

Abstract
Heterozygous mutations of the Hfe gene have been proposed as cofactors in the development and progression of nonalcoholic fatty liver disease (NAFLD). Homozygous Hfe deletion previously has been shown to lead to dysregulated hepatic lipid metabolism and accentuated liver injury in a dietary mouse model of NAFLD. We sought to establish whether heterozygous deletion of Hfe is sufficient to promote liver injury when mice are exposed to a high‐calorie diet (HCD). Eight‐week‐old wild‐type and Hfe +/− mice received 8 weeks of a control diet or HCD. Liver histology and pathways of lipid and iron metabolism were analyzed. Liver histology demonstrated that mice fed a HCD had increased NAFLD activity score (NAS), steatosis, and hepatocyte ballooning. However, liver injury was unaffected by Hfe genotype. Hepatic iron concentration (HIC) was increased in Hfe +/− mice of both dietary groups. HCD resulted in a hepcidin‐independent reduction in HIC. Hfe +/− mice demonstrated raised fasting serum glucose concentrations and HOMA‐IR score, despite unaltered serum adiponectin concentrations. Downstream regulators of hepatic de novo lipogenesis (pAKT, SREBP‐1, Fas, Scd1) and fatty acid oxidation (AdipoR2, Pparα, Cpt1) were largely unaffected by genotype. In summary, heterozygous Hfe gene deletion is associated with impaired iron and glucose metabolism. However, unlike homozygous Hfe deletion, heterozygous gene deletion did not affect lipid metabolism pathways or liver injury in this model.
Keywords: Diabetes, iron, nonalcoholic fatty liver disease, steatohepatitis
Introduction
Nonalcoholic fatty liver disease (NAFLD) is increasingly common in the developed and developing world, affecting around 30% of many adult populations (Amarapurkar et al. 2007; Vernon et al. 2011). The advanced form of the disease, nonalcoholic steatohepatitis (NASH), can lead to life‐threatening complications including liver failure and liver cancer (Anstee et al. 2013). At present, effective treatment strategies to halt or reverse the natural history of NASH are lacking. Cofactors such as type II diabetes mellitus and iron overload have been implicated in NASH pathogenesis and represent readily treatable therapeutic targets (Dongiovanni et al. 2011; Smith and Adams 2011). A greater understanding of the mechanisms by which such cofactors promote NASH disease progression is essential in order to develop effective treatments.
Iron overload due to homozygous p.C282Y mutation of Hfe is responsible for the majority of cases of hereditary hemochromatosis seen worldwide (Bacon et al. 2011). Heterozygous p.C282Y mutations are found in approximately 11% of Caucasian populations and are associated with increased iron stores, but not with liver disease in the absence of an additional cofactor (Allen et al. 2008). In a large meta‐analysis, Hfe gene mutations have been shown to convey an increased risk of nonalcoholic steatohepatitis (Ellervik et al. 2007). Among individuals with NAFLD, the presence of the heterozygous p.H63D mutation of Hfe has been shown to be associated with more advanced histological injury as assessed by the NAFLD activity score (Nelson et al. 2012). An overview of the role of HFE in mammalian iron metabolism is summarized in Figure 1.
Figure 1.
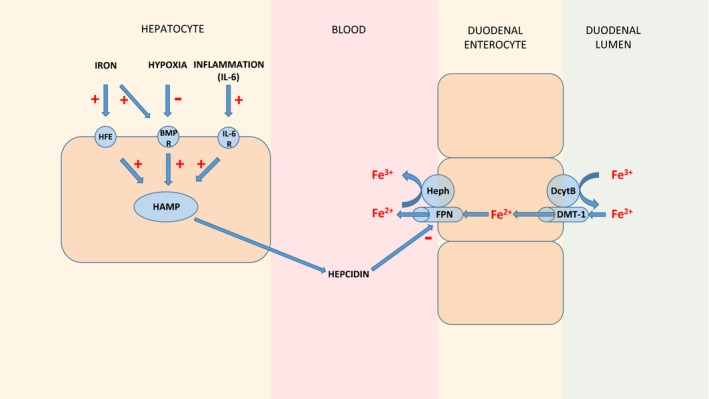
Overview of the regulation of mammalian iron homeostasis. The regulation of mammalian iron homeostasis is predominantly achieved via the control of duodenal absorption of iron by the hormone hepcidin. Hepcidin causes the internalization and degradation of the iron‐exporter ferroportin, thus reducing the passage of iron from enterocytes into the circulation. Other key proteins involved in duodenal iron absorption, divalent metal transporter 1 (DMT‐1), and the two oxidoreductases, hephaestin (Heph) and duodenal cytochrome B (DcytB), are shown. The HAMP gene encodes hepcidin and is regulated by a number of factors including iron, via the HFE and bone morphogenetic protein 6 (BMP‐6) pathway, hypoxia, via the BMP‐6 pathway, and inflammation, via an interleukin‐6 receptor (IL‐6) pathway. R denotes receptor. Adapted from Fleming et al. (Fleming and Ponka 2012) and Chua et al. (Chua et al. 2007).
Liver injury in both hemochromatosis and NASH is characterized by the presence of oxidative stress (Bacon et al. 2011; Rolo et al. 2012). Insulin resistance, itself associated with oxidative stress, is commonly observed both in individuals with NASH and also in those with hemochromatosis (Smith and Adams 2011; Simcox and McClain 2013). Given the prevalence of Hfe gene mutations and shared pathogenic mechanisms with NASH, they have received intense interest in recent years as potential cofactors in NAFLD disease progression.
Previous work from our group has demonstrated that mice with homozygous Hfe deletion which are fed a high‐calorie diet (HCD) develop steatohepatitis and early fibrosis (Tan et al. 2011). This effect of diet is not seen in wild‐type controls which develop only simple steatosis. In this model, Hfe null mice demonstrated upregulation of gene expression of de novo lipogenesis pathways and downregulation of fatty acid oxidation pathways. This imbalance of fatty acid synthesis and oxidation may explain the liver injury seen in these Hfe null mice. However, the mechanisms by which Hfe deletion might dysregulate hepatic lipid metabolism remain to be defined.
These findings led us to consider whether a partial deficiency of functional HFE, as seen in those with heterozygous C282Y and H63D mutations, might be sufficient to dysregulate hepatic lipid metabolism and promote liver injury in NAFLD. In this article, we sought to explore the mechanisms of interaction between heterozygous Hfe gene mutations and nonalcoholic fatty liver disease. We hypothesized that heterozygous Hfe deletion promotes dysregulated hepatic lipid metabolism as seen in the homozygous model. To test this hypothesis, we applied the 8‐week high‐calorie diet model of NAFLD, previously used in Hfe homozygous null mice, to mice with and without heterozygous deletion of Hfe.
Materials and Methods
Experimental animals
Eight‐week‐old male C57BL6/J Hfe +/− mice (bred at the QIMR Berghofer Medical Research Institute, Brisbane, Australia) and wild‐type (WT) littermate controls were assigned to receive a control diet (CD) or a high‐calorie (40.5% sucrose, 23.5% fat, 0.19% cholesterol by weight) diet (HCD) for 8 weeks (n = 10 per group). The constituents of these diets are summarized in Table 1. Both diets contained 1.3 μmol/g iron. The high‐calorie diet is analogous to a “Western” style diet, containing a high content of fat, simple carbohydrates, and cholesterol (Tan et al. 2011). Both diets were supplied by Specialty Feeds, Glen Forrest, Western Australia. All animals were cared for in accordance with the NHMRC code for the care and use of animals for scientific purposes and with approval of the Animal Ethics Committee of the QIMR Berghofer Medical Research Institute. Mice were housed in a temperature controlled environment (23°C) with a 12:12‐h light:dark cycle. Mice had ad libitum access to diet and water.
Table 1.
Major components of experimental diets
| Dietary component | Control diet | High‐calorie diet |
|---|---|---|
| Protein (% weight) | 19.4% | 19.4% |
| Total fat (% weight) | 7.0% | 23.0% |
| Total carbohydrate (% weight) | 61.7% | 50% |
| Digestible energy (MJ/kg) | 16.1 | 20 |
| Cholesterol (% weight) | 0% | 0.19% |
| Casein (acid) (g/kg) | 200 | 200 |
| Sucrose (g/kg) | 100 | 405 |
| Canola oil (g/kg) | 70 | 50 |
| Cellulose (g/kg) | 50 | 50 |
| Wheat starch (g/kg) | 404 | 50 |
| Dextrinized starch (g/kg) | 132 | 0 |
| Cocoa butter (g/kg) | 0 | 50 |
| Hydrogenated vegetable oil (g/kg) | 0 | 131 |
Sixteen‐week‐old mice were sacrificed by general anesthesia with intraperitoneal injection of ketamine and xylazine, following a 5‐h fast. Blood was collected by cardiac puncture and serum was stored at −80°C. Livers were excised, and pieces of tissue were either dried at 100°C for 72 h for hepatic iron concentration determination, fixed in formalin for histology, or snap frozen in liquid nitrogen and stored at −80°C. Small bowel enterocytes were collected as described previously (Chen et al. 2003; Fuqua et al. 2014). In brief, 10‐cm sections of proximal small bowel were cut longitudinally then washed in ice‐cold phosphate‐buffered saline (PBS). Samples were then gently rotated for 30 min at 4°C in 1.5 mmol/L ethylenediaminetetraacetic acid (EDTA) in PBS. The gut tissue was removed and the remaining sample containing enterocytes was centrifuged at 500 g to pellet the enterocytes.
Histological assessment
Formalin‐fixed samples were embedded in paraffin. Sections were stained with hematoxylin and eosin (H&E) for the assessment of liver injury. Sirius red staining was used to detect the presence of hepatic fibrosis. Histological scoring was performed by an expert histopathologist blinded to the study groups according to criteria described by Kleiner et al. (Kleiner et al. 2005).
Hepatic iron concentration
Oven dried liver samples weighing 4–9 mg were added to 300 μL of concentrated nitric acid. Duplicate samples for each liver were then digested in a heated sand bath. Chromogen reagent was prepared (0.1% banthophenanthroline sulfate, 1% mercaptoacetic acid). One part of chromogen reagent was added to five parts of saturated sodium acetate to make working chromogen reagent immediately prior to use. When fully digested, the volume of each sample was determined by pipette and 25 μL was added to 125 μL working chromogen reagent. Absorbance was measured at 540 nm on a plate reader, Tecan infinite F200, Tecan, Switzerland. The iron concentration by dry weight was determined with reference to an iron standard (Iron standard for AAS, Sigma, St. Louis).
Serum analysis
Serum alanine aminotransferase (ALT) was measured on a Beckman (DxC800) General Chemistry Automated Analyser (Beckman Coulter, Fullerton). Serum glucose and insulin were measured on a Cobas Integra 400 Chemistry Automated Analyser (Roche Diagnostics, Basel, Switzerland).
RNA extraction and real‐time quantitative PCR (RT‐qPCR)
RNA was extracted from tissue homogenates using QIAZOL reagent (Qiagen, Hilden, Germany). After treatment with deoxyribonuclease 1, cDNA was synthesized from 1 μg RNA, using superscript III reverse transcriptase (Invitrogen, Mulgrave, Australia). RT‐qPCR was performed in a ViiA 7 real‐time PCR machine (Invitrogen) with a Quantifast SYBR Green master mix (Qiagen) and thermal cycling as follows: 95°C for 5 min then 40 cycles at 95°C for 10 sec followed by 60°C for 30 sec prior to a melt curve analysis for validation. Relative mRNA expression was determined by calibration of Ct values to a standard curve of dilutions of a pooled mix of cDNA samples. Expression was then normalized to geometric mean of three reference genes glyceraldehyde‐3‐phosphate dehydrogenase (Gapdh), basic transcription factor‐3 (Btf3), and beta‐2‐microglobulin (B2‐mg). Primer sequences that were used are shown in Table 2.
Table 2.
RT‐qPCR primer sequences (5′ to 3′)
| Forward primer | Reverse primer | |
|---|---|---|
| Hamp1 | TTGCGATACCAATGCAGAAG | GGATGTGGCTCTAGGCTATGTT |
| Dmt1 | CCAGCCAGTAAGTTCAAGGATC | GCGTAGCAGCTGATCTGGG |
| Hephaestin | CCGACCTTACACCATTCACC | GGACAGAATCATCCGCTTTC |
| Fas | TACCAAGCCAAGCACATTCG | TGGCTTCGGCATGAGA |
| AdipoR2 | TACACACAGAGACGGGCAAC | TGGCTCCCAAGAAGAACAAG |
| Pparα | CATGTGAAGGCTGTAAGGGCTT | TCTTGCAGCTCCGATCACACT |
| Cpt1 | AGACCGTGAGGAACTCAAACCTA | TGAAGAGTCGCTCCCACT |
| Gapdh | TCCTGCACCACCAACTGCTTAGC | GCCTGCTTCACCACCTTCTTGAT |
| Btf3 | TGGCAGCAAACACCTTCACC | AGCTTCAGCCAGTCTCCTTAAAC |
| B2‐mg | CTGATACATACGCCTGCAGAGTTAA | ATGAATCTTCAGAGCATCATGAT |
Western blotting
Serum adiponectin levels were determined by western blotting. Samples of 1:1000 serum (5 μL) were electrophoresed in 2% Agarose (in Tris‐Glycine) gels (Lonza, Basel, Switzerland) at 70 V for 60 min. Protein levels of phosphoAKT (pAKT) and sterol regulatory element binding protein‐1 (SREBP‐1) were determined using western blotting of liver tissue extracts. Protein concentration was quantified using a Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford). Thirty micrograms of protein from whole liver protein extracts (pAKT) and 10 μg of protein from liver nuclear extracts (SREBP‐1) were electrophoresed in 10% sodium dodecyl sulfate–10% polyacrylamide gels for 10 min at 75 V, then 50 min at 150 V. Samples were then transferred onto polyvinylidene fluoride (PVDF) membranes (BioRad, Hercules) at 100 V for 60 min. Membranes were blocked in 5% skim milk before immunostaining with primary antibodies. Secondary antibody binding was performed using horseradish peroxidase (HRP) antibodies. Visualization was performed using a standard chemiluminescent kit (Supersignal West Femto, Thermo Scientific, Waltham) on a 4000MM pro Image Station (Carestream Health, Inc., New York). pAKT and SREBP‐1 band net intensity were normalized to the reference proteins GAPDH and Histone‐H3, respectively. Primary and secondary antibodies used for western blots were as follows: adiponectin (MAB3608, Millipore) 1:10,000, goat anti‐mouse HRP (Invitrogen) 1:200,000; pAKT (sc‐7985‐R, Santa Cruz) 1:1000, goat anti‐rabbit HRP (Invitrogen) 1:200,000; SREBP‐1 (sc‐367, Santa Cruz) 1:500, goat anti‐rabbit HRP (Invitrogen) 1:100,000; GAPDH (MAB374, Millipore) 1:150,000, goat anti‐mouse HRP (Invitrogen) 1:100,000; histone‐H3 (FL‐136, sc10809, Santa Cruz) 1:100, goat anti‐rabbit HRP (Invitrogen) 1:6000. All samples were processed concurrently using three gels with a minimum of three samples per group on each gel.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6 software (GraphPad, San Diego, CA). For normally distributed continuous data, groups were compared using two‐way ANOVA based on diet and genotype. For instances in which a significant interaction existed between diet and genotype, two predefined post hoc comparisons to evaluate the effect of genotype for each diet were performed using Sidak's multiple comparisons test. These comparisons were (2008) WT CD versus Hfe +/− CD and (2007) WT HCD versus Hfe +/− HCD. In cases in which a significant interaction did not occur, P values relating to overall effect by two‐way ANOVA of diet and genotype are reported. As initial body weight was measured prior to dietary intervention, an analysis between the two genotypes (n = 20 per group) was performed using an unpaired Student's t‐test.
For continuous data that were not normally distributed, a Kruskal–Wallis test was performed. When the results of this test were significant (P < 0.05), post hoc comparisons to evaluate the effect of genotype were performed using Dunn's multiple comparisons test (group comparisons as for Sidak's, above). Data for continuous variables are represented graphically with box and whisker plots, demonstrating the maximum, minimum, 25th and 75th centile and median values. Categorical data were analyzed by Fisher's exact test and represented in tabular form.
Results
Weight gain was largely comparable between genotypes
Weight gain between 8 and 16 weeks of age was largely comparable between genotypes except for a small increase in WT HCD‐fed mice compared to their Hfe +/− counterparts (13.1 g vs 11.0 g, P = 0.04, Sidak's multiple comparison test after two way ANOVA). This effect may be related to a higher initial weight at 8 weeks in WT mice. (28.4 g vs. 26.0 g, P = 0.016). The higher initial weight is unlikely to be a true genotype effect as a difference was not seen between the two CD groups. All four groups of mice (CD and HCD) had been fed only CD up until this stage. Mean initial weight for all WT mice (n = 20) was 27.5 g, compared to 26.9 g for Hfe +/− mice (n = 20), P = 0.39, Student's t‐test (Fig. 2A and B).
Figure 2.
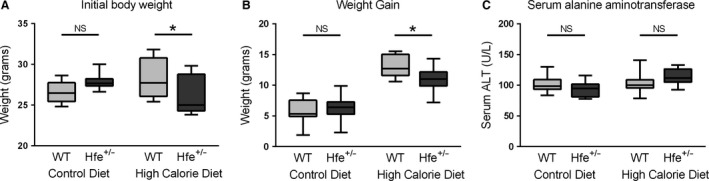
Body weight and serum ALT. (A) Initial body weight (8 weeks). An interaction between diet and genotype was present (P = 0.0044, two‐way ANOVA). Post hoc analysis found increased weight in WT mice fed a HCD compared to Hfe +/− mice (P = 0.016, Sidak's multiple comparisons test). No significant changes were seen in CD mice. (B) Weight gain (8–16 weeks). An interaction between diet and genotype was present (P = 0.040, two‐way ANOVA). Post hoc analysis found increased weight gain in WT mice fed a HCD compared to Hfe +/− mice (P = 0.040, Sidak's multiple comparisons test). No significant changes were seen in CD mice. (C) Serum alanine aminotransferase (ALT) is not influenced by diet or genotype. A significant interaction by two‐way ANOVA was present (P = 0.047). However, differences between genotype (Sidak's multiple comparisons test) were not significant for either mice fed control diet or HCD (n = 9–10 per group).
Diet, but not Hfe genotype influences liver injury in this model
There was no observed genotype effect on serum ALT (Fig. 2C). Mice fed HCD of both genotypes developed steatosis without overt steatohepatitis (Fig. 3). Table 3 shows the histological scoring of liver sections. NAFLD activity score (NAS) (P = 0.003), steatosis (P < 0.001), and hepatocyte ballooning (P = 0.038) were associated with experimental group by Fisher's exact test. These associations evidently relate to diet rather than genotype. No more than minimal fibrosis was seen in any of the groups (data not shown).
Figure 3.
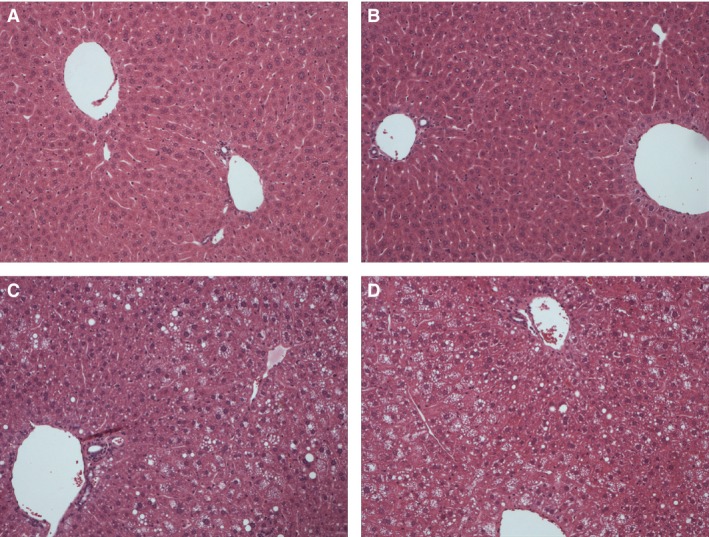
High‐calorie diet leads to increased hepatic steatosis and ballooning degeneration in Hfe +/− mice. Light microscopy of representative liver sections stained with hematoxylin and eosin are shown (original magnification ×100). (A) WT CD. (B) Hfe +/− CD. (C) WT HCD. (D) Hfe +/− HCD.
Table 3.
High‐calorie diet, but not heterozygous Hfe gene deletion leads to increased hepatic steatosis, ballooning degeneration, and NAS score
| Control diet | High calorie diet | P‐value | |||
|---|---|---|---|---|---|
| WT | Hfe +/− | WT | Hfe +/− | ||
| NAS (≥2) | 0 (0%) | 0 (0%) | 5 (50%) | 5 (50%) | 0.003 |
| Steatosis (Yes) | 0 (0%) | 0 (0%) | 8 (80%) | 8 (80%) | <0.001 |
| Lobular Inflammation (Yes) | 4 (40%) | 3 (30%) | 6 (60%) | 6 (60%) | 0.54 |
| Ballooning (Yes) | 0 (0%) | 0 (0%) | 4 (40%) | 2 (20%) | 0.038 |
Number of mice (%) with NAS (≥2), any macrovesicular steatosis (≥ grade 1), lobular inflammation (≥ grade 1), or ballooning (≥ grade 1). P‐value is the result of Fisher's exact test. (n = 10 per group).
Hepatic iron concentration (HIC) is increased in Hfe +/− mice in both dietary groups, consistent with the expected phenotype
Hamp1 is the gene encoding hepcidin, the master regulator of iron homeostasis (Fig. 4A). When Hamp1 is expressed as a ratio of HIC, expression was found to be approximately 50% lower in Hfe +/− mice across both dietary groups (Fig. 4C). This observation is consistent with haploinsufficiency of the HFE protein in Hfe +/− mice and supports HFE being the predominant regulator of Hamp1 expression in this model.
Figure 4.
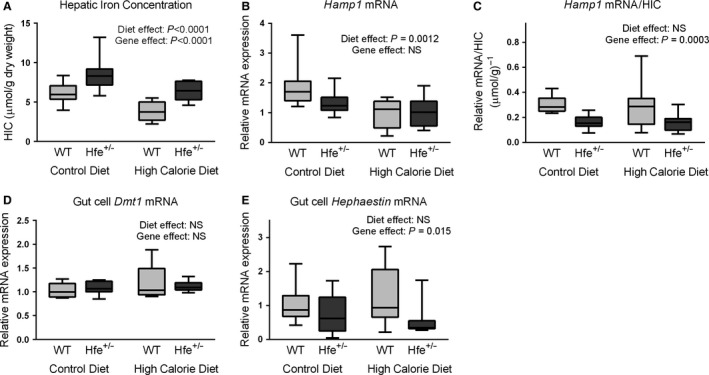
High‐calorie diet‐induced reduction in hepatic iron concentration (HIC) occurs independently of hepcidin. (A) Hepatic iron concentration. HCD was associated with reduced HIC (P < 0.0001, two‐way ANOVA). Hfe +/− mice had increased HIC compared to WT mice (P < 0.0001, two‐way ANOVA). (B) Hepatic Hamp1 mRNA expression. Hamp1 was lower in animals fed HCD (P = 0.0012, two‐way ANOVA). Gene effect was not significant (NS). (C) Hepatic Hamp1 mRNA/HIC. Hfe +/− mice had lower Hamp1/HIC ratios (P = 0.0003, two‐way ANOVA). Diet effect was NS. (D) Gut cell Dmt1 mRNA expression. Diet and gene effects were both NS (two‐way ANOVA). (E) Gut cell Hephaestin mRNA expression. Hfe +/− mice had lower Hephaestin expression than WT mice (P = 0.015). Diet effect was NS (two‐way ANOVA). (n = 6–10 per group).
High‐calorie diet‐induced reduction of HIC occurs independently of hepcidin
HIC was significantly reduced by HCD in both genotypes (P < 0.0001, two‐way ANOVA) (Fig. 4A). The explanation for this is unclear. Hamp1 mRNA expression was lower in HCD‐fed mice (diet effect P = 0.0012, two‐way ANOVA, Fig. 4B). The Hamp1/HIC results demonstrate that Hamp1 expression when normalized to HIC is unaffected by diet (Fig. 4C). As the hepcidin–ferroportin axis did not appear to be the cause of the reduced HIC, we sought to evaluate two further regulators of intestinal iron absorption, the divalent metal transporter‐1 (DMT1) and hephaestin. There was no significant diet effect at the mRNA level for either of these by two‐way ANOVA (Fig. 4D and E). Hfe +/− mice demonstrated a reduction in hephaestin mRNA levels. Given the increased iron loading in these mice, this response is consistent with previous work which has shown hephaestin mRNA levels to be regulated by iron (Anderson et al. 2002).
Hfe +/− mice display impaired glucose homeostasis
As glucose and insulin are established drivers of hepatic de novo lipogenesis (Leavens and Birnbaum 2011), we measured their fasting serum concentrations. Serum glucose was significantly higher in Hfe +/− than WT animals (genotype effect P = 0.0007, two‐way ANOVA); 8.1 mmol/L versus 7.3 mmol/L in CD‐fed mice and 8.0 mmol/L versus 7.2 mmol/L in HCD‐fed mice (Fig. 5A). A trend toward a similar effect was seen for serum insulin although this did not reach statistical significance (Fig. 5B). HOMA‐IR score, which is the product of serum glucose, serum insulin, and a constant, has been shown to be a useful static measure of insulin resistance in the fasting state (Matthews et al. 1985). HOMA‐IR was significantly higher in Hfe +/− mice fed both CD (P = 0.038) and HCD (P= 0.039) using Dunn's multiple comparison test after a significant Kruskal–Wallis test (P = 0.0024) (Fig. 5C). Serum adiponectin has previously been shown to be regulated by iron in mice and is a known key determinant of insulin resistance (Gabrielsen et al. 2012). However, we did not find any significant effect of genotype on total serum adiponectin (Fig. 5D and E). Moreover, when we looked specifically at the active form, high‐molecular‐weight (HMW) adiponectin, we again found no genotype effect irrespective of whether we analyzed absolute HMW adiponectin serum concentration or HMW adiponectin as a fraction of total adiponectin.
Figure 5.
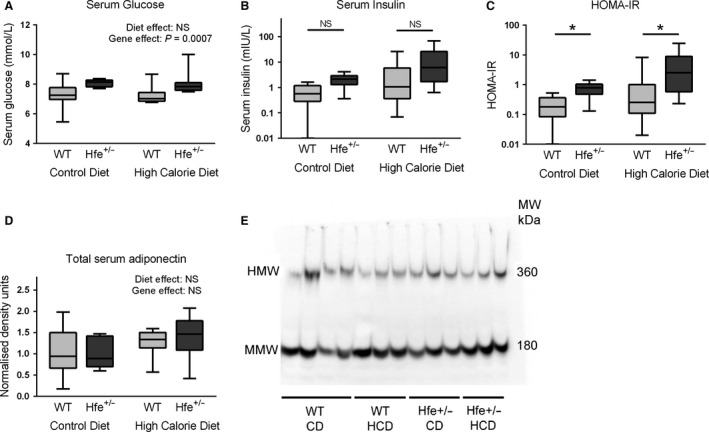
Hfe +/− mice have impaired glucose homeostasis irrespective of diet. (A) Serum glucose. Serum glucose was higher in Hfe +/− mice compared to WT mice (P = 0.0007), diet effect was NS (two‐way ANOVA). (B) Serum insulin (mIU/L), presented on logarithmic (base 10) scale. A difference between groups was present P = 0.004 (Kruskal–Wallis test). Post hoc comparisons by genotype for CD and HCD mice were both NS (P = 0.057, P = 0.084, respectively, by Dunn's multiple comparison test). (C) HOMA‐IR, presented on logarithmic (base 10) scale. A difference between groups was present (P = 0.0024) (Kruskal–Wallis test). Hfe +/− was associated with increased HOMA‐IR score for mice fed CD (P = 0.038) and HCD (P = 0.039) (Dunn's multiple comparison test). Immunoblotting for total serum adiponectin, (D) densitometry, (E) representative blots. HMW, high‐molecular‐weight adiponectin. MMW, medium molecular weight adiponectin. Diet and gene effects were both NS (two‐way ANOVA). (n = 9–10 per group).
Despite increased serum glucose and HOMA‐IR score, downstream regulators of de novo lipogenesis and fatty acid oxidation are largely unaffected by heterozygous Hfe deletion
The active form of serine/threonine kinase AKT, phospho AKT, which is a central regulator of insulin signaling to lipogenesis pathways, was entirely unaltered by diet or genotype (Fig. 6A and C). Similarly, nuclear extract quantities of sterol regulatory element binding protein‐1 (SREBP‐1), the main transcription factor responsible for regulation of de novo lipogenesis, were also unaffected by genotype (Fig. 6B and C). HCD, however, was associated with an increase in nuclear SREBP‐1 protein, which may be substrate driven. mRNA quantities of fatty acid synthase (Fas) and stearoyl CoA desaturase‐ 1 (Scd1), which are enzymes involved in hepatic lipogenesis and are downstream targets of SREBP‐1, were also unaffected by genotype (Fig. 6D and E). Scd1 mRNA, however, was upregulated by HCD, consistent with the observation that Scd1 is transcriptionally regulated by SREBP‐1 (Mauvoisin and Mounier 2011). Given the lack of genotype effect on liver histology or SREBP‐1 protein levels and that SREBP‐1 in known to act on Fas and Scd1 by regulation of transcription, we focussed on mRNA rather than protein levels of these enzymes (Latasa et al. 2003; Mauvoisin and Mounier 2011). Similarly, there was no effect of Hfe heterozygosity on mRNA quantities of three key regulators of fatty acid oxidation: adiponectin receptor‐2 (Adipo R2), peroxisome proliferator‐activated receptor alpha (Pparα), and carnitine palmitoyl transferase‐1 (Cpt1), except for a small increase in Cpt1 expression in Hfe +/− mice fed a HCD (P = 0.040, Sidak's multiple comparisons test) (Fig. 7A–C).
Figure 6.
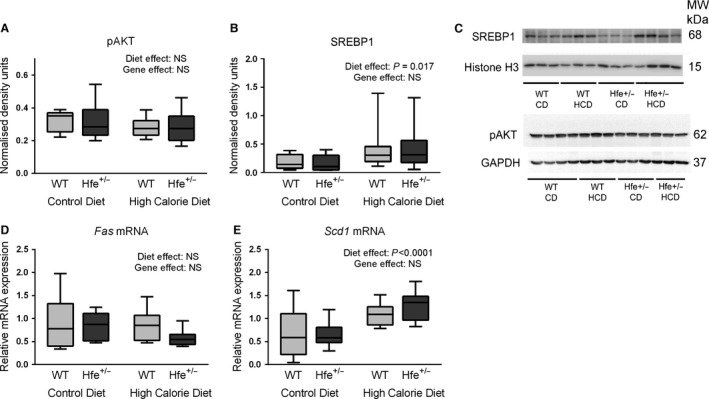
Hepatic de novo lipogenesis pathways are not upregulated despite hyperglycemia. (A) Immunoblotting densitometry of pAKT (whole liver protein extracts) normalized to GAPDH. Diet and genotype effects were both NS (two‐way ANOVA). (B) Immunoblotting densitometry of SREBP‐1 (nuclear protein extracts) normalized to Histone‐H3. HCD was associated with increased nuclear SREBP‐1 (P = 0.017). Gene effect was NS (two‐way ANOVA). (C) Representative immunoblots for pAKT, GAPDH, SREBP‐1, Histone‐H3. (n = 9–10 per group). (D) Hepatic fatty acid synthase (Fas) mRNA expression. Diet and genotype effects were both NS (two‐way ANOVA). (E) Hepatic stearoyl CoA desaturase‐1 (Scd1) mRNA expression. HCD was associated with increased Scd1 expression. Gene effect was NS (two‐way ANOVA).
Figure 7.
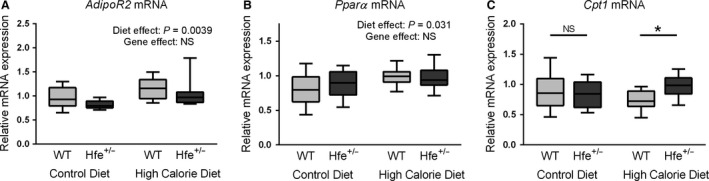
Hepatic lipid metabolism mRNA. (A) Hepatic adiponectin receptor‐2 (Adipo R2) mRNA expression. HCD was associated with increased AdipoR2 expression (P = 0.0039). Gene effect was NS (two‐way ANOVA). (B) Hepatic peroxisome proliferator‐activated receptor alpha (Pparα) mRNA expression. Pparα expression was increased by HCD. Gene effect was NS. (C) Hepatic carnitine palmitoyl transferase‐1 (Cpt1) mRNA expression. There was a significant interaction between diet and genotype (P = 0.045). Post hoc analysis found increased Cpt1 expression due to Hfe +/− in HCD‐fed mice (P = 0.040). In CD‐fed mice the results are NS (Sidak's multiple comparisons test) (n = 10 per group).
Discussion
In this study, we have demonstrated that heterozygous Hfe gene deletion in our mouse model of NAFLD leads to impaired glucose homeostasis in the fasted state, characterized by raised serum glucose concentrations and HOMA‐IR scores. Despite this, the dysregulation of hepatic lipid metabolism and histological evidence of increased liver injury seen previously in Hfe −/− mice were not seen in Hfe +/− mice.
Increased serum glucose in Hfe +/− mice is a notable finding and is significant in relation to HFE's role in insulin sensitivity, type II diabetes mellitus, and NAFLD (Nelson et al. 2012; Rong et al. 2012). This study, however, was not primarily designed to investigate glucose homeostasis. Undoubtedly, dynamic measures of glucose homeostasis such as glucose and pyruvate tolerance testing with hyperinsulinemic–euglycemic clamp studies would help future studies to characterize the extent and specific location of insulin resistance in this model.
The links between iron, insulin resistance, and type II diabetes have been extensively studied (Simcox and McClain 2013). Two large longitudinal cohort studies have demonstrated increased risk of diabetes associated with hyperferritinemia (Ford and Cogswell 1999; Montonen et al. 2012). These associations remained valid even after accounting for known confounders such as inflammation. Furthermore, therapeutic phlebotomy as a method of iron depletion has been shown in small studies to improve glycemic control in nondiabetic, prediabetic, and diabetic subjects (Facchini 1998; Fernandez‐Real et al. 2002; Houschyar et al. 2012).
Somewhat counterintuitively, previous studies have suggested enhanced insulin sensitivity in homozygous Hfe null mice (Huang et al. 2007, 2011). This has been proposed to be due to a lack of internalization of adipocyte ferroportin, subsequent adipocyte iron depletion, and upregulation of the expression of the insulin sensitizing adipokine, adiponectin (Gabrielsen et al. 2012). This effect, however, was not seen in our model, as heterozygous Hfe +/− deletion did not appear to be sufficient to interfere with serum adiponectin levels.
The findings of our study are relevant if one considers the human data regarding Hfe mutations and type II diabetes mellitus risk. A large meta‐analysis that reviewed studies describing Hfe gene polymorphisms and the risk of type II diabetes concluded that the heterozygous H63D mutation was associated with increased risk of diabetes (Rong et al. 2012). In the context of this human data, our study provides a clear framework and suitable model for future animal studies exploring the mechanistic links between HFE and type II diabetes.
Among individuals with NAFLD, the presence of heterozygous H63D mutation has been associated with higher steatosis grades and NAS score (Nelson et al. 2012). Given that H63D has less iron loading potential than the C282Y mutation, this raises the intriguing possibility of an iron‐independent role for the HFE protein in macronutrient metabolism that might protect against diabetes and NASH.
Heterozygous Hfe deletion did not appear to influence liver injury in this model, but it may be possible that histological differences between Hfe +/− and WT mice would only become evident in older mice with a more prolonged exposure to HCD and greater hepatic iron loading. Given the lack of lobular inflammation seen on liver histology, inflammatory pathways have not been explored in this model. Although it is possible that such pathways may be early markers of liver injury, analysis of these pathways would be better suited to a model with more prolonged exposure to HCD.
An interesting finding in this study was the effect of both diet and genotype on hepatic iron concentration (HIC). Increased hepatic iron in Hfe +/− mice is consistent with reduced stimulation of hepcidin–ferroportin axis in the setting of HFE deficiency. Humans with heterozygous C282Y mutations have been shown to have increased levels of serum ferritin, a marker of tissue iron stores, when compared to wild‐type controls (Allen et al. 2008). The lower HIC observed with HCD is consistent with other previous studies (Tan et al. 2011; Sonnweber et al. 2012). However, the lack of dietary effect on Hamp1 mRNA when normalized to HIC suggests that hepcidin response remains appropriate in this model. Similarly, we found no dietary effect on the mRNA levels of two other regulators of iron absorption, the enterocyte iron transporter DMT1 and the ferroxidase, hephaestin, despite a previous report of dysregulation of the mRNA transcripts of these genes in response to a high‐fat diet (Sonnweber et al. 2012).
In conclusion, we have demonstrated impaired glucose and iron homeostasis with heterozygous Hfe gene deletion in a mouse model of NAFLD. The genetic defect, however, was not associated with increased liver injury or impaired hepatic lipid metabolism.
Conflict of Interest
None declared.
Britton L., Jaskowski L., Bridle K., Santrampurwala N., Reiling J., Musgrave N., Subramaniam V. N., Crawford D.. Heterozygous Hfe gene deletion leads to impaired glucose homeostasis, but not liver injury in mice fed a high‐calorie diet. Physiol Rep, 4 (12), 2016, e12837, doi: 10.14814/phy2.12837
Funding Information
Funding for this work was supported by a National Health and Medical Research Council (NHMRC) Grant APP1029574. Dr. Laurence Britton is supported by scholarships from the Gallipoli Medical Research Foundation and the Gastroenterological Society of Australia.
References
- Allen, K. J. , Gurrin L. C., Constantine C. C., Osborne N. J., Delatycki M. B., Nicoll A. J., et al. 2008. Iron‐overload‐related disease in HFE hereditary hemochromatosis. N. Engl. J. Med. 358:221–230. [DOI] [PubMed] [Google Scholar]
- Amarapurkar, D. N. , Hashimoto E., Lesmana L. A., Sollano J. D., Chen P. J., and Goh K. L.; Asia‐Pacific Working Party on NAFLD . 2007. How common is non‐alcoholic fatty liver disease in the Asia‐Pacific region and are there local differences? J. Gastroenterol. Hepatol. 22:788–793. [DOI] [PubMed] [Google Scholar]
- Anderson, G. J. , Frazer D. M., McKie A. T., and Vulpe C. D.. 2002. The ceruloplasmin homolog hephaestin and the control of intestinal iron absorption. Blood Cells Mol. Dis. 29:367–375. [DOI] [PubMed] [Google Scholar]
- Anstee, Q. M. , Targher G., and Day C. P.. 2013. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 10:330–344. [DOI] [PubMed] [Google Scholar]
- Bacon, B. R. , Adams P. C., Kowdley K. V., Powell L. W., and Tavill A. S.. 2011. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54:328–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. , Su T., Attieh Z. K., Fox T. C., McKie A. T., Anderson G. J., et al. 2003. Systemic regulation of Hephaestin and Ireg1 revealed in studies of genetic and nutritional iron deficiency. Blood 102:1893–1899. [DOI] [PubMed] [Google Scholar]
- Chua, A. C. , Graham R. M., Trinder D., and Olynyk J. K.. 2007. The regulation of cellular iron metabolism. Crit. Rev. Clin. Lab. Sci. 44:413–459. [DOI] [PubMed] [Google Scholar]
- Dongiovanni, P. , Fracanzani A. L., Fargion S., and Valenti L.. 2011. Iron in fatty liver and in the metabolic syndrome: a promising therapeutic target. J. Hepatol. 55:920–932. [DOI] [PubMed] [Google Scholar]
- Ellervik, C. , Birgens H., Tybjaerg‐Hansen A., and Nordestgaard B. G.. 2007. Hemochromatosis genotypes and risk of 31 disease endpoints: meta‐analyses including 66,000 cases and 226,000 controls. Hepatology 46:1071–1080. [DOI] [PubMed] [Google Scholar]
- Facchini, F. S. 1998. Effect of phlebotomy on plasma glucose and insulin concentrations. Diabetes Care 21:2190. [PubMed] [Google Scholar]
- Fernandez‐Real, J. M. , Penarroja G., Castro A., Garcia‐Bragado F., Hernandez‐Aguado I., and Ricart W.. 2002. Blood letting in high‐ferritin type 2 diabetes: effects on insulin sensitivity and beta‐cell function. Diabetes 51:1000–1004. [DOI] [PubMed] [Google Scholar]
- Fleming, R. E. , and Ponka P.. 2012. Iron overload in human disease. N. Engl. J. Med. 366:348–359. [DOI] [PubMed] [Google Scholar]
- Ford, E. S. , and Cogswell M. E.. 1999. Diabetes and serum ferritin concentration among U.S. adults. Diabetes Care 22:1978–1983. [DOI] [PubMed] [Google Scholar]
- Fuqua, B. K. , Lu Y., Darshan D., Frazer D. M., Wilkins S. J., Wolkow N., et al. 2014. The multicopper ferroxidase hephaestin enhances intestinal iron absorption in mice. PLoS ONE 9:e98792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielsen, J. S. , Gao Y., Simcox J. A., Huang J., Thorup D., Jones D., et al. 2012. Adipocyte iron regulates adiponectin and insulin sensitivity. J. Clin. Invest. 122:3529–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houschyar, K. S. , Ludtke R., Dobos G. J., Kalus U., Broecker‐Preuss M., Rampp T., et al. 2012. Effects of phlebotomy‐induced reduction of body iron stores on metabolic syndrome: results from a randomized clinical trial. BMC Med. 10:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J. , Gabrielsen J. S., Cooksey R. C., Luo B., Boros L. G., Jones D. L., et al. 2007. Increased glucose disposal and AMP‐dependent kinase signaling in a mouse model of hemochromatosis. J. Biol. Chem. 282:37501–37507. [DOI] [PubMed] [Google Scholar]
- Huang, J. , Jones D., Luo B., Sanderson M., Soto J., Abel E. D., et al. 2011. Iron overload and diabetes risk: a shift from glucose to Fatty Acid oxidation and increased hepatic glucose production in a mouse model of hereditary hemochromatosis. Diabetes 60:80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner, D. E. , Brunt E. M., Van Natta M., Behling C., Contos M. J., Cummings O. W., et al. 2005. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41:1313–1321. [DOI] [PubMed] [Google Scholar]
- Latasa, M. J. , Griffin M. J., Moon Y. S., Kang C., and Sul H. S.. 2003. Occupancy and function of the ‐150 sterol regulatory element and ‐65 E‐box in nutritional regulation of the fatty acid synthase gene in living animals. Mol. Cell. Biol. 23:5896–5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavens, K. F. , and Birnbaum M. J.. 2011. Insulin signaling to hepatic lipid metabolism in health and disease. Crit. Rev. Biochem. Mol. Biol. 46:200–215. [DOI] [PubMed] [Google Scholar]
- Matthews, D. R. , Hosker J. P., Rudenski A. S., Naylor B. A., Treacher D. F., and Turner R. C.. 1985. Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419. [DOI] [PubMed] [Google Scholar]
- Mauvoisin, D. , and Mounier C.. 2011. Hormonal and nutritional regulation of SCD1 gene expression. Biochimie 93:78–86. [DOI] [PubMed] [Google Scholar]
- Montonen, J. , Boeing H., Steffen A., Lehmann R., Fritsche A., Joost H. G., et al. 2012. Body iron stores and risk of type 2 diabetes: results from the European Prospective Investigation into Cancer and Nutrition (EPIC)‐Potsdam study. Diabetologia 55:2613–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, J. E. , Brunt E. M., and Kowdley K. V.; Nonalcoholic Steatohepatitis Clinical Research Network . 2012. Lower serum hepcidin and greater parenchymal iron in nonalcoholic fatty liver disease patients with C282Y HFE mutations. Hepatology 56:1730–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolo, A. P. , Teodoro J. S., and Palmeira C. M.. 2012. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 52:59–69. [DOI] [PubMed] [Google Scholar]
- Rong, Y. , Bao W., Rong S., Fang M., Wang D., Yao P., et al. 2012. Hemochromatosis gene (HFE) polymorphisms and risk of type 2 diabetes mellitus: a meta‐analysis. Am. J. Epidemiol. 176:461–472. [DOI] [PubMed] [Google Scholar]
- Simcox, J. A. , and McClain D. A.. 2013. Iron and diabetes risk. Cell Metab. 17:329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, B. W. , and Adams L. A.. 2011. Nonalcoholic fatty liver disease and diabetes mellitus: pathogenesis and treatment. Nat. Rev. Endocrinol. 7:456–465. [DOI] [PubMed] [Google Scholar]
- Sonnweber, T. , Ress C., Nairz M., Theurl I., Schroll A., Murphy A. T., et al. 2012. High‐fat diet causes iron deficiency via hepcidin‐independent reduction of duodenal iron absorption. J. Nutr. Biochem. 23:1600–1608. [DOI] [PubMed] [Google Scholar]
- Tan, T. C. , Crawford D. H., Jaskowski L. A., Murphy T. M., Heritage M. L., Subramaniam V. N., et al. 2011. Altered lipid metabolism in Hfe‐knockout mice promotes severe NAFLD and early fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 301:G865–G876. [DOI] [PubMed] [Google Scholar]
- Vernon, G. , Baranova A., and Younossi Z. M.. 2011. Systematic review: the epidemiology and natural history of non‐alcoholic fatty liver disease and non‐alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 34:274–285. [DOI] [PubMed] [Google Scholar]