Abstract
Native PKD2-L1 channel subunits are present in primary cilia and other restricted cellular spaces. Here we investigate the mechanism for the channel's unusual regulation by external calcium, and rationalize this behavior to its specialized function. We report that the human PKD2-L1 selectivity filter is partially selective to calcium ions (Ca2+) moving into the cell, but blocked by high internal Ca2+concentrations, a unique feature of this transient receptor potential (TRP) channel family member. Surprisingly, we find that the C-terminal EF-hands and coiled-coil domains do not contribute to PKD2-L1 Ca2+-induced potentiation and inactivation. We propose a model in which prolonged channel activity results in calcium accumulation, triggering outward-moving Ca2+ ions to block PKD2-L1 in a high-affinity interaction with the innermost acidic residue (D523) of the selectivity filter and subsequent long-term channel inactivation. This response rectifies Ca2+ flow, enabling Ca2+ to enter but not leave small compartments such as the cilium.
DOI: http://dx.doi.org/10.7554/eLife.13413.001
Research Organism: Human
eLife digest
Most of our cells have a single tiny-hair like structure called a primary cilium that projects outwards from the cell surface. Many cilia contain an ion channel protein called PKD2-L1 that allows calcium ions to pass through the membrane that surrounds each cell.
There are many different calcium channels and they are found in a variety of locations in cells to control the levels of calcium ions within various cell compartments. Channels on the cell surface allow calcium ions from the external environment to enter a compartment called the cytoplasm. Under normal conditions, calcium ions always flow into cells because they are much more abundant outside the cell than inside. Despite the absence of a membrane barrier between cilia and cytoplasm, calcium ions are maintained at a higher level in the cilium than in the cytoplasm. How is this difference maintained?
DeCaen, Liu et al. now show that PKD2-L1 is first stimulated and then inactivated by calcium ions inside the cilia. Under controlled conditions, calcium ions exiting PKD2-L1 block the channel and trigger its long-term inactivation. This inactivation can be overcome by preventing calcium ions from accumulating inside the cell.
Further experiments show that cytoplasmic regions of the PKD2-L1 protein that are able to bind to calcium ions are not responsible for this unusual channel behavior. DeCaen, Liu et al. propose that the strange effect of calcium ions on the channel acts to maintain calcium ions in cilia at higher levels than those found in the cytoplasm. Future challenges include understanding how PKD2-L1 is stimulated by calcium ions and to find out the consequences of this activity.
Introduction
Polycystic kidney disease proteins (PKDs), or polycystins (PC), are divided into two distinct gene families. The four PKD1 members (PKD1, PKD1-L1, PKD1-L2, PKD1-L3) are large proteins (~1700–4300 amino acids) with 11 putative transmembrane segments (TM) and a large autocleaved N-terminal extracellular domain. In contrast, the three PKD2 members are often included in the TRP ion channel family because they have 6 TM domains and a putative selectivity filter loop between TM5 and TM6 (Ramsey et al., 2006). These include PKD2, (PC2, TRPP1; formerly TRPP2), PKD2-L1 (PC2-L1, TRPP2; formerly TRPP3) and PKD2-L2 (PC2-L2, TRPP3, formerly TRPP5) (Wu et al., 2010). Members of the PKD1 and PKD2-subfamilies are often reported to associate in the plasma membrane, although the nature of these complexes is not understood. PKD1 + PKD2 were purported to form mechanosensitive ion channels in the primary cilia of kidney collecting duct epithelia (Nauli and Zhou, 2004; Nauli et al., 2003), a hypothesis that has recently been challenged (DeCaen et al., 2013; Delling et al., 2016). In humans, autosomal dominant polycystic kidney disease (ADPKD) is associated with loss-of-function mutations in genes that encode for either PKD1 or PKD2 (Wu and Somlo, 2000). Complete loss of either Pkd1 or Pkd2 in mice results in embryonic lethality with defects in formation of the kidney, pancreas and heart (Boulter et al., 2001; Lu et al., 1997; Kim et al., 2000; Wu et al., 2000). As in human ADPKD, adult Pkd2WS25/– mice have kidney cysts, renal failure, and die early (Wu et al., 2000). PKD1-L1 + PKD2-L1 heteromers form a calcium channel complex in the primary cilia from embryonic fibroblasts and retinal pigmented epithelial cells (DeCaen et al., 2013). Mice lacking Pkd2-L1 exhibit a form of heterotaxy (intestinal malrotation) in ~50% of offspring, suggesting that the channel modulates the ciliary Sonic Hedgehog (SHh) pathway during early development (Delling et al., 2013). A putative PKD1-L3 + PKD2-L1 channel was reported in mouse taste buds and proposed to be the acid receptor required for sour taste (Kawaguchi et al., 2010), but Pkd2-L1 knockout mice do not appear to have a deficit in sour taste perception (Nelson et al., 2010).
Of all the members of the PKD family, we found that only PKD2-L1 forms a functional homotetrameric channel when heterologously expressed on the plasma membrane (DeCaen et al., 2013). Information about the physiological function of the endogenous homomeric PKD2-L1 is sparse. Emerging evidence suggests that single channel openings of homomeric PKD2-L1 channels are sufficiently large (>100 pS) to generate action potentials in the medullo-spinal cerebrospinal fluid contacting neurons (CSF-cNs) located in the ependymal layer of the brainstem (Orts-Del'Immagine et al., 2014; Orts-Del'immagine et al., 2012). Since channels containing PKD2-L1 subunits are constitutively-active and calcium-permeant, dysregulation of their activity could lead to aberrant cytoplasmic calcium regulation, especially in smaller compartments such as the primary cilia of epithelial cells (DeCaen et al., 2013), the dendritic bulb of cerebrospinal fluid-contacting neurons (Orts-Del'Immagine et al., 2014) and the apical processes of the type III taste receptors (Huang et al., 2006; Ishimaru et al., 2010). Based on results using GCaMP3-targeted calcium sensors and current clamp recordings, primary cilia membranes were depolarized and had higher resting calcium concentrations (~ −20 mV, [Ca2+]in ~700 nM) compared to the cell body (-50 mV, [Ca2+]in < 100 nM) (Delling et al., 2013). Interestingly, the endogenous PKD1-L1 + PKD2-L1 channel complex inactivates in response to high cytoplasmic [Ca2+] when measured directly from the primary cilium (IC50 =540 nM) (DeCaen et al., 2013). When measured through heterologous co-expression, the PKD1-L3 + PKD2-L1 channel was initially activated, then inactivated by extracellular Ca2+ (5–10 mM) (Chen et al., 2015), which was also observed when homomeric PKD2-L1 channels were expressed in in Xenopus oocytes (Chen et al., 1999). It is unclear if calcium regulates the channels composed of PKD2-L1 directly or indirectly through an internal or external binding site.
Here we have taken the reductionist approach of measuring the PKD2-L1 homomeric channel in a heterologous system in order to understand its mechanism of calcium-dependent inactivation. We first confirm previous studies that PKD2-L1 is modestly voltage-dependent (Shimizu et al., 2009) and show that this voltage dependence is independent of any divalent ion blocking mechanism. We then demonstrate that PKD2-L1 inactivation is initiated by the accumulation of internal Ca2+. Surprisingly, we find that removal of the cytoplasmic EF-hands and coiled-coil motifs do not alter PKD2-L1 inactivation. We observe that Ca2+ ions conduct inwardly through the selectivity filter of PKD2-L1, whereas outward Ca2+ conductance blocks the channel, followed by long-term inactivation. This type of Ca2+-dependence appears to be unique to PKD2-L1 among TRP channels tested to date and is triggered by Ca2+ coordination with D523 in the selectivity filter, which is also the site responsible for divalent metal and trivalent lanthanide metal block.
Results
PKD2-L1 channels are voltage-dependent, even in the absence of divalent ions
The voltage dependence of murine PKD2-L1 (previously named TRPP2, now called TRPP1) was previously characterized under conditions of high (11 mM) internal magnesium (Shimizu et al., 2009). To separate rectification due to divalent ion block (Voets et al., 2003; Nadler et al., 2001; Lucas et al., 2003; Topala et al., 2007) from intrinsic voltage dependence, we first examined human PKD2-L1 in symmetrical 144 mM [Na+] in the absence of divalent ions (free Ca2+ and Mg2+ <1 nM, Figure 1A). From a holding potential of 0 mV, we applied a +100 mV prepulse and then measured tail currents after immediate hyperpolarization to varying potentials (Figure 1B,C). Plots of the instantaneous maximum tail currents (Max. Itail) were nearly linear (ohmic) at all potentials. However, the steady-state current (Iss) measured after the tail current decay outwardly rectified at potentials above +50 mV. At negative potentials PKD2-L1 appears to simply deactivate as estimated by the rate constants of tail current decay (τ = 20–60 ms, Figure 1 B). In the on-cell configuration, the decay of ensemble-averaged single channel events closely matched macroscopic tail current decay (Figure 1—figure supplement 1).
Figure 1. PKD2-L1 channel deactivation is voltage-dependent.
(A) Diagram depicting symmetrical divalent free (DVF) conditions. (B) Top, voltage protocol used to generate tail currents. Representative currents from untransfected HEK cells (gray traces) and those expressing PKD2-L1 channels (black traces). Tail currents were fit to a single exponential (magenta trace), averaged and plotted relative to voltage (Inset, Error ± SEM, N = 4–7 cells). (C) Sodium current density-voltage relationships measured at the maximum of the tail current and at steady state. Results from repeated trials of the voltage protocol are indicated (Error ± SEM, N = 4–7 cells). (D) Right, Representative TRPM7 DVF Na+ currents recorded under (black traces) and after the addition of 1.8 mM CaCl2 (blue traces). Left, Resulting TRPM7 current density-voltage relationships (Error ± SEM, N = 8 cells).

Figure 1—figure supplement 1. Decay of hyperpolarization-induced tail currents from PKD2-L1 channels.

We considered the possibility of voltage or time-dependent monovalent ion block of the pore in the absence of divalent ions. However, single channel amplitudes appear to remain constant over the time course of the pulse, indicating a lack of fast (flicker) block (Figure 1—figure supplement 1). Also, replacing Na+ by K+ did not change the current amplitude or rate of decay (Figure 1C). Although we cannot rule out a site that binds and blocks all inward monovalent cations, we argue that the most likely interpretation is that the channel simply deactivates after hyperpolarization (see Discussion). It is important to note that the onset of the PKD2-L1 tail current is very fast (<120 μs, data not shown), without an obvious ‘hook’ to indicate overlapping activation and inactivation time courses as observed in delayed rectifying potassium channels (hERG and Kv3.1b) (Labro et al., 2015; Vandenberg et al., 2012). The tail currents of PKD2-L1 are unusual compared to other cation-nonselective TRP channels. For example, currents conducted by TRPM7 are ohmic and do not decay upon membrane hyperpolarization (Figure 1D). However, TRPM7 becomes outwardly rectifying when calcium is added to the external sodium solution (and, as is well-known for TRPM6/7, blocked by internal Mg2+) (Topala et al., 2007; Li et al., 2007), indicating that TRPM7 rectifies in the presence of a physiological level of extracellular calcium, and possibly other divalent ions.
Taken together, these data demonstrate that human PKD2-L1 channels are voltage-dependent, as previously described for the murine orthologue by Nilius and colleagues (Shimizu et al., 2009). Thus, PKD2-L1 voltage dependence which is independent of cation block is likely shared between murine and human orthologs and may be a unique among the TRP family of ion channels. The following experiments focus on another unusual feature of PKD2-L1 we call ‘long-term inactivation’ which is dependent on the accumulation of internal calcium ions.
PKD2-L1 channels are blocked by Ca2+ exiting the cell
High external Ca2+ (10 mM) potentiates human (Chen et al., 1999) but not murine PKD2-L1 (Shimizu et al., 2009). To examine calcium’s modulation of the current under physiological Ca2+ conditions, we expressed human PKD2-L1 and measured PKD2-L1 currents under voltage clamp in 2 mM [Ca2+]ex. As with high external calcium, PKD2-L1 current was initially potentiated but, interestingly, was then completely and apparently irreversibly inactivated over 8 min (Figure 2—figure supplement 1A,B). During the voltage ramp, we observed a negative shift of Erev from 8 to 0 mV, reflecting a loss in the available calcium-permeable PKD2-L1 channels and/or a shift in the ion composition of the patched cells (Figure 2—figure supplement 1C). Similar shifts in equilibrium potentials due to accumulation of internal cations have been reported for cells overexpressing P2X2 channels (Li et al., 2015). Since potentiation (4 min) and inactivation (8 min) were slow, we hypothesized that internal accumulation of Ca2+ could account for the delayed onset of both. We integrated the tail currents triggered by −60 mV repolarizations to estimate the number of calcium ions accumulating within the cell (Figure 2—figure supplement 1A,C). We found that Ca2+ moving through PKD2-L1 saturates the internal calcium buffers (5 mM BAPTA and intrinsic) and accumulates to concentrations >10 μM (Figure 2—figure supplement 1D). The time course of BAPTA saturation by PKD2-L1-mediated increases in Ca2+ occurs after 4 min, suggesting that internal calcium accumulation is responsible for channel inactivation. Consistent with these observations, we varied internal [Ca2+] and observed that cells patched with high initial [Ca2+] (≥ 450 nM) inactivates 1–4 min sooner than cells patched with lower internal free-Ca2+ (≤ 100 nM; Figure 2A,B). Finally, when the buffering strength is increased (15 mM BAPTA, 5 mM EGTA), PKD2-L1 currents potentiate, but do not inactivate, over a 10 min time course (black trace, Figure 2B).
Figure 2. High intracellular Ca2+ irreversibly inactivates PKD2-L1.
(A) Representative PKD2-L1 currents captured at 1, 4 and 8 min time points recorded with the indicated buffered [free- Ca2+]; 2 mM Ca2+ in the external solution. (B) Time course of outward peak current density recorded under the indicated conditions (Error ± SEM, N = 7–10 cells).

Figure 2—figure supplement 1. The time course of PKD2-L1 inactivation correlates with internal Ca2+ accumulation.

To determine whether internal Ca2+ could alter PKD2-L1 channel activity when the endogenous buffering conditions were undisturbed, we patch-clamped cells expressing PKD2-L1 in the on-cell configuration and measured the outward single channel events in response to uncaged internal calcium (Figure 3). Immediately upon uncaging Ca2+-bound NP-EGTA by a 1 s, 405 nm laser pulse, PKD2-L1 open probability (Po) was reduced by 27-fold. The channel was identified as PKD2-L1 since its conductance was consistent with PKD2-L1 homomers (outward 198 pS, inward 121 pS (DeCaen et al., 2013), the conductance was not observed in untransfected HEK-293T cells, and the single channel conductance and macroscopic whole cell currents were blocked by dibucaine (Figure 1—figure supplement 1). These results demonstrate that PKD2-L1 is long-term-inactivated at high internal [Ca2+] and that this process can occur independent of detectable channel potentiation.
Figure 3. Uncaging internal Ca2+ blocks outward single channel openings.

(A) Outward PKD2-L1 single channel events measured in the on-cell configuration; holding potential = 80 mV. Ca2+ dependent fluorescence was measured using Fluo-3; internal Ca2+ was caged with NP-EGTA. Blue arrows indicate the time points at which Ca2+ was uncaged using a 1 s 405 nm UV pulse. Images before and after uncaging are shown above the single channel record. (B) Expanded time scales of the record in A, illustrating the rapidity of current block. Right, Normalized open probability histograms measured in control (gray) and after uncaging cytosolic Ca2+ (red). The open probability (Po) of PKD2-L1 was significantly reduced after the UV pulse (N= 4 cells, Error ± SEM; asterisk indicates p<0.005; Prior to UV pulse, Po = 0.0057 ± 0.001; after UV pulse Po = 0.0002 ± 0.0002).
PKD2-L1 channel C-terminal EF-hand domains do not mediate inactivation
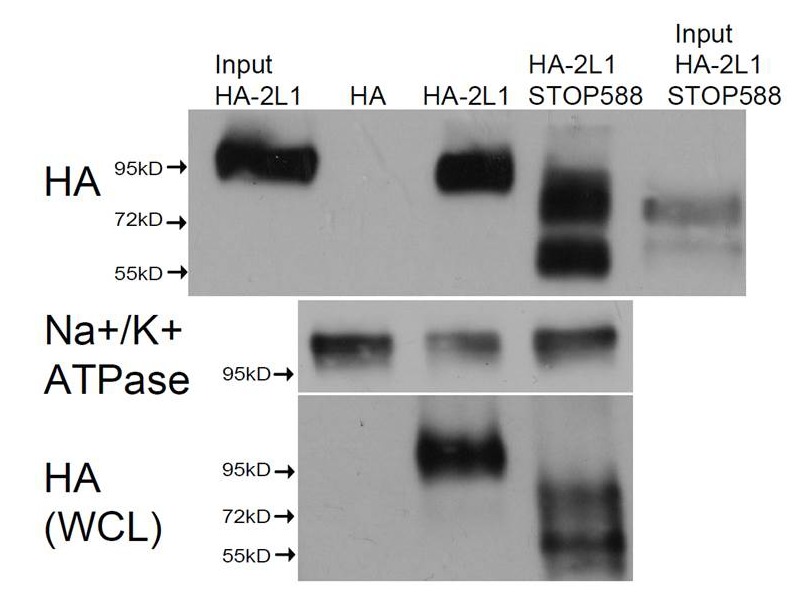
What is the mechanism by which internal Ca2+ inactivates the PKD2-L1 channel? The C-terminus of PKD2-L1 contains an oligomerization domain (OD, 588–608) (Chen et al., 2015), putative EF-hands (633–665) (Li et al., 2002) and a coiled-coil domain (CC, 690–737) (Yu et al., 2012). We generated truncations above and below each of the C-terminal motifs to determine their effect on channel function (Figure 4A). We found that the most drastic truncation, located before the first coiled-coil domain (Stop-588), abolished PKD2-L1 channel function (Figure 4C). Surface expression of the 588-Stop C-terminal cleavage mutant was unaltered in comparison to Wt PKD2-L1, as assayed by membrane biotinylation (Figure 4—figure supplement 1). The intact OD might be critical for channel function or be a subunit oligomerization site, as previously proposed (Zheng et al., 2015). However, truncating the PKD2-L1 channel just after the OD domain (Stop-608), or before and after the EF-hands and coiled-coil domain, did not alter normal current density or voltage dependence (Figure 4B–E). With the exception of the 508-stop truncation, all truncations were potentiated, then inactivated in normal intracellular [Ca2+] (Figure 4B). Thus the EF-hands and coiled-coil domain do not appear to be required for PKD2-L1’s calcium-dependent potentiation or inactivation (nor for potentiation by calmidazolium, see below).
Figure 4. PKD2-L1 channel C-terminal truncations do not alter Ca2+-dependent inactivation.
(A) Cartoon depicting the locations of the C-terminal truncation mutants relative to the putative intracellular motifs. (B) Representative PKD2-L1 currents and the time courses of the potentiation and inactivation for the truncation mutants relative to the Wt channel. Currents were recorded in physiological [Ca2+] (same conditions as described in Figure 2). (C) Representative currents activated by +10 mV voltage steps from -100 to +100 mV from the Wt and truncated PKD2-L1 channel. (D) Corresponding voltage dependence of the kinetics of channel opening (τpeak) and channel closure (τtail). (E) The average PKD2-L1 channel current density measured at +100 mV.

Figure 4—figure supplement 1. Cell surface expression of the non-functional PKD2-L1 truncation mutants.
PKD2-L1 channels are inactivated after Ca2+ block
The results above exclude the possibility that PKD2-L1’s cytoplasmic EF-hands and the coiled-coil domains are involved in inactivating its current or in trafficking the channel to the membrane. When measuring the voltage dependence of the PKD2-L1 channel under physiological Ca2+ conditions, we observed that PKD2-L1 channels exhibit a biphasic current-voltage relationship, in which the outward current decreases at potentials beyond the reversal potential for calcium (ECa) (Figure 5A). By altering extracellular calcium ([Ca2+]ex), we noted that net nonselective outward current always declined positive to ECa (Figure 5A). Repetition of the protocol demonstrated that PKD2-L1 channels had completely inactivated; that is, PKD2-L1 currents could no longer be elicited during the recording period (Figure 5—figure supplement 1). This data suggests that outward PKD2-L1 currents were stably blocked and/or inactivated by outward moving Ca2+ ions. We tested TRPM7, TRPV1, and TRPV3 for similar behavior and found that none of their outward currents exhibited this biphasic dependence (Figure 5—figure supplement 1, TRPV1 data not shown). Comparing the PKD2-L1 outward current to the inward tail current elicited by repolarization to −60 mV (Figure 5—figure supplement 1), we observed that the outward current was blocked, whereas the inward tail current remained relatively constant. These observations suggest that Ca2+ induced block and subsequent inactivation of all outward current, while inward flux was unimpeded. To examine this phenomenon, we used a voltage protocol designed to capture the rates of Ca2+- dependent block and inactivation (Figure 5B,C). A series of voltage clamp steps 10–30 mV negative to ECa were applied so that inward tail currents and outward currents were stable over time. Thus, inward Ca2+ could permeate PKD2-L1 without blocking or inactivating the channel. Then, the depolarization step was increased to 28–42 mV more positive than ECa, where the outward currents were immediately blocked by outwardly moving calcium ions. In contrast, tail currents triggered by repolarization to -60 mV were not immediately blocked, but progressively decreased after each depolarization positive to ECa, indicating that the inward PKD2-L1 currents were not long-term- inactivated until after the outward current was blocked by Ca2+. By plotting the sum of the durations of the depolarization steps positive to ECa against the tail current magnitude (Figure 5B,C), we estimated the rate of inactivation, τinact ≈ 2 s (1.8 ± 0.2 s; 2.1 ± 0.2 s; 1.7 ± 0.2 s for the [Ca2+]ex conditions 10 μM, 1 mM and 20 mM respectively).
Figure 5. Block of PKD2L-1 by outward Ca2+ triggers channel inactivation.
(A) Left, Exemplar currents measured with the indicated [Ca2+]ex (inset). Right, resulting outward current densities at the indicated ECa (N = 6–7, Error ± SEM). (B) PKD2-L1 currents recorded from separate cells activated in the presence of 10 μM, 1mM and 20 mM [Ca2+]ex by a series of depolarizations (0.2 Hz) hyperpolarized and depolarized relative to ECa. (C) Top, resulting normalized peak and tail currents (Error ± SEM, N =4–5 cells). Bottom, PKD2-L1 inactivation rate in each [Ca2+]ex condition (10 μM, 1 mM and 20 mM) was determined by single exponential fitting of the decay of the tail current amplitude (τinact. = 1.8 ± 0.2 s; 2.1 ± 0.2 s; 1.7 ± 0.2 s, respectively).

Figure 5—figure supplement 1. Outward PKD2-L1 current is blocked at membrane potentials positive to ECa.

Selectivity filter residues mediating selectivity and block
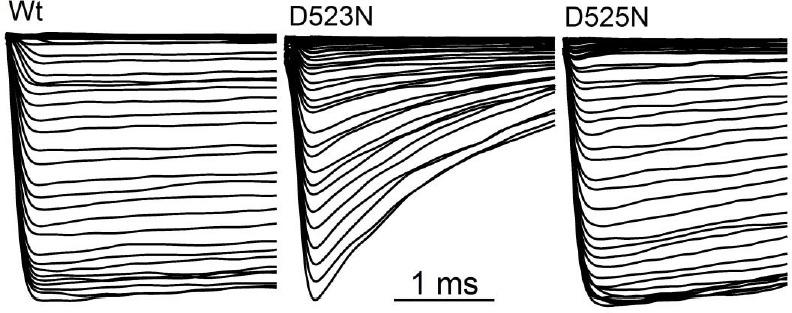
The above results suggest that outward-going calcium ions through PKD2-L1 block the pore and induce a long period of channel inactivation. Thus, we sought to reduce the affinity for the Ca2+ coordination sites within the selectivity filter to relieve the outward current block by Ca2+ and thus reduce inactivation. We previously reported that the PKD2-L1 conductance could be abolished by double serine or alanine mutations, to Asp523 and Asp525 (D523S:D525S or D523A:D525A), whereas mutation of Asp530 (D530N) did not alter the conductance (DeCaen et al., 2013). We also tested individual alanine substitutions to either position (data not shown), which also abolished the PKD2-L1 conductance. These data suggest that residues D523 and D525 are critical positions within the PKD2-L1 selectivity filter. Since D523 is the most conserved residue among the PKD2 family members, we propose that it is essential for block, inactivation, and its modest Ca2+ selectivity (Figure 6A). Since asparagine (Asn, N) has a similar volume (≈ 11 nM (Nauli and Zhou, 2004)) but its carboxamide side chain is not as electronegative as the carboxylate found in the native aspartate (Asp, D), we generated individual substitutions of Asn for D523 and D525. We found that at similar current densities, D523N mutant-associated currents did not inactivate over an 8 min time course, whereas D525N currents first potentiated and then inactivated similar to the Wt channel (Figure 6B). To determine if the D523N mutations altered outward Ca2+-dependent inactivation, we used the same depolarization protocol as used in Figure 5B and observed that the outward peak and inward tail currents were stable over time and potential, indicating that Ca2+ does not block or inactivate the D523N current (Figure 6C,D). Furthermore, the voltage dependence of the D523N outward current is not biphasic at potentials more positive to ECa (Figure 6—figure supplement 1).
Figure 6. Filter mutant D523N is not blocked by outward Ca2+ and is not inactivated.
(A) Amino acid alignment of the pore regions of PKD2 family members. The black bar indicates the selectivity filter. (B) Representative currents and time course of the average tail and peak current densities for the selectivity filter mutants (Error ± SEM, N =7 cells). (C) Left, representative D523N currents activated by a series of depolarizations (0.2 Hz) negative (+120 mV) and positive (+180 mV) to ECa (138 mV). Right, resulting normalized peak and tail current amplitudes (Error ± SEM, N =5 cells). (D) The relative permeability (Px/PCs) of Na+, K+ and Ca2+ for Wt and mutant channels. These values were calculated using the measured reversal potentials from the steady state voltage-current relationships in Figure 6—figure supplement 1, and tabulated in Figure 6—source data 1). (E) Top, representative tail currents activated by repolarizing -60 mV from the indicated potentials. Bottom, Corresponding voltage dependence of the channel closing (deactivation) kinetics (τtail;Error ± SEM, N=6–8).
DOI: http://dx.doi.org/10.7554/eLife.13413.012

Figure 6—figure supplement 1. The current-voltage relationships of PKD2-L1 channels in the presence of different extracellular cations.

These data demonstrate that Ca2+ does not block the D523N current in the outward direction and thus the ensuing inactivation does not occur. The selectivity for Ca2+ of the filter mutant channels D523N and D525N are reduced 19- and 8- fold (respectively) compared to the wild type channel (PCa/PCs = 7), as estimated by the change in Erev when measured in Na+, K+, Ca2+ and Cs+ extracellular conditions (Figure 6D, Figure 6—figure supplement 1, Figure 6—source data 1). The speed of tail current decay of the D523N channel was enhanced ≈ 20-fold (measured at −100 mV, Wt τTail = 43 ms ± 3; D523N τTail = 2.3 ms ± 0.2), whereas those from the D525N mutation were much less affected (D525N τTail = 37 ms ± 2; Figure 6E).
Despite dramatically altering PKD2-L1 kinetics and selectivity, the D523 mutation retains sensitivity to modulation by the membrane-permeant calmodulin antagonist, calmidazolium (Wt EC50 = 2.6 μM ± 6; D523N EC50 = 9 μM ± 7; also see DeCaen et al. [DeCaen et al., 2013]) and block by the amide local anesthetic, dibucaine (Wt IC50 = 31 μM ± 5; D523N IC50 = 23 μM ± 3; Figure 7—figure supplement 1, Figure 7—source data 1). These results suggest that modulation by calmidazolium and block by dibucaine are preserved in the D523N mutant, suggesting that these compounds alter PKD2-L1 function through sites allosterically coupled to the filter. Together, these results demonstrate that while both D523 and D525 participate in Ca2+-selectivity of the pore, position D523 is the most critical to Ca2+- inactivation.
Since Asp523 apparently forms the high affinity coordination site for Ca2+ that is responsible for Ca2+-selectivity and initiates outward Ca2+-dependent inactivation, we hypothesized that it might also be the binding site for divalent metals and trivalent metal antagonists. The transition metals, cadmium and zinc, blocked the PKD2-L1 Wt current (IC50 = 25 μM ± 5; IC50 = 156 μM ± 15) and the D525N filter mutant (IC50 = 40 μM ± 8; IC50 = 209 μM ± 28), but were ineffective antagonists of the D523N channel at concentrations below 1 mM (Figure 7A,B; Figure 7—source data 1). Trivalent cations, such as lanthanum and gadolinium, are commonly used non-specific blockers of Ca2+-permeable channels. We found that the PKD2-L1 channel is 20-times more sensitive to block by Gd3+ (IC50 = 9 μM ± 3) and La3+ (IC50 = 3 μM ± 4) than reported for members of the TRPV and TRPA families (IC50 ≥ 200 μM, Figure 7C,D) (Leffler et al., 2007; Banke, 2011; Xu et al., 2002). This potency was preserved in the D525N filter mutant channel (IC50 = 11 μM ± 3 and 1 μM ± 4, respectively). However, the D523N channel displayed anomalous mole fraction behavior in the presence of La3+, conducting through the channel at low to mid-µM concentrations (1–100 μM), but blocking the basal current in the millimolar range (Figure 7E). These findings suggest that D523 forms a coordination site for divalent and trivalent metal cations.
Figure 7. Loss of transition metal block in selectivity filter mutant, D523N.
(A, B) Effects of Zn2+ and Cd2+ on PKD2-L1 currents. Top, Exemplar PKD2-L1 currents activated by voltage ramps at the indicated [Zn2+]. Bottom, Corresponding time courses of the Zn2+ block of the Wt and D525N PKD2-L1 channels. The internal buffer was 15 mM BAPTA and 5 mM EGTA, which prevented Ca2+ accumulation and inactivation of the PKD2-L1 currents. (B) Divalent metal concentration-dependent block of the Wt, D523N and D525N channels (Error ± SEM, N = 4–6 cells). (C–E) Effects of trivalent metal block. Left, Exemplar PKD2-L1 currents activated by voltage ramps in the presence of La3+. Right, Corresponding time courses of the La3+ block of the indicated PKD2-L1 channels. (D) Concentration-dependent block of PKD2-L1 and D525N channels by trivalent ions, and (E) potentiation of the D523N filter mutant channel (Error ± SEM, N = 4–5 cells).
DOI: http://dx.doi.org/10.7554/eLife.13413.015

Figure 7—figure supplement 1. Calmidazolium activation and dibucaine block is preserved in the D523N filter mutant channel.

Discussion
Regulation of ion channels by cytoplasmic calcium is a common theme in biology (Clapham, 2007; Yu and Catterall, 2004) and can occur through direct binding to the channel protein or through adaptor or modulatory proteins. Calcium binding proteins like calmodulin often bind to Ca2+-permeant ion channels and alter their function (Ben-Johny et al., 2015). Calcium coordinating motifs within channel proteins, such as cytoplasmic EF-hands found in voltage gated CaV channels, alter channel inactivation based on localized calcium accumulation. Here, we have shown a direct mechanism of calcium regulation of the PKD2-L1 channel, in which outward Ca2+ binds in the selectivity filter and initiates inactivation of the channel. Calcium and other ions pass inwardly through the PKD2-L1 channel as long as the electrochemical gradient permits; when channel densities are high, this is sufficient to significantly shift internal ion concentrations as seen by the slowly shifting Erev. However, depolarizations above ECa lead to channel block and inactivation (as proposed in Figure 8A). To our knowledge, this is the only reported ion channel with Ca2+-dependent rectification due to pore block-inactivation, and so far is unique to PKD2-L1.
Figure 8. Proposed kinetic scheme of PKD2-L1 channel states and a hypothetical model of Ca2+ coordination sites in the selectivity filter.
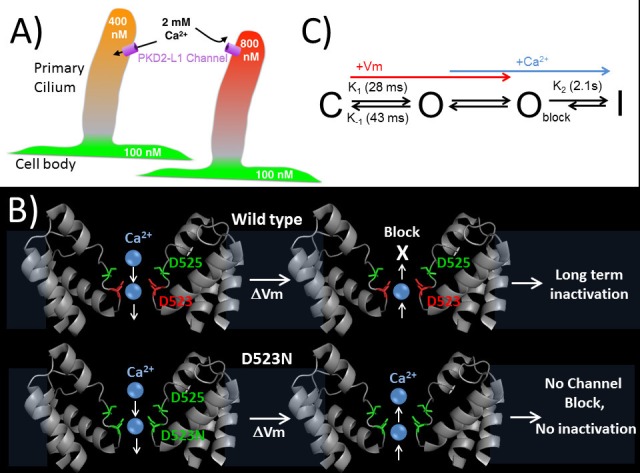
(A) Calcium clamp of restricted spaces, such as primary cilia, by PKD2-L1. Resting [Ca2+] ≅ 500–700 nM in primary cilia. PKD2-L1 channels in the cilia membrane are potentiated before inactivation by [Ca2+] < ~500 nM, but simply inactivated at [Ca2+] > ~700 nM. (B) Model of Wt and D523N filters with proposed Ca2+ movement in response to membrane potential. The PKD2-L1 low affinity Ca2+ (D525) and high affinity (D523) binding sites are colored yellow and red, respectively, corresponding to residues N921 and D918 in the TRPA1 structure (PDB: 3J9P) as a substitute for the undetermined PKD2-L1 structure. (C) Proposed four channel state scheme C = closed; O = open; Oblock = open channels blocked by outwardly moving Ca2+ and I = inactivated (long-term). Corresponding rate constants (K) are indicated between the three channel states (K1 = opening, K-1 = closure, K2 = inactivation). The colored arrows indicate the direction of channel state stimulus by either membrane depolarization (+Vm) or outwardly moving calcium ions (Ca2+). The recovery from calcium-dependent inactivation is shown as a shortened arrow since inactivation is irreversible on the time scale of the experiments (<20 min).
Many members of the TRPA, C, M and V families are potentiated and inactivated by Ca2+ dependent mechanisms (Gordon-Shaag et al., 2008; Zhu, 2005). Thus far, the proposed mechanism(s) by which Ca2+ inhibits TRP channels are indirect, such as with PI(4,5)P2-depletion, Ca2+-regulated kinases, phosphatases, and phospholipases, via both calmodulin-dependent and independent processes (Gordon-Shaag et al., 2008; Zhu, 2005). We showed that Ca2+ occupancy in the filter blocks the outward PKD2-L1 current and triggers its long-term inactivation. Although mutating the selectivity filter of TRPA1 (D918) can abolish extracellular Ca2+-dependent potentiation and inactivation, the effect of extracellular Ca2+ on these processes follows elevation of intracellular calcium and is proposed to operate through an indirect, unknown mechanism (Wang et al., 2008). For PKD2-L1, outward Ca2+ current inactivates the channel after binding within the selectivity pore. Although we cannot rule out allosteric modulation by calmodulin or kinases during inactivation, involvement of the C-terminal EF-hand and coiled-coil are unlikely since no effect was observed on inactivation when these motifs were removed. Likewise, for the TRPA1 channel, removing the EF-hands did little to alter the onset of potentiation and inactivation (Wang et al., 2008). Our observations from our PKD2-L1 EF-hands truncation are in agreement with results from the PKD2-L1 splice variants expressed in the liver, where a naturally occurring C-terminal EF-hand truncation is found to have no effect on Ca2+-dependent inactivation (Li et al., 2002). Thus if C-terminal modulation is involved, the location for binding of a modulator would have to be located above the oligomerization domain, which appears to be critical for PKD2-L1 function, as the trafficking of the Stop-588 mutant was the same as the full length channel.
We have shown that even potent (BAPTA, 5 mM) cytoplasmic Ca2+ buffers can be saturated when PKD2-L1 channels are overexpressed. Once occupied by outwardly moving calcium ions, the PKD2-L1 channels irreversibly inactivates in seconds (τ = 2.1 s). Thus our results explain the inactivation phenomenon observed in Chen et al. (Chen et al., 1999) and describe a direct level of Ca2+ feedback inhibition of the PKD2-L1 channel. Given that cytoplasmic [Ca2+] ranges from 50 nM to as high as 10 µM (near mouths of channels), what is the physiological relevance of outward calcium current regulation in PKD2-L1 channels? Because of the cell’s large volume (typically 1–3 pL), it is unlikely that the concentration of calcium in the cytosol would ever reach sufficient concentrations to move outwardly through PKD2-L1 channel and trigger inactivation. However, because PKD2-L1 channels have large inward conductances (PKD2-L1: 120–150 pS and PKD1-L1 +PKD2-L1 = 86 pS) (DeCaen et al., 2013; Chen et al., 1999; Shimizu et al., 2009) with modest selectivity for Ca2+ (PCa/PCs PKD2-L1 and PKD1-L1 +PKD2-L1 = 6–15) (DeCaen et al., 2013; Shimizu et al., 2009), the expression of just a few channels (1–10) in small cellular compartments could result in large changes in local Ca2+ concentrations. For example, the homomeric PKD2-L1 is enriched in the membrane of the dendritic nob of CSF contacting neurons, which have small volumes (113 fL) (Orts-Del'Immagine et al., 2014; Orts-Del'Immagine et al., 2016). Here, PKD2-L1’s alkaline-pH-stimulated channel activity (Po = 0.02) would increase the dendritic nob cytoplasmic calcium by ≈ 22 μM per channel opening. In smaller compartments, like the primary cilium (0.5 fL), where roughly 30 heteromeric PKD1-L1 +PKD2-L1 channels are expressed, ATP stimulation (Po = 0.01) would increase the cilioplasmic calcium by ≈ 31 μM per channel opening. Thus, prolonged PKD2-L1 (or PKD1-L1 +PKD2-L1) channel openings could result in increases in cellular compartment [Ca2+] up to the external [Ca2+]. Most important, we hypothesize that PKD2-L1 maintains Ca2+ concentration in the 500 -700 nM range observed in primary cilia (Nelson et al., 2010; Wu et al., 2000). As shown in Figure 2, entering Ca2+ transiently potentiates PKD2-L1 up to ~750 nM [Ca2+]In - above this level, inhibition dominates. This behavior would serve as [Ca2+] clamp, a feedback mechanism that counteracts Ca2+ loss via diffusion into the cytoplasm (Figure 8B). This mechanism would ensure that the primary cilium is a specialized calcium compartment, even though no calcium diffusion barrier exists between cilium and cytoplasm (Nelson et al., 2010). However, the Ca2+ exchangers and intrinsic buffers present in each compartment are not known, limiting a more detailed interpretation of this mechanism.
We have established that D523 within the filter is responsible for Ca2+-selectivity as ions pass inward, and for block by Ca2+ as it moves out of the cell (Figure 6, Figure 6-figure supplement 1). Independent substitutions of the aspartate filter residues 523 and 525 with asparagine resulted in a significant reduction of Ca2+-selectivity, while independent alanine substitutions for either residue resulted in a non-conducting channel. Based on these observations we propose that this unique calcium conductance is achieved with two ion coordinating sites involving filter residues D523 and D525. We speculate that Ca2+ unidirectional conductance is achieved by a ‘knock-on’ ionic interaction, where the outer residue (D525) coordinates Ca2+ with weaker affinity than the inner residue (D523). In this model, the interaction of Ca2+ with D523 is broken by repulsion by a second Ca2+ occupying the outer D525. When the membrane potential is more depolarized than ECa, Ca2+ occupies the high-affinity binding site (D523). Since there is no inner Ca2+ coordinating site to electrostatically ‘knock-off’ Ca2+ from D523, it is not displaced from this site and outward current is blocked. Since Ca2+ does not block the D523N channel, we propose that Ca2+ is able to move in both directions in the mutant channel’s selectivity filter (Figure 8B). This effect can be explained by the loss of the high-affinity binding site within the innermost portion of the selectivity filter. Since D523 is conserved within the PKD2 family, it is possible that other members of PKD2 family may share this same unidirectional calcium conductance, although the selectivities of homomeric PKD2 and PKD2-L2 channels have yet to be defined.
We established that D523 is the metal binding site for divalent and trivalent ions within the selectivity filter of PKD2-L1. The potency (IC50) of PKD2-L1 block by Gd3+ was shifted from 9 μM to greater than 1 mM in the D523N channel, which reflects a loss of affinity >6.7 kcal/mol. Conversely, the D523N mutant conducts the smaller trivalent La3+ at µM concentrations, whereas the Wt and D525N channels are blocked within this concentration range. These seeming incompatible observations are reminiscent of La3+ anomalous mole fraction effects reported in Wt members of the TRPC family (C3, C4, C5 and C6) (Jung et al., 2003; Strübing et al., 2001; Hofmann et al., 1999). Thus, whether trivalent and divalent metals permeate or block TRP channels is likely determined by the strength of their electrostatic interactions with acidic residues that line the selectivity filter.
A simple preliminary four state model of PKD2-L1 is shown in Figure 8C. We have defined the resting state found at negative potentials prior to depolarization as the closed state (C), although PKD2-L1 has some of constitutive activity even at −100 mV (PO = 0.007). After depolarization, our analysis of macroscopic and single channel currents demonstrates that channel openings are transiently increased upon hyperpolarization (Figure 1—figure supplement 1). We proposed that these large, slowly decaying tail currents represent the population of channels transitioning from the open (O) to closed (C) state. The closed to open transition (K1) can be measured by the kinetics of the peak currents during membrane depolarization (<10 mV). While it is possible that PKD2-L1 gating may have voltage dependent inactivation properties akin to some delayed rectifying potassium channels (IKr) (Warmke and Ganetzky, 1994; Trudeau et al., 1995), our experiments did not capture this kinetic property. Further biophysical characterization will be necessary to test this possibility. The PKD2-L1 channel opening (K1) and closure (K-1) are reversible and modestly voltage dependent, increasing 1.5x and 2.2x with 100 mV membrane potential shifts, respectively. Outwardly moving Ca2+ rapidly block the open channel state (Oblock) and trigger a slower long-term inactivation process (K2). The rate of inactivation (K2) is dependent on outward Ca2+ current and is irreversible on the time scale of the experiments. Interestingly, the enhanced onset of channel opening and closing found in the D523N mutant may be related to the lack of Ca2+ selectivity and Ca2+-dependent inactivation. These observations demonstrate that the rates of PKD2-L1 gating can be altered by mutating positions within the selectivity filter, suggesting that permeant ion coordinating sites are partly involved. This feature has precedence in BK channels, where permeant ions alter the opening and closing transitions but do not alter the calcium- or voltage-dependent activation pathways (Thompson and Begenisich, 2012; Piskorowski and Aldrich, 2006). It is possible that PKD2-L1 pore blockade by outwardly moving Ca2+ ion(s) are displaced by inwardly Na+ current and a slow Ca2+ unbinding process prolongs the time course of channel closure. That is, hyperpolarization helps clear Ca2+ ions from the PKD2-L1 selectivity filter. This effect may be analogous to displacement of quaternary ammonium ion(s) by inward K+ current conducted by KV channels found in the squid giant axon (Armstrong, 1971).
Materials and methods
Whole-cell voltage-clamp experiments
HEK 293T cells were transiently transfected with the mammalian cell expression plasmid pTracer CMV2 containing the human PKD2-L1 gene (isoform 1). Cells were seeded onto glass coverslips and placed in a perfusion chamber, enabling changes in extracellular conditions. Data generated from cells patched in the whole cell configuration with <1 GΩ of resistance and >90 pA of leak current (Vmem =-60 mV) were not analyzed due to insufficient voltage control. Cell-attached single channel data with seal resistance <8 GΩ were not analyzed. Unless otherwise indicated, the pipette electrode solution contained (in mM): CsMES (80), NaCl (20), HEPES (10), MgCl2 (2), Cs4-BAPTA (5); CaCl2 was added to achieve 90 nM free Ca2+ and pH was adjusted to 7.4 with CsOH (MaxChelator (Bers et al., 2010)). The standard bath solution contained NaCl (150), HEPES (10), CaCl2 (2) and pH was adjusted with NaOH. When testing the relative permeability of monovalent cations, the bath solution contained (in mM): X-Cl (150), HEPES (10) and the pH was adjusted with X-OH; X is the indicated monovalent cation. When testing the relative permeability of Ca2+, the bath solution contained: NMDG (100), CaCl2 (20), HEPES (10), and pH was adjusted with CaOH2. All saline solutions were adjusted to 300 mOsm (±5) with mannitol, if needed. Data was analyzed by Igor Pro 7.00 (Wavemetrics, Lake Oswego, OR). The reversal potential, Erev was used to determine the relative permeability of monovalent cation X to Cs+ (PX/PCs) according to the following equation:
where Erev, α, R, T and F are the reversal potential, effective activity coefficients for cation x (i, internal and e, external), the universal gas constant, absolute temperature, and the Faraday constant, respectively. Based on out measurements of the Erev under our semi-bi-ionic conditions, the PKD2-L1 permeability of Cs+ and NMDG+ ions are equivalent, but are much less permeant to other cations tested (Ca+, K+ and Na+). The effective activity coefficients (αx) were calculated using the following equation:
where γx is the activity coefficient and [X] is the concentration of the ion. For calculations of membrane permeability, activity coefficients (γ) were calculated using the Debye-Hückel equation: 0.74, 0.72, 0.69, and 0.29 correspond to Na+, K+, Cs+ and Ca2+, respectively. To determine the relative permeability of divalent cations to Cs+, the following equation was used:
Erev for each cation condition was corrected to the measured liquid junction potentials (-4.4 to 3.4 mV). Single channel events (Figure 1—figure supplement 1 and Figure 3) were measured with standard extracellular saline in the pipette and high potassium saline in the bath (in mM): KCl (125), NaCl (20), HEPES (10) and CaCl2 (2) to neutralize the resting membrane potential. To determine the time course of channel opening (τpeak) and closure (τtail) the peak current during the depolarization (△ mV) and the tail currents during repolarization (−60 mV) were fit using the following exponential equation:
where τ is the time constant. The internal accumulation of calcium during experiments in Figure 2—figure supplement 1 was estimated using the following equation:
where Q is the cumulative integrated Itail current and r (0.199) is the product of the extracellular calcium (2 mM) to sodium (150 mM) ratio multiplied by the relative permeability of calcium for sodium ions (PCa/PNa = 14.9). With a starting condition of 100 nM free Ca2+, the cumulative [free Ca2+] was estimated using MaxChelator (Bers et al., 2010), where 5 mM BAPTA was used as an internal Ca2+ buffer. We assume that the 5 mM BAPTA perfusing the cell was the dominant buffer over intrinsic mobile and immobile buffers which ‘typically’ bind Ca2+ in the range of 40 bound/1 free (Zhou and Neher, 1993). HEK293 cells have an average radius of 8 μM and a volume of ~2100 μM (Nauli and Zhou, 2004), (2.1×10–12 L). Single channel current magnitude and open time was estimated at the resting membrane potentials for PKD2-L1 channel in CSF-contacting neurons (−55 mV) (Orts-Del'Immagine et al., 2016), PKD2-L1 (DeCaen et al., 2013) and PKD1-L1 +PKD2-L1 channels from primary cilia (−17 mV) (Delling et al., 2013).
Ca2+ uncaging experiments
PKD2-L1 transfected HEK cells were incubated with extracellular o-nitrophenyl EGTA (NP-EGTA; 4 mM) and Fluo-3-AM (4 mM) for 30 min at room temperature. Cells were visualized with an Olympus FV1000 confocal microscope equipped with an SIM scanner. After establishing a high resistance seal using electrodes with 3–5 mΩ pipette resistance in the on-cell configuration, the cell membrane potential was held at +80 mV and cytoplasmic calcium was uncaged by a 500 ms, 405 nm laser pulse. All images were analyzed using ImageJ (NIH) and IgorPro 7 (Wavemetrics).
Surface biotinylation assay
Biotinylation of cell-surface proteins was performed using EZ-Link Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. In brief, 48 hr after transfection, HEK 293T cells were washed with PBS, and EZ-Link Sulfo-NHS-SS-Biotin applied to living cultured cells expressing HA, HA-2L1, HA-2L1 (Stop-588) for 30 min at 4°C. Cells were lysed, and the biotinylated proteins were precipitated using streptavidin agarose beads. The eluted proteins were analyzed by immunoblotting.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Funding Information
This paper was supported by the following grants:
Howard Hughes Medical Institute to David E Clapham.
National Institutes of Health Pathway to Independence (PI) Award (K99/R00) to Paul G DeCaen.
Additional information
Competing interests
DEC: Reviewing editor, eLife.
The other authors declare that no competing interests exist.
Author contributions
PGD, Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting or revising the article, Contributed unpublished essential data or reagents.
XL, Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting or revising the article, Contributed unpublished essential data or reagents.
SA, Conception and design, Drafting or revising the article, Contributed unpublished essential data or reagents.
DEC, Conception and design, Analysis and interpretation of data, Drafting or revising the article.
References
- Armstrong CM. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. The Journal of General Physiology. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG. The dilated TRPA1 channel pore state is blocked by amiloride and analogues. Brain Research. 2011;1381:21–30. doi: 10.1016/j.brainres.2011.01.021. [DOI] [PubMed] [Google Scholar]
- Ben-Johny M, Dick IE, Sang L, Limpitikul WB, Kang PW, Niu J, Banerjee R, Yang W, Babich JS, Issa JB, Lee SR, Namkung H, Li J, Zhang M, Yang PS, Bazzazi H, Adams PJ, Joshi-Mukherjee R, Yue DN, Yue DT. Towards a Unified Theory of Calmodulin Regulation (Calmodulation) of Voltage-Gated Calcium and Sodium Channels. Current Molecular Pharmacology. 2015;8:188–205. doi: 10.2174/1874467208666150507110359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM, Patton CW, Nuccitelli R. A practical guide to the preparation of Ca(2+) buffers. Methods in Cell Biology. 2010;99:1–26. doi: 10.1016/B978-0-12-374841-6.00001-3. [DOI] [PubMed] [Google Scholar]
- Boulter C, Mulroy S, Webb S, Fleming S, Brindle K, Sandford R. Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:12174–12179. doi: 10.1073/pnas.211191098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XZ, Vassilev PM, Basora N, Peng JB, Nomura H, Segal Y, Brown EM, Reeders ST, Hediger MA, Zhou J. Polycystin-L is a calcium-regulated cation channel permeable to calcium ions. Nature. 1999;401:383–386. doi: 10.1038/43907. [DOI] [PubMed] [Google Scholar]
- Chen P, Wu JZ, Zhao J, Wang P, Luo J, Yang W, Liu XD. PKD2L1/PKD1L3 channel complex with an alkali-activated mechanism and calcium-dependent inactivation. European Biophysics Journal. 2015;44:483–492. doi: 10.1007/s00249-015-1040-y. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- DeCaen PG, Delling M, Vien TN, Clapham DE. Direct recording and molecular identification of the calcium channel of primary cilia. Nature. 2013;504:315–318. doi: 10.1038/nature12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delling M, DeCaen PG, Doerner JF, Febvay S, Clapham DE. Primary cilia are specialized calcium signalling organelles. Nature. 2013;504:311–314. doi: 10.1038/nature12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delling M, Indzhykulian AA, Liu X, Li Y, Xie T, Corey DP, Clapham DE. Primary cilia are not calcium-responsive mechanosensors. Nature. 2016;531:656–660. doi: 10.1038/nature17426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon-Shaag A, Zagotta WN, Gordon SE. Mechanism of Ca(2+)-dependent desensitization in TRP channels. Channels. 2008;2:125–129. doi: 10.4161/chan.2.2.6026. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Huang AL, Chen X, Hoon MA, Chandrashekar J, Guo W, Tränkner D, Ryba NJ, Zuker CS. The cells and logic for mammalian sour taste detection. Nature. 2006;442:934–938. doi: 10.1038/nature05084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimaru Y, Katano Y, Yamamoto K, Akiba M, Misaka T, Roberts RW, Asakura T, Matsunami H, Abe K. Interaction between PKD1L3 and PKD2L1 through their transmembrane domains is required for localization of PKD2L1 at taste pores in taste cells of circumvallate and foliate papillae. FASEB Journal. 2010;24:4058–4067. doi: 10.1096/fj.10-162925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Mühle A, Schaefer M, Strotmann R, Schultz G, Plant TD. Lanthanides potentiate TRPC5 currents by an action at extracellular sites close to the pore mouth. The Journal of Biological Chemistry. 2003;278:3562–3571. doi: 10.1074/jbc.M211484200. [DOI] [PubMed] [Google Scholar]
- Kawaguchi H, Yamanaka A, Uchida K, Shibasaki K, Sokabe T, Maruyama Y, Yanagawa Y, Murakami S, Tominaga M. Activation of polycystic kidney disease-2-like 1 (PKD2L1)-PKD1L3 complex by acid in mouse taste cells. The Journal of Biological Chemistry. 2010;285:17277–17281. doi: 10.1074/jbc.C110.132944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Drummond I, Ibraghimov-Beskrovnaya O, Klinger K, Arnaout MA. Polycystin 1 is required for the structural integrity of blood vessels. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1731–1736. doi: 10.1073/pnas.040550097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labro AJ, Priest MF, Lacroix JJ, Snyders DJ, Bezanilla F. Kv3.1 uses a timely resurgent K(+) current to secure action potential repolarization. Nature Communications. 2015;6:10173. doi: 10.1038/ncomms10173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler A, Linte RM, Nau C, Reeh P, Babes A. A high-threshold heat-activated channel in cultured rat dorsal root ganglion neurons resembles TRPV2 and is blocked by gadolinium. The European Journal of Neuroscience. 2007;26:12–22. doi: 10.1111/j.1460-9568.2007.05643.x. [DOI] [PubMed] [Google Scholar]
- Li Q, Liu Y, Zhao W, Chen XZ. The calcium-binding EF-hand in polycystin-L is not a domain for channel activation and ensuing inactivation. FEBS Letters. 2002;516:270–278. doi: 10.1016/S0014-5793(02)02513-9. [DOI] [PubMed] [Google Scholar]
- Li M, Du J, Jiang J, Ratzan W, Su LT, Runnels LW, Yue L. Molecular determinants of Mg2+ and Ca2+ permeability and pH sensitivity in TRPM6 and TRPM7. The Journal of Biological Chemistry. 2007;282:25817–25830. doi: 10.1074/jbc.M608972200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Toombes GE, Silberberg SD, Swartz KJ. Physical basis of apparent pore dilation of ATP-activated P2X receptor channels. Nature Neuroscience. 2015;18:1577–1583. doi: 10.1038/nn.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Peissel B, Babakhanlou H, Pavlova A, Geng L, Fan X, Larson C, Brent G, Zhou J. Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nature Genetics. 1997;17:179–181. doi: 10.1038/ng1097-179. [DOI] [PubMed] [Google Scholar]
- Lucas P, Ukhanov K, Leinders-Zufall T, Zufall F. A diacylglycerol-gated cation channel in vomeronasal neuron dendrites is impaired in TRPC2 mutant mice: mechanism of pheromone transduction. Neuron. 2003;40:551–561. doi: 10.1016/S0896-6273(03)00675-5. [DOI] [PubMed] [Google Scholar]
- Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM, Fleig A. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 2001;411:590–595. doi: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nature Genetics. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Zhou J. Polycystins and mechanosensation in renal and nodal cilia. BioEssays. 2004;26:844–856. doi: 10.1002/bies.20069. [DOI] [PubMed] [Google Scholar]
- Nelson TM, Lopezjimenez ND, Tessarollo L, Inoue M, Bachmanov AA, Sullivan SL. Taste function in mice with a targeted mutation of the pkd1l3 gene. Chemical Senses. 2010;35:565–577. doi: 10.1093/chemse/bjq070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orts-Del'Immagine A, Kastner A, Tillement V, Tardivel C, Trouslard J, Wanaverbecq N. Morphology, distribution and phenotype of polycystin kidney disease 2-like 1-positive cerebrospinal fluid contacting neurons in the brainstem of adult mice. PLoS One. 2014;9:e13413. doi: 10.1371/journal.pone.0087748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orts-Del'Immagine A, Seddik R, Tell F, Airault C, Er-Raoui G, Najimi M, Trouslard J, Wanaverbecq N. A single polycystic kidney disease 2-like 1 channel opening acts as a spike generator in cerebrospinal fluid-contacting neurons of adult mouse brainstem. Neuropharmacology. 2016;101 doi: 10.1016/j.neuropharm.2015.07.030. [DOI] [PubMed] [Google Scholar]
- Orts-Del'immagine A, Wanaverbecq N, Tardivel C, Tillement V, Dallaporta M, Trouslard J. Properties of subependymal cerebrospinal fluid contacting neurones in the dorsal vagal complex of the mouse brainstem. The Journal of Physiology. 2012;590:3719–3741. doi: 10.1113/jphysiol.2012.227959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskorowski RA, Aldrich RW. Relationship between pore occupancy and gating in BK potassium channels. The Journal of General Physiology. 2006;127:557–576. doi: 10.1085/jgp.200509482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annual Review of Physiology. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Janssens A, Voets T, Nilius B. Regulation of the murine TRPP3 channel by voltage, pH, and changes in cell volume. PflüGers Archiv : European Journal of Physiology. 2009;457:795–807. doi: 10.1007/s00424-008-0558-6. [DOI] [PubMed] [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/S0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Thompson J, Begenisich T. Selectivity filter gating in large-conductance Ca(2+)-activated K+ channels. The Journal of General Physiology. 2012;139:235–244. doi: 10.1085/jgp.201110748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topala CN, Groenestege WT, Thébault S, van den Berg D, Nilius B, Hoenderop JG, Bindels RJ. Molecular determinants of permeation through the cation channel TRPM6. Cell Calcium. 2007;41:513–523. doi: 10.1016/j.ceca.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Trudeau M, Warmke J, Ganetzky B, Robertson G. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K(+) channels: structure, function, and clinical significance. Physiological Reviews. 2012;92:1393–1478. doi: 10.1152/physrev.00036.2011. [DOI] [PubMed] [Google Scholar]
- Voets T, Janssens A, Prenen J, Droogmans G, Nilius B. Mg2+-dependent gating and strong inward rectification of the cation channel TRPV6. The Journal of General Physiology. 2003;121:245–260. doi: 10.1085/jgp.20028752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YY, Chang RB, Waters HN, McKemy DD, Liman ER. The nociceptor ion channel TRPA1 is potentiated and inactivated by permeating calcium ions. The Journal of Biological Chemistry. 2008;283:32691–32703. doi: 10.1074/jbc.M803568200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Somlo S. Molecular genetics and mechanism of autosomal dominant polycystic kidney disease. Molecular Genetics and Metabolism. 2000;69:1–15. doi: 10.1006/mgme.1999.2943. [DOI] [PubMed] [Google Scholar]
- Wu G, Markowitz GS, Li L, D'Agati VD, Factor SM, Geng L, Tibara S, Tuchman J, Cai Y, Park JH, van Adelsberg J, Hou H, Kucherlapati R, Edelmann W, Somlo S. Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nature Genetics. 2000;24:75–78. doi: 10.1038/71724. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Sweet TB, Clapham DE. International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacological Reviews. 2010;62:381–404. doi: 10.1124/pr.110.002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Ramsey IS, Kotecha SA, Moran MM, Chong JA, Lawson D, Ge P, Lilly J, Silos-Santiago I, Xie Y, DiStefano PS, Curtis R, Clapham DE. TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature. 2002;418:181–186. doi: 10.1038/nature00882. [DOI] [PubMed] [Google Scholar]
- Yu FH, Catterall WA. The VGL-Chanome: A Protein Superfamily Specialized for Electrical Signaling and Ionic Homeostasis. Science Signaling. 2004;2004:re15. doi: 10.1126/stke.2532004re15. [DOI] [PubMed] [Google Scholar]
- Yu Y, Ulbrich MH, Li MH, Dobbins S, Zhang WK, Tong L, Isacoff EY, Yang J, Li Ming-hui. Molecular mechanism of the assembly of an acid-sensing receptor ion channel complex. Nature Communications. 2012;3:1252. doi: 10.1038/ncomms2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Hussein S, Yang J, Huang J, Zhang F, Hernandez-Anzaldo S, Fernandez-Patron C, Cao Y, Zeng H, Tang J, Chen XZ. A novel PKD2L1 C-terminal domain critical for trimerization and channel function. Scientific Reports. 2015;5:9460. doi: 10.1038/srep09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Neher E. Mobile and immobile calcium buffers in bovine adrenal chromaffin cells. The Journal of Physiology. 1993;469:245–273. doi: 10.1113/jphysiol.1993.sp019813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu MX. Multiple roles of calmodulin and other Ca(2+)-binding proteins in the functional regulation of TRP channels. PflüGers Archiv : European Journal of Physiology. 2005;451:105–115. doi: 10.1007/s00424-005-1427-1. [DOI] [PubMed] [Google Scholar]