

Abstract
Adeno-associated viral (AAV) vectors are currently being tested in multiple clinical trials for liver-directed gene transfer to treat the bleeding disorders hemophilia A and B and metabolic disorders. The optimal viral capsid for transduction of human hepatocytes has been under active investigation, but results across various models are inconsistent. We tested in vivo transduction in “humanized” mice. Methods to quantitate percent AAV transduced human and murine hepatocytes in chimeric livers were optimized using flow cytometry and confocal microscopy with image analysis. Distinct transduction efficiencies were noted following peripheral vein administration of a self-complementary vector expressing a gfp reporter gene. An engineered AAV3 capsid with two amino acid changes, S663V+T492V (AAV3-ST), showed best efficiency for human hepatocytes (~3-times, ~8-times, and ~80-times higher than for AAV9, AAV8, and AAV5, respectively). AAV5, 8, and 9 were more efficient in transducing murine than human hepatocytes. AAV8 yielded the highest transduction rate of murine hepatocytes, which was 19-times higher than that for human hepatocytes. In summary, our data show substantial differences among AAV serotypes in transduction of human and mouse hepatocytes, are the first to report on AAV5 in humanized mice, and support the use of AAV3-based vectors for human liver gene transfer.
Introduction
Among various genetic diseases, adeno-associated viral (AAV) vectors are currently being used in multiple phase 1/2 clinical trials for the treatment of the X-linked bleeding disorder hemophilia by in vivo hepatic gene transfer.1,2 For the treatment of plasma protein deficiencies such as hemophilia, the goal is to achieve and maintain therapeutic systemic levels of the transgene product, which is synthesized and secreted by transduced hepatocytes. The viral capsid is a major determinant of the tropism, and therefore also transduction efficiency, of the AAV vector.3 In the initial trial on F9 gene transfer with AAV, the originally developed serotype 2 (AAV2) was infused into the hepatic artery of patients with severe hemophilia B (factor IX, FIX, deficiency).4 Since much higher liver transduction efficiency was seen in preclinical mouse studies with AAV serotype 8 (AAV8) compared to AAV2, AAV8 has been used in three more recent or still ongoing trials.1 Furthermore, AAV8 has strong tropism to the liver following peripheral vein infusion, so that it can simply be administered intravenously. Yet two other phase 1/2 trials are enrolling patients for hepatic F9 and F8 (encoding factor VIII, FVIII, for treatment of hemophilia A) gene transfer using AAV5 (NCT02396342 and NCT02576795). These data illustrate that several well-defined AAV serotypes are currently undergoing clinical evaluation for hepatic delivery of genes, although their relative efficiencies in patients remain poorly defined.
Over the last 15 years, besides more extensive testing of the originally discovered serotypes, many novel AAV serotypes have been isolated and characterized, and still others have been engineered using site-directed mutagenesis or capsid shuffling strategies.5 Thus, a number of candidate vectors have emerged for gene transfer to various tissues. Importantly, in vitro studies using primary cells or cell lines are not necessarily predictive of in vivo transduction efficiencies. In addition, for any given combination of serotype and target tissue, efficacy has not always been consistent between species.6 This raises the question of the optimal strategy for selection of a capsid for testing in humans. Long-term liver-derived transgene expression has been achieved with multiple serotypes in canine and non-human primate studies.7,8,9 In murine studies, liver transduction with AAV2 (used in the initial FIX clinical trial) is generally limited to about 5% of hepatocytes (albeit this is not known for human liver). Relative to AAV2, 1–2 × log10 higher levels of transgene expression can be accomplished with AAV8 in mice, with nearly all hepatocytes being transduced.10 In clinical studies, however, a traditional single-stranded ssAAV2 vector administered through the hepatic artery accomplished similar FIX transgene product levels as a self-complementary scAAV8 vector administered via peripheral vein.11 As mentioned, one advantage of AAV8 over AAV2 is its strong liver tropism allowing peripheral vein injection, by which means AAV2 typically loses fivefold or more transduction efficacy compared to portal vein or hepatic artery administration.12 Similar to clinical data, in non-human primates, FIX expression was found to be similar between AAV2 (given via portal vein) and AAV8 (given via peripheral vein).13,14 Altogether these data suggest that AAV8, despite maintaining strong liver tropism after peripheral vein injection, does not have the superior transduction efficiency in human and non-human primate liver that is seen in mice. While AAV5 shows reduced hepatic transduction efficiency after tail vein injection in mice, both portal and peripheral vein injections yielded FIX levels similar to AAV8 in non-human primates.7,15 Although non-human primates are evolutionary closer to humans than mice or dogs, they may still not be entirely predictive of performance in human hepatocytes either.6,16
AAV vectors, which achieve sustained transgene product expression in animal models, elicit in humans a cellular immune response to the vectors' capsid antigens, which upon hepatic transfer is linked to raises in transaminases accompanied by loss of transgene expression presumably due to immune-mediated destruction of AAV-transduced hepatocytes. AAV capsid-specific immune responses are dependent on the AAV vector dose and have thus far mainly been observed at the high doses needed to achieve therapeutic levels of the transgene product. One would expect, although this remains to be confirmed experimentally, that AAV vectors that transduce hepatocytes more efficiently may allow for sufficient dose-sparing to blunt this detrimental T-cell response.
Models are needed that are more predictive of in vivo gene transfer to human hepatocytes. Human liver chimeric mice harbor xenografted human hepatocytes and allow for the study of human hepatocyte biology in laboratory animals. These mice support engraftment of human hepatocytes upon elimination of a portion of the endogenous mouse hepatocytes. They need to be immunodeficient to prevent xenorejection. One widely used liver chimeric model was developed by Grompe and colleagues and is based on mice that are fumarylacetoacetate hydrolase deficient (fah-/-).17 Liver injury in these mice is controlled by administration of 2-(2-nitro-4-trifluoro-methyl-benzoyl)-1, 3 cyclohexanedione (NTBC). Since NTBC inhibits accumulation of toxic metabolites in fah-/- hepatocytes, its withdrawal results in a strong selective advantage for transplanted FAH-expressing human hepatocytes in the liver. In this human liver chimeric mouse model, AAV8 demonstrated a >1 × log10 higher transduction efficiency in murine compared to human hepatocytes.16 In addition, using the model to select for a vector with optimal transduction of human hepatocytes from an AAV capsid library yielded a capsid (LK03) with a viral protein 3 (VP3) protein sequence identical to that of AAV3 and other viral protein sequences derived from several serotypes.16 AAV3 had long been ignored as a potential vector because of its poor in vivo transduction of the murine liver. However, more recent work by Srivastava and colleagues revealed the ability of AAV3 to transduce human and non-human primate hepatocytes in vitro and in vivo, likely owing to the use of human hepatocyte growth factor receptor for cellular entry.18,19,20 Performance of the AAV3 capsid was further improved by elimination of one serine and one threonine residue (AAV3-ST containing mutations S663V and T492V).18,20 Using a human liver chimeric mouse model, others identified AAVrh10 and AAV9 as potentially good candidates for liver gene transfer in humans as well.21
Here, we sought to (i) develop methodology to accurately determine the percent of human hepatocytes transduced by AAV vectors in chimeric livers, and (ii) to compare the optimized AAV3 vector with AAV9, and with capsids currently being tested in clinical trials (AAV5 and AAV8).
Results
In vivo rAAV hepatocyte transduction and analysis by flow cytometry
In order to directly compare the relative AAV transduction efficiencies of mouse versus human hepatocytes, we tested green fluorescent protein (GFP) encoding AAV vectors in the fah-/- human liver chimeric mouse model. Extensive liver humanization was supported after crossing the fah-/- animals with Recombination-activating gene 2 (rag2-/-) and IL-2 Receptor, common γ-chain deficient mice (il2rgnull), so called FRG mice.22,23 We have previously observed that fah-/- mice crossed to NRG animals, designated FNRG mice, allow for superior human hepatocytes engraftment compared to the originally described FRG mice.22,24 We therefore used FNRG mice for these studies.
After transplantation of human pediatric hepatocytes or human fetal hepatoblasts into FNRG mice, we induced mouse liver damage by intermittent withdrawal of the protective drug NTBC and followed engraftment levels over time by measuring human albumin (hAlb) levels in the sera. Others have shown that hAlb levels correlate with human chimerism in the liver.24 We almost exclusively used mice that were highly engrafted, i.e., hAlb levels of ~1 mg/ml or higher, which corresponds to at least 10% human chimerism, and injected them with scAAV-GFP vectors of different serotypes (at a dose of 1 × 1011 vector genomes (vg)/mouse given i.v.). Included in this comparison were AAV3-ST, AAV5, AAV8, and AAV9 (Table 1). Two weeks after gene transfer, we isolated the liver cells and used flow cytometry to differentiate mouse from human hepatocytes and to analyze GFP reporter transgene expression in these cell populations (Figure 1a). Phosphate buffered saline (PBS) vehicle-injected mice served as negative controls.
Table 1. Human serum albumin levels and percent transduction efficiency for individual humanized mice.
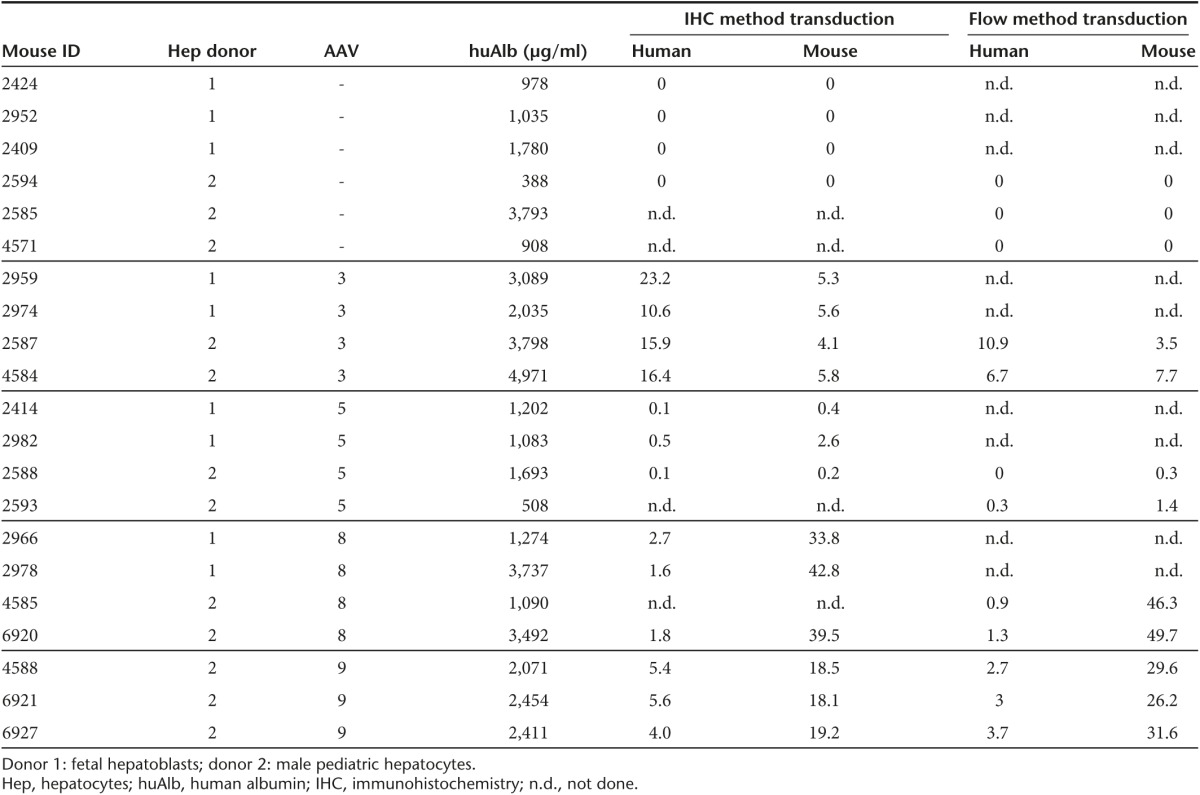
Figure 1.

Transduction of chimeric mouse livers with scAAV-GFP vectors and measurement of percent transduced human and murine hepatocytes by flow cytometry. (a) Outline of human-liver chimeric FNRG mouse production by infusing human liver-derived cells, followed by 2-(2-nitro-4-trifluoro-methyl-benzoyl)-1, 3 cyclohexanedione cycling. Upon humanization, mice can be challenged with AAV and transduction efficiencies in both human and mouse populations analyzed. (b) Example of the flow gating strategy used to exclude doublets, differentiate human from mouse hepatocytes, and calculate the amount of GFP expressing cells in each population. (c) Relative transduction based on GFP positivity, of human versus mouse hepatocytes of different AAV serotypes measured by flow cytometry. These data were obtained from a single donor (referred to as Donor 2 in Figure 3).
For flow cytometry, we excluded doublets by the area-to-width ratio in the forward and sideward scatter. We used the presence of FAH to identify human hepatocytes, and exploited the expression of mouse CD81 with a species-specific antibody to visualize mouse hepatocytes. The amount of GFP-expressing cells was then quantified in each population using FACSDiva Software. Figure 1b shows an example of this flow gating strategy applied to hepatocytes derived from an AAV3-ST transduced mouse. In total, 22 highly engrafted human-liver chimeric mice were randomized over five study groups (n = 6, 4, 4, 5, and 3 for PBS-, AAV3-ST-, AAV5-, AAV8-, and AAV9-injected mice, respectively), of whom 12 were analyzed using flow cytometry. The in vivo transduction efficiencies of AAV serotypes 3, 5, and 8 were each determined by flow cytometry in two human-liver chimeric mice, whereas AAV9 and the PBS control were tested in three mice each (Figure 1c). AAV5 infections led to few GFP-positive events in both the human and mouse hepatocyte populations (mean of 0.1 and 0.9%, respectively) indicating poor overall in vivo transduction efficiency. For AAV8 and 9, however, GFP-positive mouse hepatocytes were very frequently observed (mean of 48.0 and 29.1%, respectively), while human hepatocyte transduction was much lower (1.1 and 3.1%, respectively). The highest transduction efficiency of human hepatocytes was observed in AAV3-ST-injected mice (on average 8.8%). This was also the only capsid that yielded lower transduction of murine hepatocytes (~5.6%) compared to human hepatocytes.
Analysis of transduction efficiency using optimized immunostaining and image analysis
Transduced livers were prepared for immunohistochemistry as outlined in Figure 2. To obtain quantitative data, immunofluorescence image acquisition was performed using a confocal laser scanning microscope. A region of interest (ROI) was scanned sequentially at 0.5–1 μm z-intervals. The resulting stack was compressed based on brightest pixel in order to minimize false positives from overlapping cells. The settings for contrast, brightness, pinhole, acquisition mode, and scanning time were maintained throughout the work. Quantitative determination of vector driven GFP expression from mouse and human hepatocytes was based on the relative ROI area and pixel color as determined by an algorithm uniformly applied by the image analysis software. The resulting values are based on the average 3–5 ROI from each of the three to four individual mice per AAV serotype.
Figure 2.

Preparation and immunostaining of chimeric liver tissue for subsequent image acquisition and analysis. (a) Preparation of liver tissue for immunohistochemistry. (b) Treatment of liver sections and staining protocol for GFP and fumarylacetoacetate hydrolase.
Similar to the flow cytometry data, immunofluorescence microscopy and confocal microscopy combined with above-described image analyses showed differences in transduction efficiency between the serotypes (Figure 3a). AAV5 poorly transduced hepatocytes (mostly below 1%) but was somewhat more effective in mouse compared to human hepatocytes (Figure 3b–d). While AAV8 transduced mouse hepatocytes most efficiently (38.7 ± 2.6%) followed by AAV9 (18.6 ± 0.3%; Figure 3c), their human transduction efficiencies were reduced by 19 (2 ± 0.3%) and 3.7 times (5 ± 0.5%, Figure 3a). AAV3-ST performed best in human hepatocytes (16.5 ± 2.6% GFP+) and was 3.3-, 8-, and 82-times better than AAV9, 8, and 5, respectively (Figure 3d). Transduction of murine hepatocytes with AAV3-ST (5.2 ± 0.4%) was 3.2× lower than for human hepatocytes (Figure 3b).
Figure 3.

Visualization and analysis of scAAV-GFP transduction of human and murine hepatocytes in chimeric livers by confocal microscopy and image analysis of immunostained sections. (a) Representative examples of livers transduced with the indicated serotypes. Green stain: GFP+ murine hepatocytes; red stain: human hepatocytes (FAH+) lacking GFP expression; purple cells: GFP-transduced human hepatocytes (FAH+GFP+). Original magnification: 200x. Blue color in liver sections of PBS treated mice represents nuclear staining with DAPI (4',6-Diamidino-2-Phenylindole). (b) Percent GFP+ murine and human hepatocytes as a function of serotype. (c) Comparison of percent GFP+ murine hepatocytes, arranged from serotype with the lowest (AAV5) to that with the highest efficacy (AAV8). (d) Comparison of percent GFP+ human hepatocytes, arranged from serotype with the lowest (AAV5) to that with the highest efficacy (AAV3-ST). Symbols in (d) represent two different human hepatocyte donors (Donor 1: triangles, Donor 2: circles), with Donor 2 matching data in Figure 1. Fold differences are indicated for the respective comparisons, and statistically significant differences are indicted with * for P < 0.05; **P < 0.01, ***P < 0.001, and ****P < 0.0001 (ns = not significant). FAH, fumarylacetoacetate hydrolase. AAV, adeno-associated virus.
Discussion
AAV is the most advanced vector in clinical development for in vivo gene transfer to the human liver. Recent clinical trials for hemophilia B have illustrated the potential of this vector platform to accomplish sustained therapeutic expression.1,25,26 However, doses per kg required for efficacy have been substantially higher than what is needed for the best performing serotypes in murine models. This underperformance relative to animal models poses a problem for sustained therapy as CD8+ T-cell responses against the viral capsid are vector dose-dependent.1,4,11,25,27,28 Moreover, expression of FVIII from an AAV vector is less efficient and likely requires higher vector doses than is the case for FIX.9,29,30
In search of preclinical models that better predict efficacy in humans, mice with chimeric livers harboring human hepatocytes are receiving increased use. However, reported relative performance of AAV capsids, such as AAV3 or AAV8, in human hepatocytes or between human and murine hepatocytes in such humanized mice differ substantially. While in some cases AAV8 vectors transduced human hepatocytes substantially less efficiently, others found similar rates between mouse and human hepatocytes.16,18,31
Strong impact of viral capsid and mammalian species on hepatocyte transduction
Our data suggest stark differences between in vivo transduction of mouse and human hepatocytes for serotypes currently used in or considered for clinical trials. AAV5, 8, and 9 all showed higher transduction efficiency in mouse compared to human hepatocytes. This difference was most pronounced for AAV8, showing a >1 × log10 reduced transduction rate for human hepatocytes. This is consistent with data by Lisowski et al.16 and lower than expected efficacies in clinical trials of F9 gene transfer to the human liver. We used both pediatric hepatocytes and fetal hepatoblasts as a source to humanize the liver, while Lisowski et al. used adult hepatocytes from two different donors. Both our studies found a ~ 20-fold higher percentage of mouse compared to human hepatocytes transduced with AAV8. Similarly, both studies find substantially superior transduction of human hepatocytes when using an AAV3-based capsid. Therefore, we do not believe that inconsistent results of other studies are skewed due to variations that may result from the source or specific donor of hepatocytes. With regard to AAV8 transduction, gender of the donors also does not explain why our data are consistent with those by Lisowski et al. but differ from those by Wang et al., as Lisowski et al. and Wang et al. both used human hepatocytes from female donors, while at least one of our donors was male.16,31 Similarly, the method of vector purification was identical for our study and that by Wang et al. (iodixanol gradient), while Lisowski et al. used CsCl-gradient centrifugation.
The AAV3-based capsid was the only one tested here that exhibited greater efficiency for human compared to mouse hepatocytes, which can be explained by AAV3's use of the human hepatocyte growth factor receptor (which differs from the murine homolog in key amino acid residues).19 For gene transfer to human hepatocytes AAV3 performed best followed by AAV9 that was ~3-fold less efficient, AAV8 and AAV5 that were respectively about 1 and 2 × log10 less effective. It will be interesting to address whether the observed differences in transduction efficiency are due to disparities in viral entry (such as receptor binding) or postentry events (such as translocation to the nucleus or uncoating). To understand the mechanisms behind the distinct transduction patterns, it will also be important to determine receptor usage in human vs. murine hepatocytes. Interestingly, AAV2, 3, 8, and 9 can all utilize the laminin receptor (LamR), which serves as a receptor for other viruses as well.32 Platelet-derived growth factor receptor-α and -β have been identified as receptors for AAV5.33 Whether these receptors play a role in liver gene transfer remains to be determined.
Optimized methodology is required to accurately determine human hepatocyte transduction
We used two different methods to score percent GFP+ murine and human hepatocytes. Although only two mice per serotype were analyzed in most cases by flow cytometry (therefore not allowing statistical analyses), fold-differences between the average human hepatocyte transduction rates, comparing AAV3-ST to the other capsids, were nearly identical to those obtained with image analysis (performed for three to four mice/capsid). A subtle difference between the data obtained with the two methods is that flow analysis tended to score transduction of murine hepatocytes higher and transduction of human hepatocytes lower compared to image analysis of antibody-stained tissue sections. This may reflect differences in the sensitivity of staining and specific settings (including parameters set in the respective analytical softwares), which were optimized to eliminate background and false positives. For serotypes with high mouse hepatocyte transduction, a major source of error in estimating human hepatocyte transduction can arise from doublets or overlap of nontransduced human hepatocytes with GFP-positive mouse hepatocytes during flow cytometry or microscopy, respectively. Our methods were optimized in order to minimize these events. Both methods presented here are similarly powerful in measuring rates of transduction of human hepatocytes in a chimeric mouse liver. Flow cytometry however represents a less tedious and time consuming alternative to staining and image analysis. To avoid misidentification of the species identity of a GFP+ hepatocyte when using flow cytometry, it is critical to include optimized stains for markers of both the murine and human cells and to include a step that rigorously eliminates doublets (which may also form between mouse and human cells). Finally, to obtain reliable frequencies, it is also important to only use mice with high levels of engraftment of human hepatocytes (>10%).
Consideration of immune responses to AAV capsid in humans
Immune responses to AAV capsid are a major problem in clinical gene therapy.34 For example, the prevalence of circulating neutralizing antibodies (NAB) in the human population against AAV is high. This is the case in particular for serotype 2, so that the majority of adult patients cannot be treated by injection of AAV2 vector into blood vessels.35 Moreover, humans can have memory CD8+ T cells from natural infection that respond to AAV capsid antigen.28,36 Such responses are vector dose dependent and may eliminate transduced hepatocytes after initial successful gene transfer. This can result in mild liver toxicity and loss of therapeutic expression, which may however be prevented by transient immune suppression.11,25 The duration of major histocompatibility complex I (MHC I) presentation of capsid antigen and the risk and functionality of CD8+ T-cell responses induced by AAV gene transfer are influenced by a number of parameters, which include innate sensing of the genome, serotype, and vector dose.27,37,38,39,40,41
In transduced target cells, trafficking to the nucleus following endosomal escape is limited by ubiquitination followed by proteasomal degradation.42,43,44,45 This pathway reduces efficiency of transduction and promotes MHC I presentation of capsid antigen. Elimination of surface exposed tyrosine, serine, or threonine residues that are potential sites of phosphorylation, a signal for ubiquitination of capsid, limits proteasomal degradation and therefore improves transduction and reduces MHC I presentation.12,27,46 In case of AAV3 capsid, the combination of S663V + T492V mutations was most effective in improving transduction efficiency of human hepatocyte cell lines in vitro and also increased systemic expression of a secreted transgene product in non-human primates (up to fivefold compared to wild-type AAV3 capsid).18,20 An effect of these mutations on AAV3 capsid antigen presentation, however, has not yet been formally demonstrated. Capsid-derived CD8+ T-cell epitopes are diverse and often conserved among several serotypes.36 The capsid sequence of AAV5 is more divergent from the other serotypes tested here and therefore may be less likely targeted by CD8+ T-cell epitopes.
Conclusions
In aggregate, prior published data suggest that transduction of non-human primate liver is similarly efficient for a number of serotypes, including AAV3, AAV5, AAV8, and AAVrh10.7,13,31 In contrast, our results in the human liver mouse model show rather distinct human hepatocyte transduction efficiencies. The engineered AAV3-ST capsid was nearly 1 × log10 more efficient than AAV8, and nearly 2 × log10 compared to AAV5 following peripheral vein administration. AAV9 was the serotype in our tested panel showing human hepatocyte transduction closest to AAV3-ST. However, AAV9 has a broad tissue tropism and transduces heart, skeletal muscle, and neurons in addition to liver.47 In contrast, biodistribution studies in non-human primates show strong liver tropism of AAV3 and AAV3-ST following peripheral vein administration with minimal distribution to other organs (except for the spleen, which invariably takes up viral particles).18
AAV5 vector has been used in a recent clinical trial for the rare inherited metabolic disorder porphyria.48 The vector was given via peripheral vein at high doses for hepatic gene transfer. Liver biopsies taken 1 year after vector administration showed PCR positivity for vector sequences, while efficacy remained unclear. Now, first data have been reported from scAAV5-F9 hepatic gene transfer following peripheral vein injection of 5 × 1012 vg/kg. Early FIX expression levels of ~5% of normal were obtained. When compared with published data on scAAV8-F9 (using the same expression cassette), these results suggest a ~3-fold dose advantage of AAV8 over AAV5 (http://www.clinicalleader.com/doc/uniqure-announces-preliminary-cohort-hemophilia-clinical-trial-0001). Our data showed a ~10-fold difference. Therefore, our data are consistent with the conclusion that higher doses of AAV5 will be required to achieve therapeutic levels in human liver (which could be a particular challenge for F8 gene transfer for treatment of hemophilia A), although the difference in efficacy compared to AAV8 may not be as large as in our humanized mouse model. However, one has to caution that methods of vector production (baculovirus infection of insect cells vs transfection of mammalian cells) and purification were different and that measurement of titers may also vary. Furthermore, our study determined percent transduced hepatocytes, whereas levels of transgene expression per cell may also vary between serotypes and could also contribute to overall levels of systemic FIX expression. While data from ongoing clinical trials should further clarify whether the non-human primate or the human liver mouse model is the better predictor human liver efficacy, our data presented here argue for use of AAV3-based vectors, in particular when a large percentage of hepatocytes needs to be transduced to achieve clinical efficacy.
Materials and Methods
AAV vectors. Recombinant AAV (rAAV) vectors were produced by transfection of human embryonic kidney (HEK 293) cells using liposomes. Three (or two) plasmid DNAs had to be combined to produce each recombinant AAV serotype. These plasmids included (i) pAAV-CB-scGFP, containing the expression cassette (enhanced GFP under transcriptional control of CMV enhanced chicken β-actin promoter) flanked by AAV ITRs (with one ITR modified to produce self-complementary vector genomes); (ii) a helper plasmid DNA containing rep2/cap3 or rep2/cap9 (for production of AAV3-ST and AAV9); and (iii) pHelper plasmid (Invitrogen, Carlsbad, CA) containing adenoviral helper genes. For rAAV5 and rAAV8 packaging, rep/cap and adenoviral helper genes were combined in a single construct (pXYZ5 and pDG8, respectively). HEK-293 cells were expanded in 15-cm plates. Ninety microgram of total DNA (recombinant and helpers, in equimolar amounts) per plate were applied. Cells were harvested 72 hrs after transfection, and the virus purified using the iodixanol-density protocol.49 The titer of each preparation was determined by dot blot hybridization.
Generation of human liver chimeric mice and AAV gene transfer. Cryopreserved pediatric human hepatocytes were purchased from Celsis Inc. (Chicago, IL) Fresh human hepatoblasts were isolated from human fetal livers procured from Advanced Bioscience Resources (ABR) as described.50 Under anesthesia with isoflurane, human liver cell suspensions were injected intrasplenically (0.5–1 × 106 cells per mouse) into female fah-/- NOD rag1-/-il2rgnull (FNRG) mice that were generated by 13-generation backcross of the fah-/- allele to NOD rag1-/-il2rgnull (NRG) animals, respectively provided by M. Grompe (Oregon Health & Science University) or obtained from Jackson Laboratories.17,51 Starting on the day of transplantation, mice were cycled off the liver protective drug NTBC (Yecuris, Tualatin, OR) as described by others.22,24 Human albumin levels in mouse sera were measured by enzyme-linked immunosorbent assay (Bethyl Laboratories, Montgomery, TX). AAV vectors were injected through the tail vein at 1 × 1011 vector genomes per mouse. Two weeks later, human-chimeric mouse livers were removed and processed for flow cytometry and immunohistochemistry.
Hepatocyte isolation and flow cytometry. Mice were anaesthetized by ketamine/xylazine injection after which the inferior vena cava was cannulated with an angiocath (BD Biosciences, San Jose, CA) for in situ liver perfusion. After perfusion with heparin the large liver lobe was tied off in order to prevent its digestion, removed and fixed in 10% formalin for immunohistochemistry. The remainder of the liver was then perfused with PBS with 0.5 mmol/l ethylenediaminetetraacetic acid, followed by PBS with 0.05% collagenase. The collagenase perfused liver lobes were resected and put over a 100-μm cell strainer before Percoll purification and fixation in 4% buffered formalin. After permeabilization (cytoperm/cytofix, BD Biosciences), cells were stained for human- and mouse-specific markers. Human cells were identified by the presence of human FAH (Santa Cruz Biotechnology, Dallas, TX, + AF594 conjugated rabbit-anti goat, Invitrogen), whereas mouse cells were stained with biotinylated anti-mouse CD81 (BD Pharmingen, San Jose, CA) and PECy7-conjugated streptavidin (BD Pharmingen). The frequency of rAAV-GFP-positive cells was then quantified in either population by flow cytometry (BD LSRII). Doublets were excluded by the area-to-width ratio in the forward and sideward scatter. After doublet exclusion, ~15,000 cells were gated for each sample. The proportion of GFP-expressing cells was determined using FACSDiva Software.
Tissue processing and immunofluorescent labeling. Harvested livers where cut into ~2–3 mm slices and placed in 10% buffered formalin (Sigma-Aldrich, St. Louis, MO) overnight at 4 °C. After extensive washes in PBS, tissue was cryoprotected in 30% sucrose overnight. Without washing, tissue was placed in a shallow mold and embedded in optimal cutting temperature compound and snap frozen in liquid nitrogen cooled 2-methyle-butane.52 Tissue sections were cut at 5–10 μm onto poly-L-coated slides and air dried before storing at −20 °C.
For immunofluorescent labeling, sections were equilibrated to room temperature and rehydrated in PBS. Tissue was permeabilized with 0.1% Triton X-100 for 5 minutes and washed twice in PBS. Tissue was blocked with 5% donkey serum in PBS for 30 minutes, followed by incubation with rabbit anti-GFP antibody at 1:500 (ab290, Abcam, Cambridge, MA) and α-hFAH (sc-66223, Santa Cruz Biotechnology) for 2 hours at room temperature. The sections were then washed in PBS three times and incubated for 30 minutes with Alexa Fluor 488 donkey α-rabbit IgG and Alexa Fluor 568 donkey α-goat IgG (Invitrogen) at 1:400 each for 10 minutes before being washed and mounted with or without 4, 6 diamidino-2-phenylindole. Images were captured using a Leica (Wetzlar, Germany) Confocal TCS SP5 or a Nikon (Melville, NY) Eclipse 80i fluorescence microscope and Retiga 2000R digital camera (QImaging, Surrey, BC, Canada). Images were postprocessed with Volocity 3D Image Analysis Software v6.3 (Perkin-Elmer, Waltham, MA). Statistical analysis and graphical presentation was carried out with GraphPad Prism 6 (GraphPad Software, San Diego, CA).
Author Contributions
K.V., B.E.H., I.Z., G.D.K., J.W.X., and Y.dJ. performed experiments. K.V., B.E.H., E.B.T., H.C.E., K.A.H., A.S., Y.dJ., and R.W.H. designed experiments and interpreted data. C.M.R. contributed resources. K.V., B.E.H., Y.dJ., and R.W.H. wrote the manuscript. Y.dJ. and R.W.H. directed the study.
Acknowledgments
This work was supported by NIH grants R01 AI51390 (to R.W.H.), R01 HL097088 (to A.S. and R.W.H.), P01 HL078810 (to H.C.E.), R01 DK085713 (to C.M.R.), and K08DK090576 (to Y.dJ.). K.V. is supported by a Fellowship of the Belgian American Educational Foundation. K.A.H. is a co-founder, employee, and equity holder in Spark Therapeutics. She also holds issued patents related to AAV gene therapy. R.W.H. holds issued patents related to AAV gene therapy and has been receiving royalty payments from Spark Therapeutics. A.S. holds issued patents related to AAV gene therapy that have been licensed to Applied Genetic Technology Corporation.
References
- High, KA and Anguela, XM (2016). Adeno-associated viral vectors for the treatment of hemophilia. Hum Mol Genet 25(R1): R36–R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, GL and Herzog, RW (2015). Gene therapy for hemophilia. Front Biosci (Landmark Ed) 20: 556–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asokan, A, Schaffer, DV and Samulski, RJ (2012). The AAV vector toolkit: poised at the clinical crossroads. Mol Ther 20: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno, CS, Pierce, GF, Arruda, VR, Glader, B, Ragni, M, Rasko, JJ et al. (2006). Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 12: 342–347. [DOI] [PubMed] [Google Scholar]
- Marsic, D, Govindasamy, L, Currlin, S, Markusic, DM, Tseng, YS, Herzog, RW et al. (2014). Vector design Tour de Force: integrating combinatorial and rational approaches to derive novel adeno-associated virus variants. Mol Ther 22: 1900–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay, MA (2015). Selecting the best AAV capsid for human studies. Mol Ther 23: 1800–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Rosales, C, McIntosh, J, Rastegarlari, G, Nathwani, D, Raj, D et al. (2011). Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol Ther 19: 876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer, GP, Herzog, RW, Mount, J, Arruda, VR, Tillson, DM, Hathcock, J et al. (2009). Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood 113: 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino, DE, Lange, AM, Altynova, ES, Sarkar, R, Zhou, S, Merricks, EP et al. (2011). Efficacy and safety of long-term prophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors. Mol Ther 19: 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, M, Nayak, S, Hoffman, BE, Terhorst, C, Cao, O and Herzog, RW (2009). Improved induction of immune tolerance to factor IX by hepatic AAV-8 gene transfer. Hum Gene Ther 20: 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Tuddenham, EG, Rangarajan, S, Rosales, C, McIntosh, J, Linch, DC et al. (2011). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365: 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, L, Li, B, Mah, CS, Govindasamy, L, Agbandje-McKenna, M, Cooper, M et al. (2008). Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci USA 105: 7827–7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Gray, JT, McIntosh, J, Ng, CY, Zhou, J, Spence, Y et al. (2007). Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates. Blood 109: 1414–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Gray, JT, Ng, CY, Zhou, J, Spence, Y, Waddington, SN et al. (2006). Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood 107: 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi, F, Schüttrumpf, J, Arruda, VR, Liu, Y, Liu, YL, High, KA et al. (2002). Improved hepatic gene transfer by using an adeno-associated virus serotype 5 vector. J Virol 76: 10497–10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisowski, L, Dane, AP, Chu, K, Zhang, Y, Cunningham, SC, Wilson, EM et al. (2014). Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 506: 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grompe, M, Lindstedt, S, al-Dhalimy, M, Kennaway, NG, Papaconstantinou, J, Torres-Ramos, CA et al. (1995). Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat Genet 10: 453–460. [DOI] [PubMed] [Google Scholar]
- Li, S, Ling, C, Zhong, L, Li, M, Su, Q, He, R et al. (2015). Efficient and targeted transduction of nonhuman primate liver with systemically delivered optimized AAV3B vectors. Mol Ther 23: 1867–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, C, Lu, Y, Kalsi, JK, Jayandharan, GR, Li, B, Ma, W et al. (2010). Human hepatocyte growth factor receptor is a cellular coreceptor for adeno-associated virus serotype 3. Hum Gene Ther 21: 1741–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, C, Wang, Y, Zhang, Y, Ejjigani, A, Yin, Z, Lu, Y et al. (2014). Selective in vivo targeting of human liver tumors by optimized AAV3 vectors in a murine xenograft model. Hum Gene Ther 25: 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissig-Choisat, B, Wang, L, Legras, X, Saha, PK, Chen, L, Bell, P et al. (2015). Development and rescue of human familial hypercholesterolaemia in a xenograft mouse model. Nat Commun 6: 7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma, H, Paulk, N, Ranade, A, Dorrell, C, Al-Dhalimy, M, Ellis, E et al. (2007). Robust expansion of human hepatocytes in Fah-/-/Rag2-/-/Il2rg-/- mice. Nat Biotechnol 25: 903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissig, KD, Le, TT, Woods, NB and Verma, IM (2007). Repopulation of adult and neonatal mice with human hepatocytes: a chimeric animal model. Proc Natl Acad Sci USA 104: 20507–20511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissig, KD, Wieland, SF, Tran, P, Isogawa, M, Le, TT, Chisari, FV et al. (2010). Human liver chimeric mice provide a model for hepatitis B and C virus infection and treatment. J Clin Invest 120: 924–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Reiss, UM, Tuddenham, EG, Rosales, C, Chowdary, P, McIntosh, J et al. (2014). Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 371: 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog, RW (2015). Hemophilia gene therapy: caught between a cure and an immune response. Mol Ther 23: 1411–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino, AT, Basner-Tschakarjan, E, Markusic, DM, Finn, JD, Hinderer, C, Zhou, S et al. (2013). Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood 121: 2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi, F, Maus, MV, Hui, DJ, Sabatino, DE, Murphy, SL, Rasko, JE et al. (2007). CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med 13: 419–422. [DOI] [PubMed] [Google Scholar]
- McIntosh, J, Lenting, PJ, Rosales, C, Lee, D, Rabbanian, S, Raj, D et al. (2013). Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood 121: 3335–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack, BK, Herzog, RW, Terhorst, C and Markusic, DM (2014). Development of gene transfer for induction of antigen-specific tolerance. Mol Ther Methods Clin Dev 1: 14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L, Bell, P, Somanathan, S, Wang, Q, He, Z, Yu, H et al. (2015). Comparative study of liver gene transfer with AAV vectors based on natural and engineered AAV capsids. Mol Ther 23: 1877–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akache, B, Grimm, D, Pandey, K, Yant, SR, Xu, H and Kay, MA (2006). The 37/67-kilodalton laminin receptor is a receptor for adeno-associated virus serotypes 8, 2, 3, and 9. J Virol 80: 9831–9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pasquale, G, Davidson, BL, Stein, CS, Martins, I, Scudiero, D, Monks, A et al. (2003). Identification of PDGFR as a receptor for AAV-5 transduction. Nat Med 9: 1306–1312. [DOI] [PubMed] [Google Scholar]
- Mingozzi, F and High, KA (2013). Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 122: 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi, F, Anguela, XM, Pavani, G, Chen, Y, Davidson, RJ, Hui, DJ et al. (2013). Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci Transl Med 5: 194ra92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui, DJ, Edmonson, SC, Podsakoff, GM, Pien, GC, Ivanciu, L, Camire, RM et al. (2015). AAV capsid CD8+ T-cell epitopes are highly conserved across AAV serotypes. Mol Ther Methods Clin Dev 2: 15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust, SM, Bell, P, Cutler, BJ, Ashley, SN, Zhu, Y, Rabinowitz, JE et al. (2013). CpG-depleted adeno-associated virus vectors evade immune detection. J Clin Invest 123: 2994–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino, AT, Suzuki, M, Markusic, DM, Zolotukhin, I, Ryals, RC, Moghimi, B et al. (2011). The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood 117: 6459–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, GL, Martino, AT, Zolotukhin, I, Ertl, HC and Herzog, RW (2014). Role of the vector genome and underlying factor IX mutation in immune responses to AAV gene therapy for hemophilia B. J Transl Med 12: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, GL, Suzuki, M, Zolotukhin, I, Markusic, DM, Morel, LM, Lee, B et al. (2015). Unique roles of TLR9- and MyD88-dependent and -independent pathways in adaptive immune responses to AAV-mediated gene transfer. J Innate Immun 7: 302–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, TL, Li, H, Faust, SM, Chi, E, Zhou, S, Wright, F et al. (2014). CD8+ T cell recognition of epitopes within the capsid of adeno-associated virus 8-based gene transfer vectors depends on vectors' genome. Mol Ther 22: 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, JD, Hui, D, Downey, HD, Dunn, D, Pien, GC, Mingozzi, F et al. (2010). Proteasome inhibitors decrease AAV2 capsid derived peptide epitope presentation on MHC class I following transduction. Mol Ther 18: 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C, He, Y, Nicolson, S, Hirsch, M, Weinberg, MS, Zhang, P et al. (2013). Adeno-associated virus capsid antigen presentation is dependent on endosomal escape. J Clin Invest 123: 1390–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, Z, Zak, R, Luxton, GW, Ritchie, TC, Bantel-Schaal, U and Engelhardt, JF (2002). Ubiquitination of both adeno-associated virus type 2 and 5 capsid proteins affects the transduction efficiency of recombinant vectors. J Virol 76: 2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pien, GC, Basner-Tschakarjan, E, Hui, DJ, Mentlik, AN, Finn, JD, Hasbrouck, NC et al. (2009). Capsid antigen presentation flags human hepatocytes for destruction after transduction by adeno-associated viral vectors. J Clin Invest 119: 1688–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanidi, GV, Rivers, AE, Ortiz, L, Song, L, Ling, C, Govindasamy, L et al. (2013). Optimization of the capsid of recombinant adeno-associated virus 2 (AAV2) vectors: the final threshold? PLoS One 8: e59142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows, AS, Duncan, FJ, Camboni, M, Waligura, K, Montgomery, C, Zaraspe, K et al. (2015). A GLP-compliant toxicology and biodistribution study: systemic delivery of an rAAV9 vector for the treatment of mucopolysaccharidosis IIIB. Hum Gene Ther Clin Dev 26: 228–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Avola, D, Lopez-Franco E, Harper P, Fontanellas A, Grosios N, Henrichson A, et al. (2015). Phase 1 clinical trial of liver-directed gene therapy with rAAV5/2-PBGD in acute intermittent porphyria: safety data. Hum Gene Ther 26: A26. [Google Scholar]
- Zolotukhin, S, Byrne, BJ, Mason, E, Zolotukhin, I, Potter, M, Chesnut, K et al. (1999). Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther 6: 973–985. [DOI] [PubMed] [Google Scholar]
- Andrus, L, Marukian, S, Jones, CT, Catanese, MT, Sheahan, TP, Schoggins, JW et al. (2011). Expression of paramyxovirus V proteins promotes replication and spread of hepatitis C virus in cultures of primary human fetal liver cells. Hepatology 54: 1901–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm, MA, Cuthbert, A, Yang, C, Miller, DM, DiIorio, P, Laning, J et al. (2010). Parameters for establishing humanized mouse models to study human immunity: analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the IL2rgamma(null) mutation. Clin Immunol 135: 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, GL, Hoffman BE (2012). Optimal immunofluorescent staining for human factor IX and infiltrating T cells following gene therapy for hemophilia B. J Genet Syndr Gene Ther S1. [DOI] [PMC free article] [PubMed]