, Figure S1A–C, and Figure S2). NMR relaxation experiments on native mβ2m also showed no regions of increased dynamics on the ps–ns time scale, apart from the DE loop which is known to be flexible in all β2m variants (Figure S2D–F). Thus, there is no correlation between thermodynamic or kinetic stability and amyloidogenicity of these different β2m variants.
Figure 2.
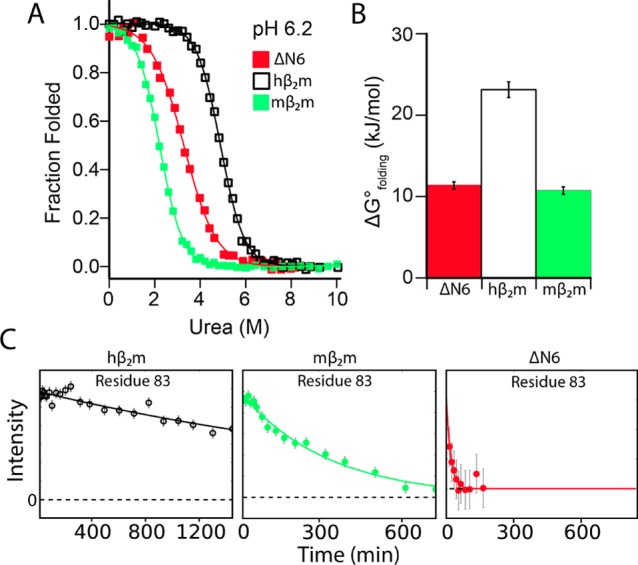
Thermodynamic and kinetic stability do not correlate with amyloid propensity. (A) Equilibrium denaturation curves for mβ2m, hβ2m, and ΔN6 monitored using tryptophan fluorescence (75 mM sodium phosphate buffer pH 6.2, 25 °C). (B) Unfolding free energies obtained by fitting data in (A) to a two state model (see Table S1). (C) Representative hydrogen exchange profiles for the amide hydrogen of residue 83 in hβ2m, mβ2m, and ΔN6 at 25 °C and pH 6.2 (see Figure S2).
Real-Time Characterization of a Transient Folding Intermediate
We next investigated whether the folding pathway of mβ2m also involves transient formation of an intermediate containing trans X-Pro 32 (known as the IT state28), the accumulation of which has been shown to correlate directly with the rate of amyloid formation of the human protein.9 For these experiments, mβ2m was unfolded either by incubating the protein at pH 2.0 or by the addition of 8 M urea at pH 6.2. Refolding was then initiated by dilution to a buffer of pH 7.2 or to a buffer lacking urea (see Methods, Figure S3), and NMR spectra were collected in real time to track the refolding reaction in residue-specific detail. In the case of hβ2m, and in accordance with previous studies,28 a well-dispersed spectrum was observed 3 min after refolding was initiated, in which only small chemical shift differences are detected in comparison with those of the native protein (Figure 3A and 3B). These results indicate that a native-like intermediate (the IT state) accumulates during folding of hβ2m, consistent with previous results.9,28
Figure 3.

Folding of hβ2m and mβ2m in real-time. (A) 1H–15N SOFAST HSQC spectrum of native hβ2m in 10 mM sodium phosphate pH 6.2. (B) The 1H–15N SOFAST HSQC spectrum of hβ2m collected 3 min after refolding was initiated by urea dilution. Peaks that are already in their native positions are colored gray, while peaks with non-native chemical shifts are shown in red. (C) The 1H–15N HSQC spectrum of ΔN6 closely resembles the spectrum of the real-time folding intermediate of hβ2m. An overlay of the spectra shown in B and C is shown as an inset. (D) 1H–15N HSQC spectrum of native mβ2m in 10 mM sodium phosphate pH 6.2. (E) The 1H–15N BEST-TROSY-HSQC of mβ2m collected 3 min after refolding was initiated by urea dilution. Peaks that are already in their native positions are colored green while peaks with non-native chemical shifts are shown in purple. (F) The 1H–15N HSQC spectrum of mβ2m at pH 3.6 closely resembles the spectrum of the real-time folding intermediate of mβ2m.
As previously reported, the spectrum of ΔN6 is very similar to that of the human IT state (Figure 3C).28 In marked contrast with the behavior of hβ2m, however, the 1H–15N HSQC spectrum of mβ2m 3 min after refolding is initiated revealed only ∼20 intense peaks with limited chemical shift dispersion in the 1H dimension which coexist with the most intense peaks of the native state (Figure 3D and 3E). Interestingly, the partially folded state of mβ2m at pH 3.6 gives rise to a 1H–15N HSQC spectrum that closely resembles the spectrum collected at pH ≈ 6.2, 3 min after refolding was initiated (Figure 3F). These results suggest that partially folded species are significantly populated during the folding of mβ2m, but that these species differ in structure and/or dynamics compared with their human counterparts.
In order to assign the real-time spectrum of the intermediate state of mβ2m, continuous, NUS NMR spectra (2D-BEST-TROSY HSQC, 3D-HNCA+, and 3D-HNCO+) were collected during refolding and the whole data set was coprocessed together, resulting in a temporal resolution of a few minutes.36 Importantly, and in contrast with other real-time studies of protein folding which consist of acquisition of sequential stand-alone spectra,40 this NUS approach requires the acquisition of only a single spectrum. Therefore, it does not require prior knowledge about the folding reaction in order to decide on the length of each individual experiment, since the time resolution can be determined in the processing step. Additionally, increased sensitivity is achieved by coprocessing less sensitive 3D NMR spectra (e.g., HNCA) with 2D experiments (e.g., HSQC).41
The same set of intense resonances shown in Figure 3E were observed in 3D real-time spectra, consistent with these residues being flexible in the folding intermediate of mβ2m. As only 20 spin systems are present in the real-time HNCA spectrum, assignment was challenging. To overcome this issue, further backbone assignment experiments were performed on mβ2m at pH 3.6 (Figure 3F) and mβ2m at pH 6.2 with 1 M urea added, both of which gave rise to spectra similar to those of the kinetic intermediate (Figure 3F and data not shown), removing the need for rapid data acquisition (see Methods). The residue-type specific information of the Cβ atoms from these equilibrium experiments greatly facilitated the assignment of the real-time HNCA spectrum.
The assignment walk on the Cα resonances of mβ2m 5 min after folding was initiated is shown in Figure 4A. The assignment revealed that all of the intense peaks shown in Figure 3E correspond to residues located in the N-terminal region, the A strand, and the AB loop of mβ2m (Figure 4B and 4C), regions of the polypeptide chain whose dynamics have been implicated in the initiation of the aggregation of the human protein.19,28 The C-terminal four residues of the protein were also detected with chemical shifts that are different (ΔδH+N > 2 ppm) from those in the native structure. The backbone assignments (N, NH, Cα, Cβ, CO atoms; Supplementary Table 2) allow the accurate prediction of the order parameter S2 and, therefore, an assessment of protein dynamics. Figure 4B and 4C show that the 20 N-terminal residues show significantly increased dynamics in the folding intermediate in comparison with the native state, while the C-terminal four residues, while visible, have conformational dynamics similar to those of the native protein.
Figure 4.
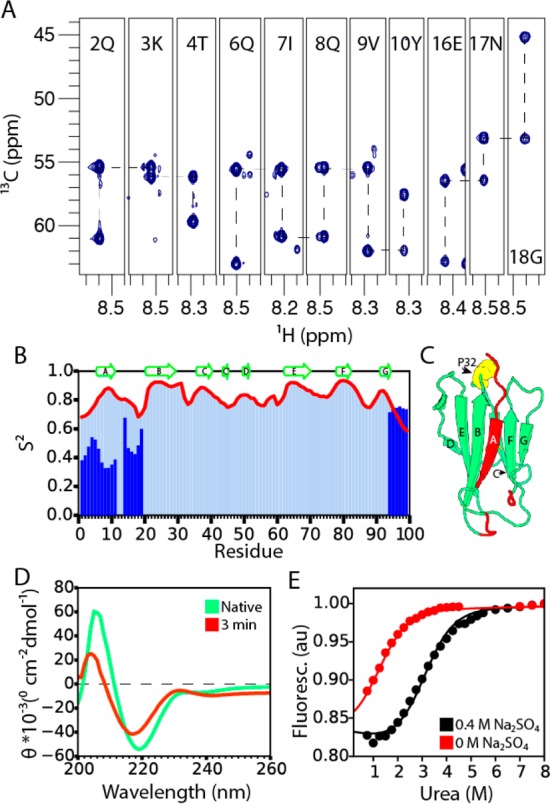
The early folding intermediate of mβ2m. (A) The assignment walk on the Cα atoms of the real-time BEST-HNCA spectrum of mβ2m collected 5 min after refolding was initiated by urea dilution at pH 6.2. (B) Predicted S2 parameters for the folding intermediate of mβ2m. The intense peaks shown in Figure 3E are shown in dark blue. The S2 parameters of the native state are shown as light blue bars and as a red line. (C) Residues observed in the real-time spectrum of the folding intermediate are highlighted in red in the structure of mβ2m. Pro 32 is shown as yellow spheres. (D) Far-UV CD spectra of native mβ2m at pH 6.2 (green) and of the folding intermediate 3 min after folding was initiated by a pH jump from pH 2.0 to pH 6.2 (red). (E) Normalized fluorescence signal at the end of each stopped-flow transient (20 s) as a function of urea concentration for mβ2m in the absence (red) or presence (black) of 0.4 M sodium sulfate. The data were fitted globally to a two-state model (see Methods).
The molten-globule-like behavior of the intermediate state (named here I1) prevents the direct observation of the majority of the resonances (apart from the 20 most flexible) in 1H-detected experiments, as they are in conformational/solvent exchange. To overcome this problem direct 13C-detection experiments were performed on mβ2m at pH 3.6 in order to obtain information about the structural properties of the rest of the protein in the I1 state. These experiments revealed ∼80 resonances, 20 of which have sharp lines which correspond to the 20 N-terminal residues assigned above. Other resonances show broader lines suggesting that they correspond to residues with a higher degree of folding (Figure S5). The far-UV CD spectrum of mβ2m obtained 3 min after the pH jump from pH 2.0 to pH 6.2 shows that in I1 73% of native β-sheet structure is already formed, as quantified by the ratio of the intensities at 219 nm (Figure 4D). These results show that mβ2m at pH 3.6 is partially structured, with the majority of residues being in a β-sheet conformation, potentially native-like. These residues undergo chemical exchange on the ms time scale (or exchange rapidly with solvent) and, therefore, cannot be observed in the 1H–15N spectrum shown in Figure 3E.
In order to estimate the stability of the I1 state, stopped-flow fluorescence experiments were used to determine the fluorescence intensity of mβ2m 20 s after folding was initiated (Figure 4E). These experiments revealed that, by contrast with the IT state of hβ2m for which the ΔG°un is −9.57 ± 0.54 kJ/mol at 37 °C,9 the I1 state of mβ2m (ΔG°un = ∼ –4.8 kJ/mol at 37 °C) is only marginally stable in solution, in accordance with the real-time NMR data (Figure 4E). Overall, the data show that mβ2m folds through a flexible/molten globule-like intermediate state in which the N-terminus and the A strand are dynamic and detached from a native-like β-sandwich fold (I1) (Figure 4C).
The flexible intermediate I1 is not aggregation-prone
The amyloid fibrils of hβ2m are composed of parallel in register β-strands,42,43 while in the native monomer the β-strands are all antiparallel (Figure 1A). Thus, a major conformational change has to occur on the pathway to fibrils. Detachment of the A strand might represent a first step toward the remodeling of the native protein, and therefore, the early intermediate of mβ2m, I1, might be expected to be highly amyloidogenic. To test this hypothesis, aggregation assays were performed using ΔN6 at pH 6.2 as a mimic of the highly aggregation-prone state IT, and mβ2m at pH 3.6, conditions which favor the less structured intermediate state (I1) of mβ2m. Consistent with previous results, these experiments showed that ΔN6 aggregates rapidly at pH 6.2 with a lag time of ∼30 h, resulting in the formation of amyloid fibrils (Figure 5A). In marked contrast, no increase in ThT fluorescence was observed for mβ2m at pH 3.6 (Figure 5A). Indeed, the majority of the murine protein remained soluble after 100 h of incubation, while ΔN6 was quantitatively converted into amyloid fibrils (Figure 5B and 5C). Interestingly, the small amount of mβ2m that was not found in the supernatant also formed short fibrils ∼300 nm in length (Figure 5C). These results show that the partially folded state of mβ2m is not highly aggregation prone. On the other hand, the specific structural features of the native-like IT intermediate of hβ2m are crucial for assembly.
Figure 5.
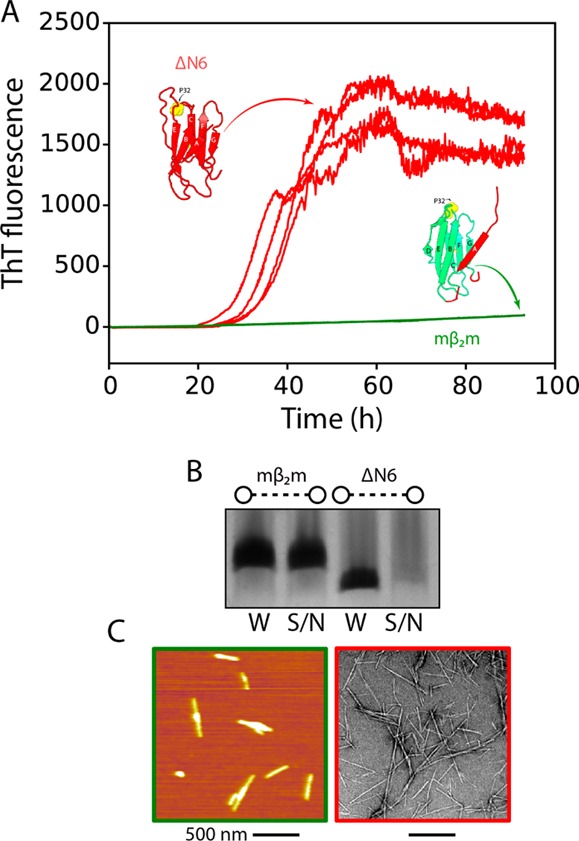
Amyloidogenicity of intermediate states of mβ2m and hβ2m. (A) ThT fluorescence assays of 60 μM ΔN6 in 10 mM sodium phosphate buffer pH 6.2 (red) or 60 μM mβ2m in 10 mM sodium phosphate, 10 mM sodium acetate pH 3.6 (green). Five replicates are shown for each protein. (B) SDS-PAGE gel of the end points shown in (A). 20 μL of the reaction were spun down using a benchtop centrifuge for 20 min and a sample of the total reaction (W) or of the supernatant (S/N) after centrifugation, was analyzed by SDS-PAGE. (C) AFM image of 60 μM mβ2m after 95 h of incubation (green in A, note that fibrils correspond to <5% of the protein added) and electron micrograph of 60 μM ΔN6 (fibrils correspond to >95% of the protein added) after 95 h of incubation (red in A). Scale bar represents 500 nm.
A native-like IT intermediate is populated on the pathway to native mβ2m
The data presented above demonstrate that the conformational properties of the dynamic I1 state of mβ2m are different from those the native-like hβ2m IT state. However, additional states could be populated after the formation of I1 and prior to the formation of native mβ2m. Indeed, as the folding time progresses a third set of peaks (apart from the native and the flexible intermediate states (Figure 3)) emerges in the real-time 1H–15N HSQC spectrum (Figure S4). These peaks show small chemical shift differences compared with the native mβ2m resonances and are generally broad (Figure S4) suggesting that additional, more native-like states are populated at later times during the folding of the protein. This observation presumably reflects an ordered assembly mechanism, in which the initially highly dynamic intermediate (I1) folds to the native state via a transiently populated more structured (IT-like) state.
To investigate this possibility further, partially folded mβ2m at pH 3.6 (which mimics the I1 state – Figures 3E and 3F) was allowed to fold by a rapid pH jump to pH 6.2 and folding was monitored in real time using NMR. The 1H–15N HSQC spectrum collected 3 min after refolding was initiated showed a well dispersed spectrum (Figure 6A) in striking and marked contrast with the molten globule-like spectrum of the I1 state shown in Figure 3E. Indeed, the spectrum obtained 3 min after the pH jump is reminiscent of that of native mβ2m with significant chemical shift differences being limited to residues in the N-terminal region (residues 1–6), the BC, DE, and FG loops (Figure 6A and 6B). Moreover, the peaks that show chemical shift differences from the native protein are remarkably similar to the third set of peaks observed in Figure S4B, consistent with folding from the partially folded state (I1) to a more native-like intermediate (presumably IT). Importantly, residues that show significant chemical shift changes are surrounded by residues whose resonances are not detected in the real-time NMR spectrum (Figure 6B). These areas reside in close spatial proximity to Pro 32 in the native structure and, therefore, are exchange-broadened. A similar scenario has been observed for the real-time folding intermediate of hβ2m (IT).40 Furthermore, ΔN6 shows chemical shift differences to native hβ2m in these same regions.28 Together, the results show that mβ2m folds through a native-like intermediate state that has similar structural properties to the amyloidogenic IT state of hβ2m. However, due to its decreased stability, this species is copopulated with an ensemble of partially folded, flexible states in which the N-terminal region is highly disordered (I1).
Figure 6.
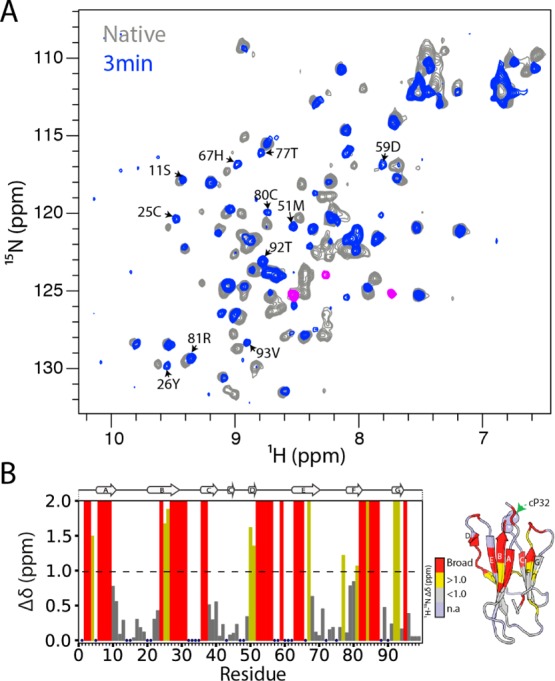
Detection of a native-like folding intermediate for mβ2m. (A) The 1H–15N SOFAST HSQC spectrum of mβ2m collected 3 min after refolding was initiated from the partially folded state at pH 3.6 (blue), overlaid with the 1H–15N SOFAST HSQC of native mβ2m (collected after 90 min) (gray). Peaks corresponding to the molten globule I1 state are colored in pink. (B) Per residue combined 1H–15N chemical shift differences between the native and the IT state of mβ2m, using the spectra displayed in (A). Blue dots represent residues for which assignments are missing in the native spectrum. Residues that show chemical shift differences greater than 1 ppm (dashed line) are colored yellow, those that show chemical shift differences less than 1 ppm are shown in gray, and residues that are broadened beyond detection in the 3 min spectrum are colored red. The strucure of mβ2m colored in the same color scheme is shown on the right.
Discussion
Characterization of Protein Energy Landscapes Using Sparse Data
The results presented above highlight the importance of determining the precise details of the folding energy landscape of a protein in order to elucidate whether one or more partially folded, or non-native states, have the potential to initiate amyloid assembly. Characterization of ensembles of interconverting non-native species that not only are lowly populated but also have a short lifetime, such as those involved in protein folding and aggregation, is a challenging task, even for the most advanced biophysical methods. While time-resolved NMR studies on proteins can be highly informative, these studies usually suffer from low resolution and poor sensitivity. By combining the use of sparsely sampled NMR and coprocessing of more complicated/less sensitive experiments with others that show increased sensitivity, we demonstrate here the detection and atomic level depiction of the early molten globule state, I1, of mβ2m, that is populated for only ∼15 min, a task that would not have been possible using standard NMR methodologies (see Supplementary Methods). The power of the real-time NMR experiments allowed us to identify conditions that stabilize the I1 state (pH 3.6 or addition of 1 M urea at pH 6.2) and to perform more detailed NMR experiments on the trapped intermediate state that led to a complete description of the folding mechanism of the murine protein. The approach is potentially applicable to other amyloidogenic proteins or proteins that fold through the accumulation of transient intermediate states.
The precise balance between folding intermediates determines the aggregation propensity of β2m
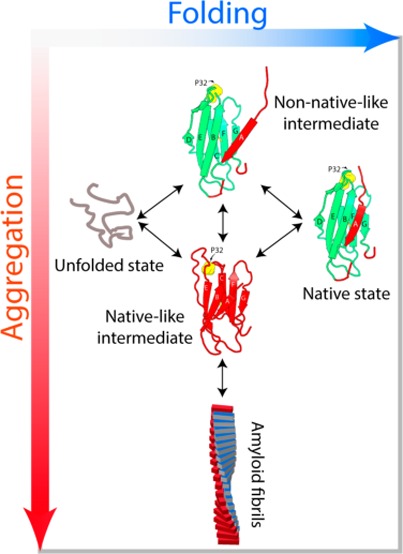
The folding pathway of hβ2m has been investigated in detail over the past decade using different protein variants and different techniques.9,28,44−48 Together, these studies have shown that the folding of hβ2m involves the formation of a native-like intermediate IT that is kinetically trapped by virtue of the non-native trans X-Pro 32 bond.9,47 This species is preceded by a less well characterized species (I1)44 that forms in the dead time of a stopped flow experiment (<3 ms) and is less structured than the IT state (Figure 7). The fine details of the exchange processes between these different folding intermediates has a dramatic effect on the propensity to aggregate. For hβ2m the structured aggregation-prone IT state is the most highly populated species during folding, accumulating, on average, to ∼4% (pH 7.0, 37 °C) at equilibrium.9,10 By contrast, the flexible I1 state represents the most highly populated intermediate state during the folding of mβ2m. Indeed, the most intense peaks of the I1 state (but not those of the IT state) are visible in the spectrum of native mβ2m, enabling estimation of its equilibrium concentration to ∼7% (Figure S6). This reduces the population of the mβ2m IT state, with the effect that aggregation no longer occurs (at least on an experimentally tractable timescale) (Figure 7). Thus, although the folding mechanisms of human and murine β2m are conserved (the same species are populated on the pathway to the native state), the precise balance between these states reduces the population of the key amyloidogenic precursor IT for mβ2m and thus defines the course of amyloid formation.
Figure 7.

Balance between protein folding and aggregation. The native-like IT state is predominantly populated during the folding of hβ2m (gray scheme, left). This allows the entrance to the aggregation landscape (red scheme) as IT shows enhanced amyloidogenicity. In the case of mβ2m (right) IT represents only a minor conformation during folding, but instead the flexible molten globule-like state in which the A strand is detached from the β-sandwich fold is the major non-native species. As I1 is not aggregation-prone, mβ2m is protected from misfolding and instead folds to the native state (the energy levels of the aggergation landscape are drawn for illustration purposes only).
Interestingly, urea denaturation experiments on a partially folded state of hβ2m formed at pH 3.6 have shown that the N-terminal six residues and the A strand are the least stable regions, while the rest of the protein forms a stable core.49 These results are consistent with the real-time NMR studies on the I1 state of mβ2m presented here, showing that the conformational properties of the early partially folded states of hβ2m and mβ2m are similar. Interestingly, neither of these states is able to form long, straight fibrils characteristic of amyloid, but instead they form short rod-like fibrils (mβ2m at pH 3.6) or worm-like fibrils (hβ2m at pH 3.6)50 (Figure 5).
A direct link between decreased native state stability and increased aggregation propensity has been observed for several proteins including lysozyme,51 transthyretin,6 and antibody light chains.52 Accordingly, destabilizing mutations enhance the rate of exchange between the native protein and partially folded non-native species, which show increased amyloidogenicity compared with the native state. Interestingly, mβ2m is less thermodynamically and kinetically stable than hβ2m and, as a consequence, the molten globule-like nonamyloidogenic I1 state is the most abundant non-native species (Figure 7). The increased conformational dynamics results in a protein that is unstable yet protected from amyloid assembly, since I1 is not able to form amyloid. These findings argue against a simple link between native state stability and amyloidogenicity (at least for β2m). Instead, they highlight the importance of the precise conformational properties of the native-like IT state that are vital for assembly.
Together, the results demonstrate that amyloid formation of β2m at neutral pH is initiated via the highly structured IT state. Hence, from the myriad of potential non-native conformations that could be populated during folding, only the IT state allows the entrance of β2m to the aggregation landscape (Figure 7). Such a finding highlights the ordered specificity in the early stages of assembly into amyloid and opens the opportunity to target a specific non-native state in order to control the onset of aggregation, for example through the development of antibodies, nanobodies, or small molecules that specifically recognize this species. The results highlight the importance of considering multiple factors in order to predict amyloid formation. Hydrophobicity, the propensity of the sequence to aggregate, the stability of the native state, solubility, transient exposure of aggregation-prone regions through protein dynamics, and the stability of the polypeptide sequence within the fully assembled fibril structure itself, may all contribute to enhanced amyloidogenicity. Although, these factors are well understood individually, the interplay between them during aggregation remains poorly explained. The results presented emphasize the importance of understanding the energy landscape of aggregation in intricate detail, from both thermodynamic and kinetic view points, in order to predict whether or not a protein will aggregate and how/why minor alterations in solution conditions/amino acid sequence can have a dramatic effect on the course of assembly, by small changes in the relative populations of amyloidogenic versus nonamyloidogenic states.
Conclusion
In this study we have used NUS NMR methods to study the relationship between protein folding and aggregation of a globular protein that forms amyloid fibrils from a structured precursor state, using β2m as the test protein. We show that the least thermodynamically stable protein is the least aggregation-prone sequence of the family of proteins studied here. Analysis of the folding energy landscape of the protein using real-time NMR revealed that the decreased stability and decreased lifetime of a precise and well-defined native-like amyloidogenic precursor (IT) are sufficient to tip the balance from aggregation to folding. The power of sparsely sampled NMR allowed us not only to detect a dynamic intermediate state (I1) of mβ2m but also to structurally characterize this species in residue-specific detail. The results reveal that the least stable protein (mβ2m) populates predominantly a flexible intermediate (I1) that is not aggregation-prone, while its more stable counterpart (hβ2m) folds through a native-like intermediate that has enhanced amyloidogenicity. Subtle changes in the folding energy landscape thus lead to dramatic changes in the aggregation outcome. The results reveal that protein stability does not correlate with aggregation propensity. Instead it is the precise balance and kinetic partitioning of intermediate states that determines whether β2m will fold to the native state or aggregate to form amyloid fibrils.
Acknowledgments
We thank Geoff Kelly for assistance with the 13C-detected experiments, Nasir Khan for technical support, David Eisenberg for the generous gift of the mβ2m plasmid, and the members of the Radford group for stimulating discussions. We acknowledge the Wellcome Trust (094232), the University of Leeds for funding the NMR instrumentation, and the Francis Crick Institute for provision of access to the MRC Biomedical NMR Centre at Mill Hill (Grant U117533887). The Francis Crick Institute receives its core funding from Cancer Research UK, the UK Medical Research Council, and the Wellcome Trust. T.K.K. and S.E.R. acknowledge funding from the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013)/ERC Grant Agreement No. 322408. T.K.K. and C.L.P. are supported by the Wellcome Trust (089311/Z/09/Z).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b02464.
Supplementary Methods, explanation of time resolved NUS NMR; Table S1, unfolding free energies of native and intermediate states; Table S2, chemical shift assignments of the I1 state; Figures S1–S2, hydrogen exchange and relaxation data for mβ2m; Figures S3–S4, refolding of mβ2m by pH jump or urea and spectra at later times during refolding; Figure S5, CON spectrum of the I1 state; and Figure S6, detection of I1 in the spectrum of native mβ2m (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sipe J. D.; Benson M. D.; Buxbaum J. N.; Ikeda S.-I.; Merlini G.; Saraiva M. J. M.; Westermark P. Amyloid 2014, 21, 221–224. 10.3109/13506129.2014.964858. [DOI] [PubMed] [Google Scholar]
- Eisenberg D.; Jucker M. Cell 2012, 148, 1188–1203. 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P. J.; Vendruscolo M.; Dobson C. M. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Baldwin A. J.; Knowles T. P. J.; Tartaglia G. G.; Fitzpatrick A. W.; Devlin G. L.; Shammas S. L.; Waudby C. A.; Mossuto M. F.; Meehan S.; Gras S. L.; Christodoulou J.; Anthony-Cahill S. J.; Barker P. D.; Vendruscolo M.; Dobson C. M. J. Am. Chem. Soc. 2011, 133, 14160–14163. 10.1021/ja2017703. [DOI] [PubMed] [Google Scholar]
- Booth D. R.; Sunde M.; Bellotti V.; Robinson C. V.; Hutchinson W. L.; Fraser P. E.; Hawkins P. N.; Dobson C. M.; Radford S. E.; Blake C. C. F.; Pepys M. B. Nature 1997, 385, 787–793. 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- Johnson S. M. S.; Connelly S. S.; Fearns C. C.; Powers E. T. E.; Kelly J. W. J. J. Mol. Biol. 2012, 421, 185–203. 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J.; Clarke A. R. Science 2007, 318, 930–936. 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- Lindberg M. J.; Tibell L.; Oliveberg M. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 16607–16612. 10.1073/pnas.262527099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn T. R.; Parker M. J.; Homans S. W.; Radford S. E. Nat. Struct. Mol. Biol. 2006, 13, 195–201. 10.1038/nsmb1058. [DOI] [PubMed] [Google Scholar]
- Mangione P. P.; Esposito G.; Relini A.; Raimondi S.; Porcari R.; Giorgetti S.; Corazza A.; Fogolari F.; Penco A.; Goto Y.; Lee Y. H.; Yagi H.; Cecconi C.; Naqvi M. M.; Gillmore J. D.; Hawkins P. N.; Chiti F.; Rolandi R.; Taylor G. W.; Pepys M. B.; Stoppini M.; Bellotti V. J. Biol. Chem. 2013, 288, 30917–30930. 10.1074/jbc.M113.498857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia G. G.; Pawar A. P.; Campioni S.; Dobson C. M.; Chiti F.; Vendruscolo M. J. Mol. Biol. 2008, 380, 425–436. 10.1016/j.jmb.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Belli M.; Ramazzotti M.; Chiti F. EMBO Rep. 2011, 12, 657–663. 10.1038/embor.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders J. C.; Young L. M.; Mahood R. A.; Jackson M. P.; Revill C. H.; Foster R. J.; Smith D. A.; Ashcroft A. E.; Brockwell D. J.; Radford S. E. Nat. Chem. Biol. 2015, 12, 94–101. 10.1038/nchembio.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulawa C. E.; Connelly S.; DeVit M.; Wang L.; Weigel C.; Fleming J. A.; Packman J.; Powers E. T.; Wiseman R. L.; Foss T. R.; Wilson I. A.; Kelly J. W.; Labaudinière R. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 9629–9634. 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarell C. J.; Karamanos T. K.; White S. J.; Bunka D. H. J.; Kalverda A. P.; Thompson G. S.; Barker A. M.; Stockley P. G.; Radford S. E. J. Biol. Chem. 2014, 289, 26859–26871. 10.1074/jbc.M114.595066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. I. A.; Arosio P.; Presto J.; Kurudenkandy F. R.; Biverstål H.; Dolfe L.; Dunning C.; Yang X.; Frohm B.; Vendruscolo M.; Johansson J.; Dobson C. M.; Fisahn A.; Knowles T. P. J.; Linse S. Nat. Struct. Mol. Biol. 2015, 22, 207–213. 10.1038/nsmb.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl F. U.; Bracher A.; Hayer-Hartl M. Nature 2011, 475, 324–332. 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Dumoulin M.; Last A. M.; Desmyter A.; Decanniere K.; Canet D.; Larsson G.; Spencer A.; Archer D. B.; Sasse J.; Muyldermans S.; Wyns L.; Redfield C.; Matagne A.; Robinson C. V.; Dobson C. M. Nature 2003, 424, 783–788. 10.1038/nature01870. [DOI] [PubMed] [Google Scholar]
- Karamanos T. K.; Kalverda A. P.; Thompson G. S.; Radford S. E. Mol. Cell 2014, 55, 214–226. 10.1016/j.molcel.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova M. I. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 10584–10589. 10.1073/pnas.0403756101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt G. W.; Radford S. E. FEBS Lett. 2009, 583, 2623–2629. 10.1016/j.febslet.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gejyo F.; Yamada T.; Odani S.; Nakagawa Y.; Arakawa M.; Kunitomo T.; Kataoka H.; Suzuki M.; Hirasawa Y.; Shirahama T.; Cohen A. S.; Schimd K. Biochem. Biophys. Res. Commun. 1985, 129, 701–706. 10.1016/0006-291X(85)91948-5. [DOI] [PubMed] [Google Scholar]
- Calabrese M. F.; Eakin C. M.; Wang J. M.; Miranker A. D. Nat. Struct. Mol. Biol. 2008, 15, 965–971. 10.1038/nsmb.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S.; Hasegawa K.; Yamaguchi I.; Goto Y.; Gejyo F.; Naiki H. Biochim. Biophys. Acta, Proteins Proteomics 2005, 1753, 34–43. 10.1016/j.bbapap.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Hasegawa K.; Tsutsumi-Yasuhara S.; Ookoshi T.; Ohhashi Y.; Kimura H.; Takahashi N.; Yoshida H.; Miyazaki R.; Goto Y.; Naiki H. Biochem. J. 2008, 416, 307–315. 10.1042/BJ20080543. [DOI] [PubMed] [Google Scholar]
- Yamamoto S.; Hasegawa K.; Yamaguchi I.; Tsutsumi S.; Kardos J.; Goto Y.; Gejyo F.; Naiki H. Biochemistry 2004, 43, 11075–11082. 10.1021/bi049262u. [DOI] [PubMed] [Google Scholar]
- Corazza A.; Rennella E.; Schanda P.; Mimmi M. C.; Cutuil T.; Raimondi S.; Giorgetti S.; Fogolari F.; Viglino P.; Frydman L.; Gal M.; Bellotti V.; Brutscher B.; Esposito G. J. Biol. Chem. 2010, 285, 5827–5835. 10.1074/jbc.M109.061168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichner T.; Kalverda A. P.; Thompson G. S.; Homans S. W.; Radford S. E. Mol. Cell 2011, 41, 161–172. 10.1016/j.molcel.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G.; Michelutti R.; Verdone G.; Viglino P.; Hernandez H.; Robinson C. V.; Amoresano A.; Dal Piaz F.; Monti M.; Pucci P.; Mangione P.; Stoppini M.; Merlini G.; Ferri G.; Bellotti V. Protein Sci. 2000, 9, 831–845. 10.1110/ps.9.5.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashley C. L.; Hewitt E. W.; Radford S. E. J. Mol. Biol. 2016, 428, 631–643. 10.1016/j.jmb.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanda P.; Kupče E̅.; Brutscher B. J. Biomol. NMR 2005, 33, 199–211. 10.1007/s10858-005-4425-x. [DOI] [PubMed] [Google Scholar]
- Zweckstetter M.; Bax A. J. Am. Chem. Soc. 2000, 122, 3791–3792. 10.1021/ja0000908. [DOI] [Google Scholar]
- Schanda P.; Brutscher B. J. Am. Chem. Soc. 2005, 127, 8014–8015. 10.1021/ja051306e. [DOI] [PubMed] [Google Scholar]
- Gil-Caballero S.; Favier A.; Brutscher B. J. Biomol. NMR 2014, 60, 1–9. 10.1007/s10858-014-9847-x. [DOI] [PubMed] [Google Scholar]
- Favier A.; Brutscher B. J. Biomol. NMR 2011, 49, 9–15. 10.1007/s10858-010-9461-5. [DOI] [PubMed] [Google Scholar]
- Mayzel M.; Rosenlöw J.; Isaksson L.; Orekhov V. Y. J. Biomol. NMR 2014, 58, 129–139. 10.1007/s10858-013-9811-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Delaglio F.; Cornilescu G.; Bax A. J. Biomol. NMR 2009, 44, 213–223. 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph M. G.; Shen L. Q.; Lamontagne S. A.; Luz J. G.; Delaney J. R.; Ge Q.; Cho B. K.; Palliser D.; McKinley C. A.; Chen J.; Wilson I. A.; Eisen H. N. J. Immunol. 2004, 172, 2994–3002. 10.4049/jimmunol.172.5.2994. [DOI] [PubMed] [Google Scholar]
- Trinh C. H.; Smith D. P.; Kalverda A. P.; Phillips S. E.; Radford S. E. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 9771–9776. 10.1073/pnas.152337399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennella E.; Cutuil T.; Schanda P.; Ayala I.; Forge V.; Brutscher B. J. Am. Chem. Soc. 2012, 134, 8066–8069. 10.1021/ja302598j. [DOI] [PubMed] [Google Scholar]
- Hiller S.; Ibraghimov I.; Wagner G.; Orekhov V. Y. J. Am. Chem. Soc. 2009, 131, 12970–12978. 10.1021/ja902012x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelouchina G. T.; Platt G. W.; Bayro M. J.; Radford S. E.; Griffin R. G. J. Am. Chem. Soc. 2010, 132, 17077–17079. 10.1021/ja107987f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladner C. L.; Chen M.; Smith D. P.; Platt G. W.; Radford S. E.; Langen R. J. Biol. Chem. 2010, 285, 17137–17147. 10.1074/jbc.M110.117234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F.; Mangione P.; Andreola A.; Giorgetti S.; Stefani M.; Dobson C. M.; Bellotti V.; Taddei N. J. Mol. Biol. 2001, 307, 379–391. 10.1006/jmbi.2000.4478. [DOI] [PubMed] [Google Scholar]
- Mukaiyama A.; Nakamura T.; Makabe K.; Maki K.; Goto Y.; Kuwajima K. J. Mol. Biol. 2013, 425, 273–291. 10.1016/j.jmb.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Rennella E.; Cutuil T.; Schanda P.; Ayala I.; Gabel F.; Forge V.; Corazza A.; Esposito G.; Brutscher B. J. Mol. Biol. 2013, 425, 2722–2736. 10.1016/j.jmb.2013.04.028. [DOI] [PubMed] [Google Scholar]
- Sakata M.; Chatani E.; Kameda A.; Sakurai K.; Naiki H.; Goto Y. J. Mol. Biol. 2008, 382, 1242–1255. 10.1016/j.jmb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Karamanos T. K.; Kalverda A. P.; Thompson G. S.; Radford S. E. Prog. Nucl. Magn. Reson. Spectrosc. 2015, 88–89, 86–104. 10.1016/j.pnmrs.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McParland V. J.; Kalverda A. P.; Homans S. W.; Radford S. E. Nat. Struct. Biol. 2002, 9, 326–331. 10.1038/nsb791. [DOI] [PubMed] [Google Scholar]
- Kad N. M.; Myers S. L.; Smith D. P.; Smith D. A.; Radford S. E.; Thomson N. H. J. Mol. Biol. 2003, 330, 785–797. 10.1016/S0022-2836(03)00583-7. [DOI] [PubMed] [Google Scholar]
- Buell A. K.; Dhulesia A.; Mossuto M. F.; Cremades N.; Kumita J. R.; Dumoulin M.; Welland M. E.; Knowles T. P. J.; Salvatella X.; Dobson C. M. J. Am. Chem. Soc. 2011, 133, 7737–7743. 10.1021/ja109620d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z.; Hu D.; Zhu M.; Fink A. L. Biochemistry 2007, 46, 3521–3531. 10.1021/bi061716v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.