

Introduction
Berardinelli-Seip syndrome or ‘Congenital generalized lipodystrophy’ is an autosomal recessive disorder characterized by generalized loss of body fat involving face, trunks and limb. Approximately 120 patients of various ethnic backgrounds have been reported [1].
Case Report
Two siblings - a 13 year old girl and her 7 year old brother, born out of second degree consanguineous marriage, presented with abnormal facial features and gradually increasing dark discoloration of skin. On examination, the girl had acromegalic features with coarse facies, large hands and feet. There was generalized loss of subcutaneous fat, hirsutism and prominent musculatures over face and limbs. Acanthosis nigricans was present over flexural areas, including neck, axilla, elbow, waist and knee. Abdomen was distended due to hepatosplenomegaly (no ascites was detected). There was mild clitoral hypertrophy. Intelligence was normal. Biochemical parameters revealed hyperlipidemia (Serum cholesterol 250 mg/dl, triglycerides 160 mg/dl, LDL 188 mg/dl, HDL 30 mg/dl). Blood glucose was normal. Her brother had similar features of generalized loss of subcutaneous fat with muscular prominence, adenoid facies with hypertrophied pharyngeal tonsils and short stature (Ht 110 cm i.e. < 5th percentile). Acanthosis nigricans was present, but to a lesser extent. Blood sugar and lipid profile were normal.
Discussion
Lipodystrophies represent a heterogeneous group of diseases characterized by generalized or partial alterations in body fat development or distribution and insulin resistance. The other cardinal clinical signs of these syndromes are acanthosis nigricans, frequent hypergonadism in females, muscular hypertrophy and liver steatosis [2]. Lipodystrophies are classified as congenital or acquired (depending on origin) and, generalized or partial (based on clinical pattern).
Congenital generalized lipodystrophy (Berardinelli-Seip syndrome) is transmitted as an autosomal recessive trait and is linked to two genetic loci, on chromosomes 9q34 and 11q13 [2]. Recently, the 11q13 locus has been identified as the BSCL2 gene, encoding seipin, a protein of unknown function mainly expressed in brain [4].
The pathophysiology of lipodystrophies is still unknown. However, murine models of lipoatrophic diabetes revealed that primary genetic alterations in fat development resulted in diabetes and dyslipidemia [5]. Leptin deficiency, caused by the absence of adipose tissue, could be an important determinant of the metabolic abnormalities since exogenous administration or transgenic overexpression of leptin has been shown to markedly improve insulin sensitivity, glycemic control, dyslipidemia and hepatic steatosis in mice. Similarly, the defect in adinopectin, another fat derived hormone, has recently been shown to be involved in insulin resistance [6].
Males and females are equally affected and the clinical manifestations are obvious from birth, because of the lack of subcutaneous fat. Fat cells are present but are reduced in number and size, containing little fat. Anabolic features are observed at birth, with enlarged visceral organs. Toddlers may have potentially dangerous hyperplasia of pharyngeal tonsils and adenoids. Lipodystrophies produce a distinctive facies. A well-defined musculature with prominent superficial veins is one of the earliest manifestations. Clitoral or penile hypertrophy has been evident at birth, but genitomegaly is not apparent after puberty. The earliest skin manifestations include acanthosis nigricans, eruptive xanthomas and hirsutism. All patients have acanthosis nigricans to some degree [7]. Acanthosis nigricans can diminish and disappear with puberty and is said to be prominent on the elbows, knees, waist, neck and axilla. Acromegalic gigantism with advanced dentition is an early and constant feature. The growth rate is most marked in the first 4 years, and these children may attain 90 percent of their adult growth within the first 10 years of their life. Growth subsequently slows and adults are normal or short stature. Females have a masculine habitus with marked muscularity. Liver disease with fatty steatosis and cirrhosis is another constant feature. Hepatosplenomegaly tends to produce a markedly protuberant abdomen. Diabetes mellitus usually begins in teenage years. The diabetes is insulin resistant and despite poor control ketosis is absent. Hyperlipidemia usually precedes the diabetes. An increased basal metabolic rate is a frequent finding. Intelligence may range from normal to subnormal. Kidneys may be enlarged without apparent histologic cause and renal failure may ensue. Cardiomegaly is frequently observed with muscular hypertrophy and ventricular dysfunction. Common causes of death renal failure and GI hemorrhage from oesophageal varices in association with hepatic failure. Patients survive into young adulthood or early middle age.
Fig. 1.
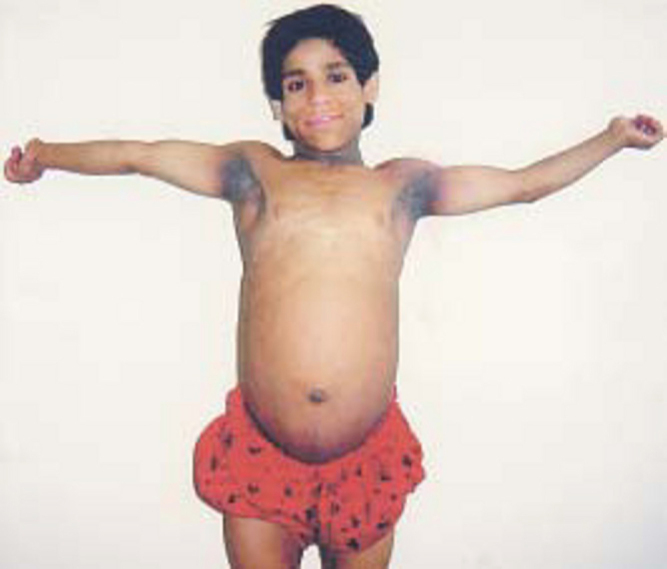
Case of Berardinelli Seip syndrome in a 13 year girl with prominent musculature, masculine habitus, acanthosis nigricans of axilla and neck, protuberant abdomen
Treatment for acanthosis nigricans can be accomplished with etretinate (beginning at 75 mg/day and increased gradually to 0.5 mg/kg/day after 10-12 weeks) [8]. Dietary fish oil may also be useful in acanthosis nigricans. Substitution of eucaloric medium chain triglycerides for long chain fatty acids can lead to improvement of chylomicronemia, xanthomatosis, hypertriglyceridemia, hepatomegaly, carbohydrate tolerance and hyperinsulinemia, but not lipoatrophy. The limited ability to store energy as fat means patients must maintain a rigid special diet with 4 regular sized meals each day and avoid large meals. Easily digestible carbohydrate should be restricted and dietary fibre is important [9].
Fig. 2.
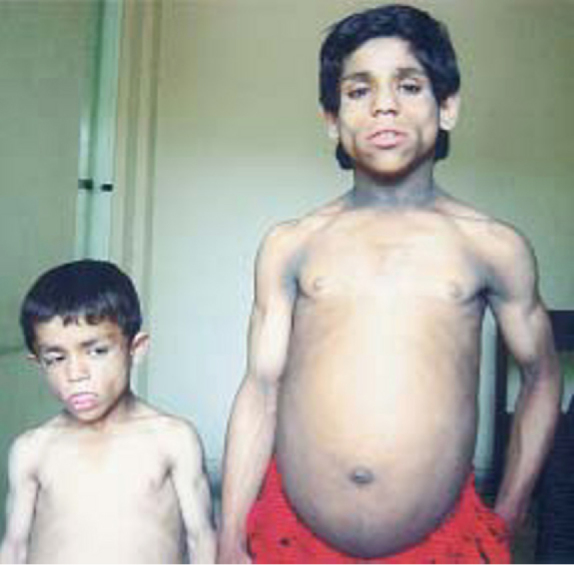
Affected cases in 2 siblings (13 years girt and 7 yr boy) of same family with consanguineous marriage in parents
References
- 1.Garg A. Lipodystrophies. Am J Med. 2000;108:143. doi: 10.1016/s0002-9343(99)00414-3. [DOI] [PubMed] [Google Scholar]
- 2.Janaki VR, Premlatha S, Rao NR, Thambiah AS. Lawrence Seip syndrome. Br J Dermatol. 1980;103(6):693–696. doi: 10.1111/j.1365-2133.1980.tb01695.x. [DOI] [PubMed] [Google Scholar]
- 4.Magre J, Delepine M, Khallouf E. Identification of the gene altered in Berardinelli Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28(4):365–370. doi: 10.1038/ng585. [DOI] [PubMed] [Google Scholar]
- 5.Van Maldergem L, Magre J, Khallouf E. Genotype phenotype relationships in Berardinelli Seip congenital lipodystrophy. J Med Genet. 2002;39(10):722–733. doi: 10.1136/jmg.39.10.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhayana S, Hegele RA. The molecular basis of genetic lipodystrophies. Clin Biochem. 2002;35(3):171–177. doi: 10.1016/s0009-9120(02)00297-7. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz RA. Acanthosis nigricans. J Am Acad Dermatol. 1994;31(1):1–19. doi: 10.1016/s0190-9622(94)70128-8. [DOI] [PubMed] [Google Scholar]
- 8.Mork NJ, Rajka G, Halse J. Treatment of acanthosis nigricans with etretinate in a patient with generalized lipodystrophy. Acta Derm Venereol. 1986;66(2):173–174. [PubMed] [Google Scholar]
- 9.Seip M, Trygstad O. Generalised lipodystrophy, congenital and acquired. Acta Paediatr Suppl. 1996;413:2–28. doi: 10.1111/j.1651-2227.1996.tb14262.x. [DOI] [PubMed] [Google Scholar]
Uncited Reference
- 3.Garg A, Wilson R, Barnea R. A gene for congenital generalized lipodystrophy maps to chromosome 9q34. J Clin Endocrinol Metab. 1999;84(9):3390–3394. doi: 10.1210/jcem.84.9.6103. [DOI] [PubMed] [Google Scholar]