

Abstract
Recombinant adeno-associated virus (rAAV) vectors are attractive vehicles for gene therapy. Gene delivery to the adipose tissue using naturally occurring AAV serotypes is less successful compared to liver and muscle. Here, we demonstrate that oral administration of an engineered serotype Rec2 led to preferential transduction of brown fat with absence of transduction in the gastrointestinal track. Among the six natural and engineered serotypes being compared, Rec2 was the most efficient serotype achieving high level transduction at a dose 1~2 orders lower than reported doses for systemic administration. Overexpressing vascular endothelial growth factor (VEGF) in brown fat via oral administration of Rec2-VEGF vector increased the brown fat mass and enhanced thermogenesis. In contrast, knockdown VEGF in brown fat of VEGF loxP mice via Rec2-Cre vector hampered cold response and decreased brown fat mass. Oral administration of Rec2 vector provides a novel tool to genetically manipulate brown fat for research and therapeutic applications.
Introduction
rAAV vectors have been used for gene therapy in genetic and acquired diseases.1 Furthermore, rAAV vectors are powerful vehicles of gene delivery for basic research due to their characteristics including transduction of both dividing and postmitotic tissues, long-lasting transgene expression, and low immunogenicity.2 The use of rAAV to genetically manipulate adipose tissues has been limited due to the low transduction efficiency of the naturally occurring AAV serotypes.3,4,5 Recently, we demonstrated that a novel engineered hybrid serotype Rec2 vector led to high transduction of adipose tissue superior to naturally occurring serotypes (AAV1 and AAV8) and other engineered serotypes (Rec1, Rec3, Rec4) when the vectors were injected directly to the fat pads.6 Rec2 vector is particularly efficient for gene delivery to the brown adipose tissue (BAT) even at a low dose that is at least 1–2 orders lower than the naturally occurring serotypes.6 To further study the tropisms of these engineered serotypes and explore their systemic application, we investigated the gene delivery efficacy to adipose tissues by oral administration, and provided proof-of-concept studies on applications to genetic manipulation of fat.
Results
Preferential transduction of BAT via oral administration of Rec2 vector
We generated the naturally occurring serotypes AAV1 and AAV8 as well as engineered hybrid serotypes Rec1, Rec2, Rec3, and Rec4 vectors with green fluorescent protein (GFP) as reporter gene.6 Equal dose (2 × 1010 vg per mouse), the similar dose we reported for direct fat pad injection, of each serotype of GFP vector was administered by oral gavage. Two weeks post-rAAV administration, tissues were harvested and GFP fluorescence was measured in fresh tissue lysates. Among the tissues collected including BAT, WAT, liver, heart, stomach, intestine, skeletal muscle, kidney, spleen, lung, and brain, only BAT, liver, and WAT showed detectable GFP fluorescence. Interestingly, BAT was the tissue showed the highest GFP fluorescence for all serotypes (Figure 1a,b). GFP fluorescence was not detected in any tissues in mice administered with AAV1 vector (data not shown). Rec2 vector showed the highest GFP fluorescence compared to all the other serotypes tested (Figure 1a,b). We also measured the rAAV vector genome copy numbers in BAT and liver (Figure 1c).
Figure 1.

Comparison of transduction efficiency of novel engineered adeno-associated virus serotypes with natural serotypes via oral administration (2 × 1010 vg per mouse). (a) Representative GFP fluorescence of BAT sections 2 weeks after oral administration. Scale bar: 100 μm. Pictures were taken with the same exposure. (b) Quantification of GFP content in tissue lysates. Values are calibrated to protein content of each sample and expressed as mean ± standard error of the mean, n = 4–6 per group. * P < 0.05 Rec2 compared to all the other serotypes in BAT or liver respectively. (c) Viral vector genomic copy numbers in BAT and liver. Values are calibrated to genomic DNA of each sample.
To further characterize the Rec2 vector via oral administration, we assessed the biodistribution of Rec2 vector (2 × 1010 vg per mouse) by measurement of GFP fluorescence (Figure 2a) and vector genome copy numbers (Figure 2b). Surprisingly, neither GFP fluorescence nor viral vector DNA was detected in the gastrointestinal track even though the vector was orally administered. In contrast, BAT showed the highest GFP content followed by liver (Figure 2a) in parallel to the viral vector DNA (Figure 2b).
Figure 2.

Biodistribution of Rec2 serotype via oral administration. (a) GFP content in selected tissues 2 weeks post-oral administration of Rec2-GFP (2 × 1010 vg per mouse, n = 4). (b) Viral vector genomic copy numbers in selected tissues. Intestine proximally from duodenum to distally ileum was cut in the middle for GFP assay and vector copy number analysis. Intestine 1: first half, and intestine 2: second half.
To test whether the administration route determined the biodistribution, equal dose of Rec2-GFP vector (2 × 1010 vg per mouse) was administered via oral gavage or tail vein injection. Intravenous injection of Rec2 vector led to transduction predominantly in the liver while oral administration showed high transduction of BAT (Figure 3a).
Figure 3.

Comparison of oral administration of Rec2 vector with intravenous injection. (a) Comparison of oral administration with tail vein injection of the equal dose of Rec2-GFP vector (2 × 1010 vg per mouse, n = 4). (b) Representative GFP fluorescence in BAT and liver of the same mouse 2 weeks post-oral administration of low-dose Rec2-GFP (5 × 109 vg per mouse). (c) GFP content in tissue lysates of mice receiving low-dose Rec2-GFP (n = 4). (d) Viral vector genomic copy numbers in selected tissues 2 weeks post-oral administration of Rec2-GFP (5 × 109 vg per mouse). (e) Incorporation of miR-122 target sequence eliminates transgene expression in liver. Woodchuck post-transcriptional regulatory element mRNA level was calibrated to Actb and expressed as fold difference between BAT and liver of each individual mouse 2 weeks after rAAV administration.
To test whether lowering the dose could further enhance the preference to BAT transduction, 5 × 109 vg per mouse of Rec2-GFP was orally administered. Figure 3b–d showed the GFP fluorescence and viral vector DNA suggesting that decreasing the dose highly favored transduction of BAT than liver. GFP content in BAT was at least 10-fold higher than that in the liver.
microRNA-122 (miR-122) is selectively expressed in the liver at high level. Incorporation of miR-122 target sequence to the rAAV vector can restrict transgene expression in the liver.7 We generated Rec2 vector with four repeats of miR-122 target sequence and tested the restriction efficacy of miR-122 by measuring transgene transcription (woodchuck post-transcriptional regulatory element (WPRE) sequence in the rAAV transgene cassette) using quantitative real-time reverse transcription-PCR. miR-122 targeting sequence largely diminished the transgene expression in the liver when the vector was either orally administered or injected directly to the inguinal WAT. GFP fluorescence in the liver was barely detectable (data not shown). When orally administered at high dose (2 × 1010 vg per mouse), transgene mRNA level in the BAT was 100~500-fold higher than in the liver of the same mouse (Figure 3e).
To confirm the efficacy of Rec2 vector via oral administration, we generated Rec2 vector to deliver another reporter gene luciferase. We previously report that Rec2 vector is highly efficient to transduce BAT when the vector is injected directly to the brown fat at the dose of 5 × 109 vg per mouse. We then compared the gene delivery efficiency of Rec2 vector between oral administration and direct fat injection. Two weeks post-oral administration of Rec2-luciferase, mice were subjected to in vivo bioluminescence measurement (Figure 4a). Quantification of luciferase activity in the dissected BAT showed dose-dependent effects of direct BAT injection (Figure 4b). Interestingly, oral administration of Rec2 vector at the dose of 5 × 109 vg per mouse led to more than threefold higher transduction than direct BAT injection at the dose of 5 × 108 vg per mouse (Figure 4b).
Figure 4.

Comparison of oral administration of Rec2 vector with direct BAT injection. (a) Representative in vivo bioluminescence imaging of luciferase 2 weeks post-oral administration (5 × 109 vg per mouse) or direct injection to the BAT at lower doses (1 × 109 vg or 5 × 108 vg per mouse). Dorsal view, radiance photons/sec/cm2/sr, color scale: Min = 8.45e3; Max = 3.90e4. (b) Luciferase activity in BAT. Relative light units were calibrated to the total protein. n = 5 per group.
Knockdown vascular endothelial growth factor (VEGF) in BAT via oral administration of Rec2-Cre vector
To investigate whether oral administration of Rec2 vector could genetically manipulate BAT functions, we targeted VEGF in BAT as a proof-of-concept study. We previously reported that direct injection of Rec2 vector harboring Cre recombinase to the BAT of insulin receptor loxP mice could knockdown insulin receptor expression in adult mice leading to dysfunction of BAT and inhibition of glucose tolerance.6 Here, the Rec2-Cre vector was orally administered to VEGF loxP mice (2 × 1010 vg per mouse) with phosphate buffered saline (PBS) as control. No difference in body weight or food intake was observed (data not shown). Four weeks post-rAAV administration, mice were subjected to a glucose tolerance test and no difference was observed (data not shown). Five weeks after oral gavage, mice were exposed to cold challenge at 4 °C for 4 hours. Mice receiving Rec2-Cre were unable to properly maintain body temperature (Figure 5a). Cre-mediated knockdown of VEGF resulted in significant decrease of BAT mass and increase of gonadal WAT (Figure 5b,c). Western blot analysis showed oral administration of Rec2-Cre reduced VEGF protein level in BAT by approximately 80% (Figure 5e,f). VEGF is the de novo angiogenic factor in BAT and transgenic mouse studies demonstrate that knockout VEGF in BAT leads to the “whitening” of BAT with characteristics such as reduced vasculature and loss of thermogenic gene UCP1.8,9,10 Oral administration of Rec2-Cre vector resulted in similar reduction of vascular marker CD31 and thermogenic marker UCP1 (Figure 5d,e). Quantitative reverse transcription-PCR showed downregulation of Cd31, Ucp1, Kdr (encoding VEGF receptor 2), and Ppargc1a (encoding PPARγ coactivator-1α, a gene involved in thermogenesis11) consistent with the dysfunction of thermogenesis (Figure 5d).
Figure 5.

Oral Rec2-Cre-mediated VEGF knockdown in BAT. (a) Body temperature at cold exposure of 4 °C. n = 8–9 per group. (b) Fat and liver mass calibrated to body weight. n = 8–9 per group. (c) Representative of BAT 6 weeks after oral Rec2-Cre administration. (d) Gene expression profile of BAT. n = 5 per group. (e) Western blot of BAT. (f) Quantification of western blot. n = 5 per group. *P < 0.05, **P < 0.01, ***P < 0.001.
Overexpession of VEGF in BAT via oral administration of Rec2-VEGF vector
Our previous study demonstrates that direct delivery of Rec2 vector harboring human VEGF to WAT increases angiogenesis and induces beige cells, i.e., “browning” of WAT.12 Here, the Rec2-VEGF vector was orally administered to wild type mice (2 × 1010 vg per mouse). Rec2-VEGF vector led to significant increase of BAT mass and decrease of subcutaneous WAT without changes of body weight or food intake (Figure 6a). Overexpression of VEGF in BAT significantly enhanced the ability to maintain body temperature at cold exposure (Figure 6b). Rec2-VEGF increased VEGF protein level in BAT by approximately 12-fold compared to control mice while CD31 and UCP1 protein level were doubled (Figure 6d,e). Gene expression profile showed upregulation of genes involved in angiogenesis and thermogenesis (Figure 6c). Human VEGF was not detectable in the serum by enzyme-linked immunosorbent assay.
Figure 6.

Oral Rec2-VEGF-mediated VEGF overexpression in BAT. (a) Fat and liver mass calibrated to body weight 6 weeks after oral Rec2-VEGF administration. n = 8 per group. (b) Body temperature at cold exposure of 4 °C. n = 8 per group. (c) Gene expression profile of BAT. n = 8 per group. (d) Western blot of BAT. (e) Quantification of western blot. n = 5 per group. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
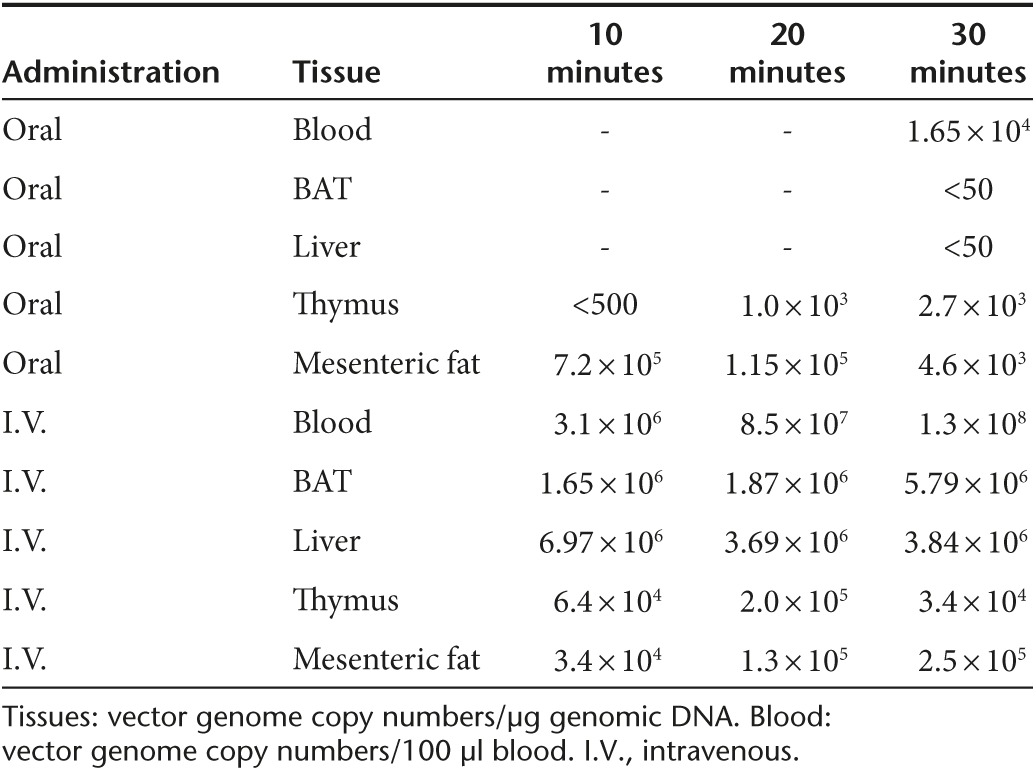
Up to date, oral administration of rAAV vectors has limited applications with the majority focusing on vaccines.12,13,14,15 Most studies on oral administration of AAV2 serotype demonstrate limited transduction of the lamina propria and epithelia of gastrointestinal track without notable transduction in other tissues.16,17 One study reports that oral administration of AAV6 vector leads to luciferase activity in the stomach and liver at the dose of 1 × 1011 vg per mouse.13 Another study shows oral administration of the AAV2-GFP at the dose of 5 × 1011 vg fails to transduce liver.17 To our knowledge, no publication is available on whether oral administration of rAAV vector can transduce adipose tissue. Our study is the first to show that an engineered hybrid serotype is highly efficient to transduce distant tissues from gastrointestinal track per oral administration. The Rec2 serotype has the following properties: (i) Oral administration leads to high transduction in BAT and liver at a dose 1~2 orders lower than commonly reported doses for systemic administration of rAAV vectors. (ii) The route of administration affects biodistribution, i.e., intravenous injection resulting in predominant liver transduction while oral administration favoring BAT transduction. (iii) Using lower dose for oral administration further shifts transduction to BAT versus liver. In addition, incorporation of miR-122 target sequence to the rAAV vector can eliminate the transgene expression in liver. Combination of these approaches can achieve selective gene delivery to the BAT via oral administration even with a strong constitutive promoter such as chicken β-actin (CBA) promoter (chicken β globin with CMV enhancer). The use of BAT-specific promoter such as UCP1 promoter can further enhance the tissue-specific transduction.18,19 The dose of oral administration to achieve the high-level expression of transgene in mouse appears comparable to direct injection to the BAT in the range of 109 vg per mouse which is approximately 2~3 orders lower than most reported doses of intravenous injection.6,20 Future studies will further characterize the Rec2 vector per oral administration including extended dose responses, long-term transgene expression in BAT, and immune responses that depend on dose, route of administration, and serotype.21,22 One interesting finding of this study is that oral administration of Rec2 vector failed to transduce gastrointestinal track but instead led to high transduction in BAT. AAV5 has been shown to transcytose gut epithelial cells.23 Based on our current results, we hypothesize that oral administration of Rec2 vector leads to transduction of BAT possibly via lymphatic system. Blood circulation is certainly involved but the lymph system might play a major role. Therefore, lower dose might favor this route further. We have some preliminary data on the presence of viral vector DNA at 10, 20, 30 minutes post-Rec2 vector injection (Table 1). Tail vein injection and oral administration showed distinctive profile. Oral administration led to very limited viral vector presence in the blood. In contrast, viral vector was detected in the thymus 10 and 20 minutes after the oral administration earlier than the blood. More interestingly, high level of viral vector was detected in the mesenteric fat that is rich in lymph nodes as early as 10 minutes after oral gavage. And furthermore, the presence of viral vector in the mesenteric fat declined overtime. Future investigations will address these fascinating questions whether Rec2 transcytoses to allow systemic delivery and how Rec2 is circulated via blood and/or possibly via lymphatic system.
Table 1. Kinetic of Rec2-GFP distribution within a period of time (10, 20, and 30 minutes) after oral gavage or tail vein injection of 5 × 109 vg/mouse.
Oral administration of Rec2 vector could knockdown VEGF in BAT leading to the decrease of BAT mass and inhibition of thermogenesis. In contrast, overexpressing VEGF resulted in the increase of BAT mass and enhanced thermogenesis at cold exposure. These examples of applications indicate that Rec2 vector is a powerful tool to genetically manipulate the BAT in adult animals. The high transduction efficiency, low dose, easy administration, selective tropism to BAT make the Rec2 vector a novel and attractive vehicle for both basic research on BAT and potential gene therapy targeting BAT for obesity and metabolic syndromes.
In addition to BAT, oral administration of Rec2 led to highly efficient gene delivery to the liver superior to published reports on naturally occurring serotypes.13,24 Thus, Rec2 vector provides an alternative route for systemic gene therapy of genetic diseases currently targeting liver such as hemophilia B, α-1 antitrypsin deficiency, ornithine transcarbamylase deficiency, phenylketonuria, and lysosomal storage disorders.25,26,27,28,29,30,31,32 Hepatic-restricted expression can be achieved by using liver-specific promoters, e.g., a minimal promoter region of α-1antitrypsin coupled with the enhancer element of the ApoE gene.33
Materials and Methods
Mice. C57Bl/6 mice, 6 weeks of age, were purchased from Charles River (Spencerville, OH). Mice bearing a Cre recombinase-conditional VEGF allele (VEGFloxP)34 on a C57Bl/6 and 129SV background were kindly provided by Dr. Andras Nagy of Mount Sinai Hospital, Toronto, Canada. Genotyping followed Dr. Nagy's protocol.9 All mice were housed in temperature (22–23 °C) and humidity controlled rooms with a 12-hour light-12-hour dark cycle, and maintained on a standard rodent diet (7912 rodent chow, Teklad) with food and water ad libitum. Female mice were used in all studies. All use of animals was approved by, and in accordance with the Ohio State University Animal Care and Use Committee.
rAAV vector construction and packaging. The rAAV plasmid contains a vector expression cassette consisting of the CMV enhancer and CBA promoter, WPRE and bovine growth hormone (bGH) poly-A flanked by AAV inverted terminal repeats. Transgenes: GFP, luciferase, Cre, and human VEGF164 were inserted into the multiple cloning sites between the CBA promoter and WPRE sequence. Naturally occurring serotypes AAV1 and AAV8, as well as engineered hybrid serotypes Rec1, Rec2, Rec3, and Rec4 vectors for GFP were packaged and purified as described previously.6 Rec2 vectors for Cre and VEGF were packaged and purified as described elsewhere.6,12,35 To generate AAV vector with miR-122 target sequence, the sense and antisense strands of four tandem repeats of miR-122 target sequence were synthesized by Sigma (Sigma, St. Louis, MO) with creation of SalI site at 5′ and 3′ end. Two complimentary strands were annealed, phosphorylated, and then cloned into SalI site positioned before bGH poly A sequence of rAAV plasmid. The sequence was listed as follows: AAV- miR-122 target sequence: sense, 5′ TCGACcaaacaccattgtcacactccaTCACcaaacaccattgtcacactccaTCACcaaacaccattgtcacactccaTCACcaaacaccattgtcacactccaG-3′; antisense, 5′-TCGACtggagtgtgacaatggtgtttgGTGAtggagtgtgacaatggtgtttgGTGAtggagtgtgacaatggtgtttgGTGAtggagtgtgacaatggtgtttgG-3′. The lower case represents miR-122 target sequence and the upper case is either SalI site (underline) or linker sequences (italic). The sequence of inserted miR-122 target sequence was verified by sequencing.
Administration of rAAV. rAAV serotype vectors carrying GFP, Rec2-Cre, and Rec2-VEGF were administered to mice, 6 weeks of age, via oral gavage after 4–6 hours fast (2 × 1010 vg in 200 µl PBS per mouse). PBS was used as a control for the vector. In the low-dose study, 5 × 109 vg per mouse was used for oral administration of Rec2-GFP. Intravenous administration of Rec2-GFP was carried out via tail vein injection (2 × 1010 vg in 50 µl PBS per mouse). rAAV injection to inguinal white fat and brown fat was described in detail previously.6
GFP content measurement. Fat pads were dissected.36 GFP content was measured according to previously published method with modifications.5 In brief, tissues were homogenized in ice-cold radioimmunoprecipitation assay buffer (25 mmol/l Tris-HCl pH 7.6, 150 mmol/l NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate) supplemented with proteinase inhibitors cocktail, followed by briefly sonication. The mixture was then spun at 13,000 rpm for 15 minutes at 4 °C. The supernatant was collected. GFP fluorescence in 100 µl supernatant was measured with microreader (Texas Instrument) using a 488-nm excitation wavelength and cut-off 515-nm/525-nm emission wavelength. GFP content was represented as readings after subtracting auto-fluorescence from GFP-null corresponding tissue and corrected by protein content in the samples.
Histology. BAT and Liver tissues were frozen in O.C.T. Compound and cryo-sectioned at 8 μm. GFP fluorescence was captured by fluorescent microscope. The fluorescence pictures were taken with the same exposure time.
Western blot. BAT tissues were homogenized by 1.5 ml pestle with cordless motor (VWR) in ice-cold radioimmunoprecipitation assay buffer containing phosphatase inhibitors and protease inhibitors cocktail (Pierce, Rockford, IL). The protein content was assayed by BCA kit (Pierce). Fifteen micrograms of protein from each sample were loaded and run in Bio-Rad precast 12% sodium dodecyl sulfate-PAGE, then transferred to nitrocellulose membranes, and blocked in PBS containing 0.1% Tween 20 and 5% nonfat dry milk for 1 hour at room temperature. Blots were incubated overnight at 4 °C with the following primary antibodies: glyceraldehyde-3-phosphate dehydrogenase(CalBiochem, Darmstadt, Germany; 1:1,000 dilution), CD31 (Abcam, Cambridge, UK; 1:1,000 dilution), VEGF (Abcam; 1:1,000 dilution), and UCP1 (Abcam; 1:7,000 dilution).
Viral vector copy number measurement. Total DNA from tissues was isolated using DNesay Blood and Tissue kit (Qiagen, Hilden, Germany). WPRE fragment was amplified to determine the copy number of viral particle. Fifty nanograms of DNA from each sample were used for real-time PCR. Mouse nucleic genomic fragment of GAPDH gene was used as control for mouse genetic DNA. The standard curve for copy number was generated from a known plasmid DNA. Viral vectors were isolated and purified from blood using High Pure Viral Nucleic Acid Kit (Roche, Mannheim, Germany).
Quantitative reverse transcription-PCR. Total RNA was isolated and purified using RNeasy mini kit plus RNase-free DNase treatment (Qiagen). First-strand cDNA was generated using TaqMan Reverse Transcription Reagent (Applied Biosystems, Grand Island, NY). Quantitative PCR was carried out using StepOnePlus Real-Time PCR System (Applied Biosystems) with the Power SYBR Green PCR Master Mix (Applied Biosystems). Primer sequences were designed to detect WPRE (5′-TGGCGTGGTGTGCACTGT-3′, 5′-GTTCCGCCGTGGCAATAG-3′) and the following mouse mRNAs: Ucp1 (5′-CGATGTCCATGTACACCAAGGA-3′, 5′-CCCGAGTCGCAGAAAAGAAG-3′), Vegf (5′-TACCTCCACCATGCCAAGTG-3′, 5′-CATGGGACTTCTGCTCTCCTTCT-3′), Cd31 (5′-CTGCCAGTCCGAAAATGGAAC-3′, 5′-CTTCATCCACCGGGGCTATC-3′), Ppargc1a (5′-AAGTGTGGAACTCTCTGGAACTG-3′, 5′-GGGTTATCTTGGTTGGCTTTATG-3′), Kdr (5′-TTTGGCAAATACAACCCTTCAGA-3′, 5′-GCAGAAGATACTGTCACCACC-3′), Actb (5′-ACCCGCGAGCACAGCTT-3′, 5′-ATATCGTCATCCATGGCGAACT-3′), Gapdh (5′-TCCCACTCTTCCACCTTCGA-3′, 5′-TGCTGTAGCCGTATTCATTGTCA-3′). Data were calibrated to endogenous control Actb or Gapdh and the relative gene expression was quantified the 2 −ΔΔCT method.37
Luciferase imaging. The luciferase imaging was carried out two weeks after delivery of Rec2-luciferase via oral administration (5 × 109 vg per mouse) or direction injection to BAT at two doses (1 × 109 vg or 5 × 108 vg per mouse). The hairs over BAT were partially removed. D-luciferin, potassium salt (Gold Biotechnology) was intraperitoneally injected into each mouse at a dose of 150 μg/g body weight. After 10 minutes, the mice were anesthetized in isoflurane chamber and then placed in a warm- and light-tight scan chamber. The bioluminescence imaging was generated using IVIS Lumina II, Caliper (Small Animal Imaging Core Facility of OSU) and presented by radiance unit of photons/second/cm2/sr, which is the number of photons per second that leave a square centimeter of tissue and radiate into a solid angle of one steradian (sr).
BAT luciferase assay. Luciferase assay was carried out using Pierce Firefly Luciferase Glow Assay Kit according to the manufacturer's instruction. BAT tissues were homogenized in buffer supplemented with protease inhibitors cocktail immediately after mice were euthanized. The luminescence was recorded with luminometer at 613 nm. The amount of luciferase activity in relative light unit of each sample was normalized to the total protein of the sample, and subtracting the count of sham BAT tissue lysate.
Glucose tolerance test. Four weeks after oral administration of rAAV, mice were injected intraperitoneally with glucose solution (1 mg glucose per gm body weight) after 4 hours fast. Blood was drawn from the tail at various time points and the blood glucose concentrations were measured with a portable glucose meter (ReliOn Ultima).
Acute cold exposure. Temperature probe (Physitemp, model BAT-12) topped with lubricant (white petroleum jelly) was used to measure the rectal temperature. Mice were placed into cages in preset 4 °C Chamber with no feed but with free access to water ad libitum. The rectal temperature was measured every hour up to 4 hours. The study was carried out 5 weeks after oral administration.
Statistical analysis. Values are expressed as mean ± standard error of the mean. We used JMP software to analyze the following: two-way analysis of variance for food intake, glucose tolerance; one-way analysis of variance for GFP content, body weight and adipose tissue weight, body temperature, quantitative reverse transcription-PCR data, western blot quantification.
Acknowledgments
This work was supported by NIH grants CA163640, CA166590, and CA178227. The authors declare no conflict of interest.
References
- Kotterman, MA and Schaffer, DV (2014). Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet 15: 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi, F and High, KA (2011). Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet 12: 341–355. [DOI] [PubMed] [Google Scholar]
- Mizukami, H, Mimuro, J, Ogura, T, Okada, T, Urabe, M, Kume, A et al. (2006). Adipose tissue as a novel target for in vivo gene transfer by adeno-associated viral vectors. Hum Gene Ther 17: 921–928. [DOI] [PubMed] [Google Scholar]
- Zhang, FL, Jia, SQ, Zheng, SP and Ding, W (2011). Celastrol enhances AAV1-mediated gene expression in mice adipose tissues. Gene Ther 18: 128–134. [DOI] [PubMed] [Google Scholar]
- Jimenez, V, Muñoz, S, Casana, E, Mallol, C, Elias, I, Jambrina, C et al. (2013). In vivo adeno-associated viral vector-mediated genetic engineering of white and brown adipose tissue in adult mice. Diabetes 62: 4012–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X, Magee, D, Wang, C, McMurphy, T, Slater, A, During, M et al. (2014). Adipose tissue insulin receptor knockdown via a new primate-derived hybrid recombinant AAV serotype. Mol Ther Methods Clin Dev (online)1. (doi:10.1038/mtm.2013.8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao, C, Yuan, Z, Li, J, He, B, Zheng, H, Mayer, C et al. (2011). Liver-specific microRNA-122 target sequences incorporated in AAV vectors efficiently inhibits transgene expression in the liver. Gene Ther 18: 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu, I, Aprahamian, T, Kikuchi, R, Shimizu, A, Papanicolaou, KN, MacLauchlan, S et al. (2014). Vascular rarefaction mediates whitening of brown fat in obesity. J Clin Invest 124: 2099–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, HK, Doh, KO, Son, JE, Park, JG, Bae, Y, Choi, S et al. (2013). Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metab 17: 61–72. [DOI] [PubMed] [Google Scholar]
- Sun, K, Kusminski, CM, Luby-Phelps, K, Spurgin, SB, An, YA, Wang, QA et al. (2014). Brown adipose tissue derived VEGF-A modulates cold tolerance and energy expenditure. Mol Metab 3: 474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver, P, Wu, Z, Park, CW, Graves, R, Wright, M and Spiegelman, BM (1998). A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92: 829–839. [DOI] [PubMed] [Google Scholar]
- During, MJ, Liu, X, Huang, W, Magee, D, Slater, A, McMurphy, T et al. (2015). Adipose VEGF links the white-to-brown fat switch with environmental, genetic, and pharmacological stimuli in male mice. Endocrinology 156: 2059–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel, JC, Di Pasquale, G, Ramlogan, CA, Patel, V, Chiorini, JA and Morris, JC (2013). Oral vaccination with adeno-associated virus vectors expressing the Neu oncogene inhibits the growth of murine breast cancer. Mol Ther 21: 680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin, KQ, Ooki, T, Mizukami, H, Hamajima, K, Okudela, K, Hashimoto, K et al. (2002). Oral administration of recombinant adeno-associated virus elicits human immunodeficiency virus-specific immune responses. Hum Gene Ther 13: 1571–1581. [DOI] [PubMed] [Google Scholar]
- Mouri, A, Noda, Y, Hara, H, Mizoguchi, H, Tabira, T and Nabeshima, T (2007). Oral vaccination with a viral vector containing Abeta cDNA attenuates age-related Abeta accumulation and memory deficits without causing inflammation in a mouse Alzheimer model. FASEB J 21: 2135–2148. [DOI] [PubMed] [Google Scholar]
- During, MJ, Symes, CW, Lawlor, PA, Lin, J, Dunning, J, Fitzsimons, HL et al. (2000). An oral vaccine against NMDAR1 with efficacy in experimental stroke and epilepsy. Science 287: 1453–1460. [DOI] [PubMed] [Google Scholar]
- Hao, ZM, Cai, M, Lv, YF, Huang, YH and Li, HH (2012). Oral administration of recombinant adeno-associated virus-mediated bone morphogenetic protein-7 suppresses CCl(4)-induced hepatic fibrosis in mice. Mol Ther 20: 2043–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassard-Doulcier, AM, Gelly, C, Bouillaud, F and Ricquier, D (1998). A 211-bp enhancer of the rat uncoupling protein-1 (UCP-1) gene controls specific and regulated expression in brown adipose tissue. Biochem J 333 (Pt 2): 243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larose, M, Cassard-Doulcier, AM, Fleury, C, Serra, F, Champigny, O, Bouillaud, F et al. (1996). Essential cis-acting elements in rat uncoupling protein gene are in an enhancer containing a complex retinoic acid response domain. J Biol Chem 271: 31533–31542. [DOI] [PubMed] [Google Scholar]
- O'Neill, SM, Hinkle, C, Chen, SJ, Sandhu, A, Hovhannisyan, R, Stephan, S et al. (2014). Targeting adipose tissue via systemic gene therapy. Gene Ther 21: 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mays, LE and Wilson, JM (2011). The complex and evolving story of T cell activation to AAV vector-encoded transgene products. Mol Ther 19: 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi, F and High, KA (2011). Immune responses to AAV in clinical trials. Curr Gene Ther 11: 321–330. [DOI] [PubMed] [Google Scholar]
- Di Pasquale, G and Chiorini, JA (2006). AAV transcytosis through barrier epithelia and endothelium. Mol Ther 13: 506–516. [DOI] [PubMed] [Google Scholar]
- Ma, H, Liu, Y, Liu, S, Xu, R and Zheng, D (2005). Oral adeno-associated virus-sTRAIL gene therapy suppresses human hepatocellular carcinoma growth in mice. Hepatology 42: 1355–1363. [DOI] [PubMed] [Google Scholar]
- Nichols, T, Whitford, MH, Arruda, VR, Stedman, HH, Kay, MA and High, KA (2014). Translational data from AAV-mediated gene therapy of hemophilia B in dogs. Hum Gene Ther Clin Dev.(online): 5–14. (doi: 10.1089/humc). [DOI] [PMC free article] [PubMed]
- Hareendran, S, Balakrishnan, B, Sen, D, Kumar, S, Srivastava, A and Jayandharan, GR (2013). Adeno-associated virus (AAV) vectors in gene therapy: immune challenges and strategies to circumvent them. Rev Med Virol 23: 399–413. [DOI] [PubMed] [Google Scholar]
- Asokan, A, Schaffer, DV and Samulski, RJ (2012). The AAV vector toolkit: poised at the clinical crossroads. Mol Ther 20: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruntman, AM and Flotte, TR (2015). Progress with recombinant adeno-associated virus vectors for gene therapy of alpha-1 antitrypsin deficiency. Hum Gene Ther Methods 26: 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L, Morizono, H, Lin, J, Bell, P, Jones, D, McMenamin, D et al. (2012). Preclinical evaluation of a clinical candidate AAV8 vector for ornithine transcarbamylase (OTC) deficiency reveals functional enzyme from each persisting vector genome. Mol Genet Metab 105: 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thöny, B (2010). Long-term correction of murine phenylketonuria by viral gene transfer: liver versus muscle. J Inherit Metab Dis 33: 677–680. [DOI] [PubMed] [Google Scholar]
- Ruzo, A, Garcia, M, Ribera, A, Villacampa, P, Haurigot, V, Marcó, S et al. (2012). Liver production of sulfamidase reverses peripheral and ameliorates CNS pathology in mucopolysaccharidosis IIIA mice. Mol Ther 20: 254–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonten, EJ, Yogalingam, G, Hu, H, Gomero, E, van de Vlekkert, D and d'Azzo, A (2013). Chaperone-mediated gene therapy with recombinant AAV-PPCA in a new mouse model of type I sialidosis. Biochim Biophys Acta 1832: 1784–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi, F, Liu, YL, Dobrzynski, E, Kaufhold, A, Liu, JH, Wang, Y et al. (2003). Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest 111: 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber, HP, Hillan, KJ, Ryan, AM, Kowalski, J, Keller, GA, Rangell, L et al. (1999). VEGF is required for growth and survival in neonatal mice. Development 126: 1149–1159. [DOI] [PubMed] [Google Scholar]
- Cao, L, Jiao, X, Zuzga, DS, Liu, Y, Fong, DM, Young, D et al. (2004). VEGF links hippocampal activity with neurogenesis, learning and memory. Nat Genet 36: 827–835. [DOI] [PubMed] [Google Scholar]
- Mann A, Thompson A, Robbins N, Blomkalns AL.(2014) Localization, identification, and excision of murine adipose depots. J Vis Exp (online) 94: (doi:10.3791/52174). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, KJ and Schmittgen, TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]