

Abstract
Pelareorep causes oncolysis in tumor cells with activated Ras. We hypothesized that pelareorep would have efficacy and immunomodulatory activity in metastatic pancreatic adenocarcinoma (MPA) when combined with carboplatin and paclitaxel. A randomized phase 2 study (NCT01280058) was conducted in treatment-naive patients with MPA randomized to two treatment arms: paclitaxel/carboplatin + pelareorep (Arm A, n = 36 evaluable patients) versus paclitaxel/carboplatin (Arm B, n = 37 evaluable patients). There was no difference in progression-free survival (PFS) between the arms (Arm A PFS = 4.9 months, Arm B PFS = 5.2 months, P = 0.6), and Kirsten rat sarcoma viral oncogene (KRAS) status did not impact outcome. Quality-adjusted Time without Symptoms or Toxicity analysis revealed that the majority of PFS time was without toxicity or progression (4.3 months). Patient immunophenotype appeared important, as soluble immune biomarkers were associated with treatment outcome (fractalkine, interleukin (IL)-6, IL-8, regulated on activation, normal T cell expressed and secreted (RANTES), and vascular endothelial growth factor (VEGF)). Increased circulating T and natural killer (NK)-cell subsets were also significantly associated with treatment outcome. Addition of pelareorep was associated with higher levels of 14 proinflammatory plasma cytokines/chemokines and cells with an immunosuppressive phenotype (Tregs, cytotoxic T lymphocyte associated protein 4 (CTLA4)+ T cells). Overall, pelareorep was safe but does not improve PFS when administered with carboplatin/paclitaxel, regardless of KRAS mutational status. Immunologic studies suggest that chemotherapy backbone improves immune reconstitution and that targeting remaining immunosuppressive mediators may improve oncolytic virotherapy.
Introduction
Pancreatic adenocarcinoma (PCA) is a leading cause of cancer death in the Western World. The incidence of PCA in the United States was 46,420 in 2014, while the 5-year survival rate was a dismal 6.7%.1 Two regimens form the mainstay of systemic therapy for metastatic pancreatic adenocarcinoma (MPA)—FOLFIRINOX (5FU, leucovorin, irinotecan, and oxaliplatin) with a median overall survival (OS) of 11.1 months,2 and gemcitabine and nab-paclitaxel with a median OS of 8.5 months.3
Ras is a guanosine triphosphate (GTP)-binding protein that regulates cell growth and survival. RAS mutations are observed in 70–90% of all PCAs.4,5,6 Reolysin® (pelareorep) is a propriety formulation of a naturally occurring nonenveloped human Reovirus Serotype3-Dearing Strain which contains live, replication-competent reovirus.7 While community-acquired reovirus infection in humans is generally mild and limited to upper respiratory and gastrointestinal tracts, pelareorep demonstrates cytotoxic effects on cancer cells with an activated Ras signaling pathway due to mutations in the RAS proto-oncogene.8,9 While its effects on transformed cells are relatively well described, the activity this virus has on the immune system of cancer-bearing individuals is less clear. Some studies suggest that it may potentiate the host antitumor immune response,10,11,12,13,14 while others show that it can further exacerbate immunosuppressive features of advanced cancer.15 Thus, investigating immune biomarkers in the context of a well-controlled clinical trial is of importance for determining whether it may complement emerging immunotherapeutic approaches for metastatic disease.
The immune reaction to cancer is complex and involves a number of soluble cytokine and chemokine mediators. The relative balance of these factors influences the phenotypic profile of systemic and tumor infiltrating immune cells and thus the antitumor immune reaction. For example, increased levels of cytokines such as IL-6 and VEGF lead to expansion of immunosuppressive lymphoid or myeloid cell subsets that contribute to immune evasion.16 Given the recent success of immunotherapy in other malignancies, it is important to understand how novel therapeutic agents such as oncolytic viruses, in combination with chemotherapy, might reverse this immunosuppression and modulate the immune response to cancer. Pelareorep was combined in with gemcitabine in a phase 1 study and was found to be safe with evidence that gemcitabine affects the humoral immune response to reovirus and attenuates the neutralizing antireolysin antibody response.17
Taxanes and platinums have been examined in advanced PCA with promising activity.18 These agents are also thought to induce some degree of antitumor immune activity, either by inducing a more immunogenic cell death or simply through the enhanced release of tumor antigen following chemotherapy-induced tumor cell death.19,20,21 Single agent docetaxel resulted in objective response rate (ORR) of 5–15% and a median OS of 5.9–8.3 months.22 Weekly paclitaxel in the second- and third-line setting following gemcitabine failure produced a complete response rate of 5%, stable disease rate of 27.7% and median OS of 4.1 months,23 while Whitehead et al.24 showed that 24-hour infusional paclitaxel given every 3 weeks resulted in ORR of 8% and median OS of 5 months. Our group performed a phase 1/2 study of carboplatin, paclitaxel and capecitabine in patients with solid tumors and noted significant activity in patients with PCA. Of 11 patients with metastatic PCA (92% of whom had failed at least 1 prior therapy (range 1–4) and >95% of whom had failed gemcitabine), 4 confirmed partial responses were seen and 8 patients had prolonged stable disease, and all of these patients had a biochemical response.25 Another study of 54 patients showed that the combination of protracted infusional 5FU (an agent with single agent ORR of 0–9%) with carboplatin in patients with advanced PCA yielded promising responses (ORR 17% with a complete response rate of 2%, median OS 5.5 months).26
The potential for synergistic activity of reovirus in combination with carboplatin and paclitaxel was suggested by prior studies in human cancer cell lines.27,28,29 Paclitaxel enhanced replication of pelareorep, resulting in enhanced apoptosis of nonsmall-cell lung cancer cells compared to either agent alone, and synergy was also seen with cisplatin.27,28 Since most pancreatic cancer cells have activated Ras, we hypothesized that MPA patients would benefit from pelareorep and that immune modulation may contribute to its efficacy. Considering the preclinical data showing synergistic activity of pelareorep with carboplatin/paclitaxel and the earlier studies cited above which showed promising activity against MPA by carboplatin and taxanes, we chose to use carboplatin and paclitaxel as the backbone for this trial rather than standard first-line therapy.
Results
Patient characteristics and clinical outcomes
Seventy-six patients were randomized between February 2011 and April 2014. Three patients withdrew from the study prior to receiving any therapy and were not considered evaluable, resulting in 73 patients evaluable for the primary endpoint of PFS: 36 on Arm A and 37 on Arm B (Figure 1). Table 1 shows the baseline characteristics for all evaluable patients. At the time the analysis data were frozen (12/31/2014), the median follow-up on patients was 10.5 months (range 8.9–23.4) and 5 of the 73 patients were progression-free and alive. Overall, 16 patients from Arm B crossed over to the pelareorep arm upon progression and received pelareorep in addition to continued carboplatin and paclitaxel.
Figure 1.

CONSORT flow diagram showing the flow of patients through the trial. Informed signed consent was obtained from all patients prior to enrolment and the study was conducted according to the Cancer Therapy Evaluation Program (CTEP) multicenter guidelines. The trial was performed after approval by a local Human Investigations Committee and in accordance with an assurance filed with and approved by the Department of Health and Human Services.
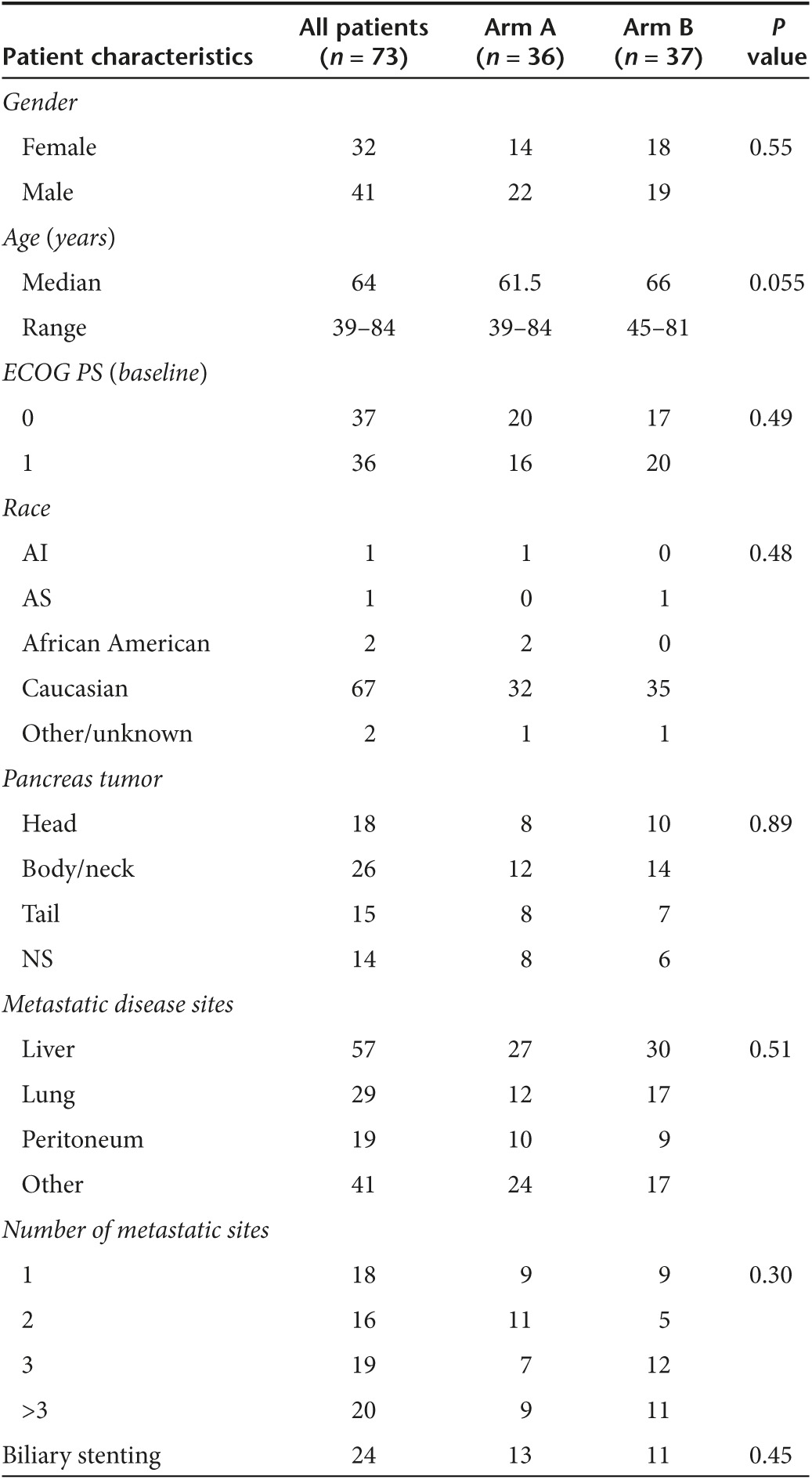
Table 1. Baseline characteristics for patients enrolled.
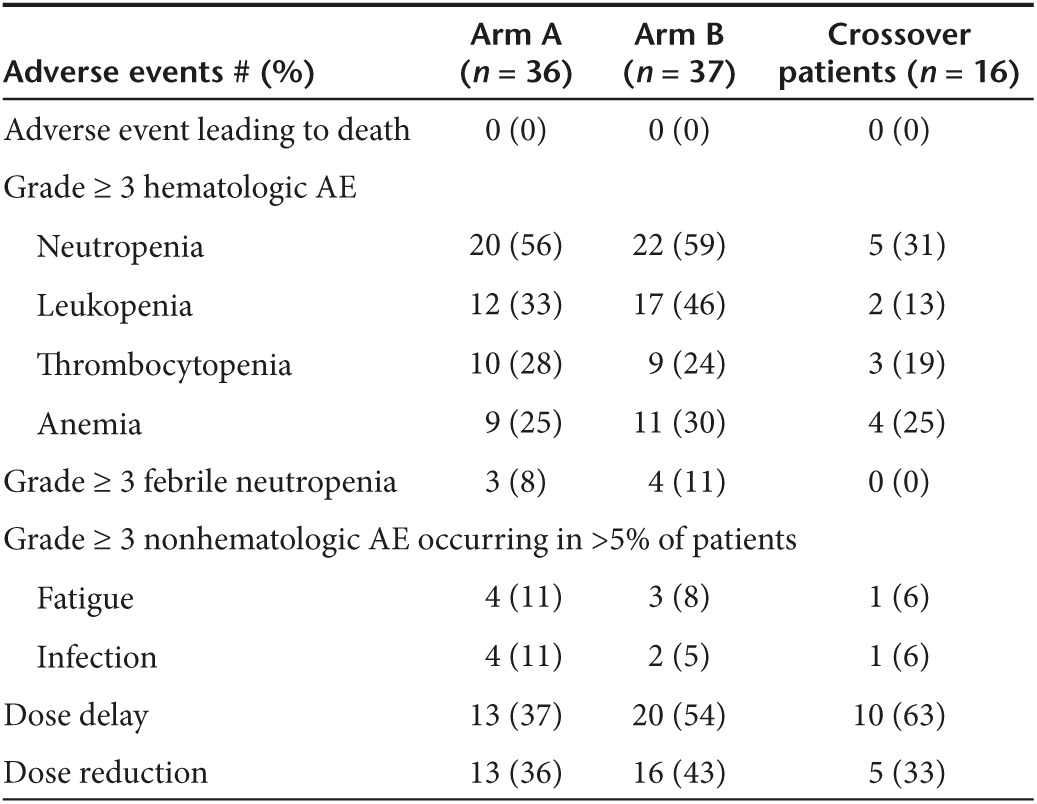
The regimen was well tolerated with no statistically significant differences in toxicity between the two arms (Table 2). The most common toxicities associated with pelareorep were grade 1 chills and grade 1 diarrhea, each occurring in 28% of patients on Arm A. There were two occurrences of lupus nephritis, which were thought to be attributable to pelareorep and both resolved following discontinuation of the virus.
Table 2. Toxicities ≥ grade 3 experienced measured using CTCAE V4.
All but five patients (68/73 = 93%) had disease progression and/or death for analysis of PFS as the primary endpoint (i.e., the randomized treatment, not including the crossover treatment). There was no significant difference in PFS between treatment arms (Arm A median PFS 4.9 months (95% CI: 3.0–6.3 months) versus 5.2 months (95% CI: 2.3–6.2 months) on Arm B, P = 0.6) (Figure 2a). Likewise, there was no significant difference in OS between the two arms (median OS on Arm A was 7.3 months, 95% CI: 4.8–11.2 months) and on Arm B was 8.8 months (95% CI: 6.6–11.8, P = 0.68, Table 3 and Supplementary Figure S2). Supplementary Table S1 shows the hazard ratios from univariate and multivariate Cox models for PFS and OS. Forty-four percent of patients on Arm A and 35% on Arm B received second-line therapy, most commonly gemcitabine- or 5FU-based (see Supplementary Table S2).
Figure 2.

Progression-free survival. (a) Progression-free survival. There was no difference in median progression-free survival between Arm A and Arm B. (b) KRAS status did not influence progression-free survival.
Table 3. Summary of survival, response, KRAS status and CA 19-9 trends.
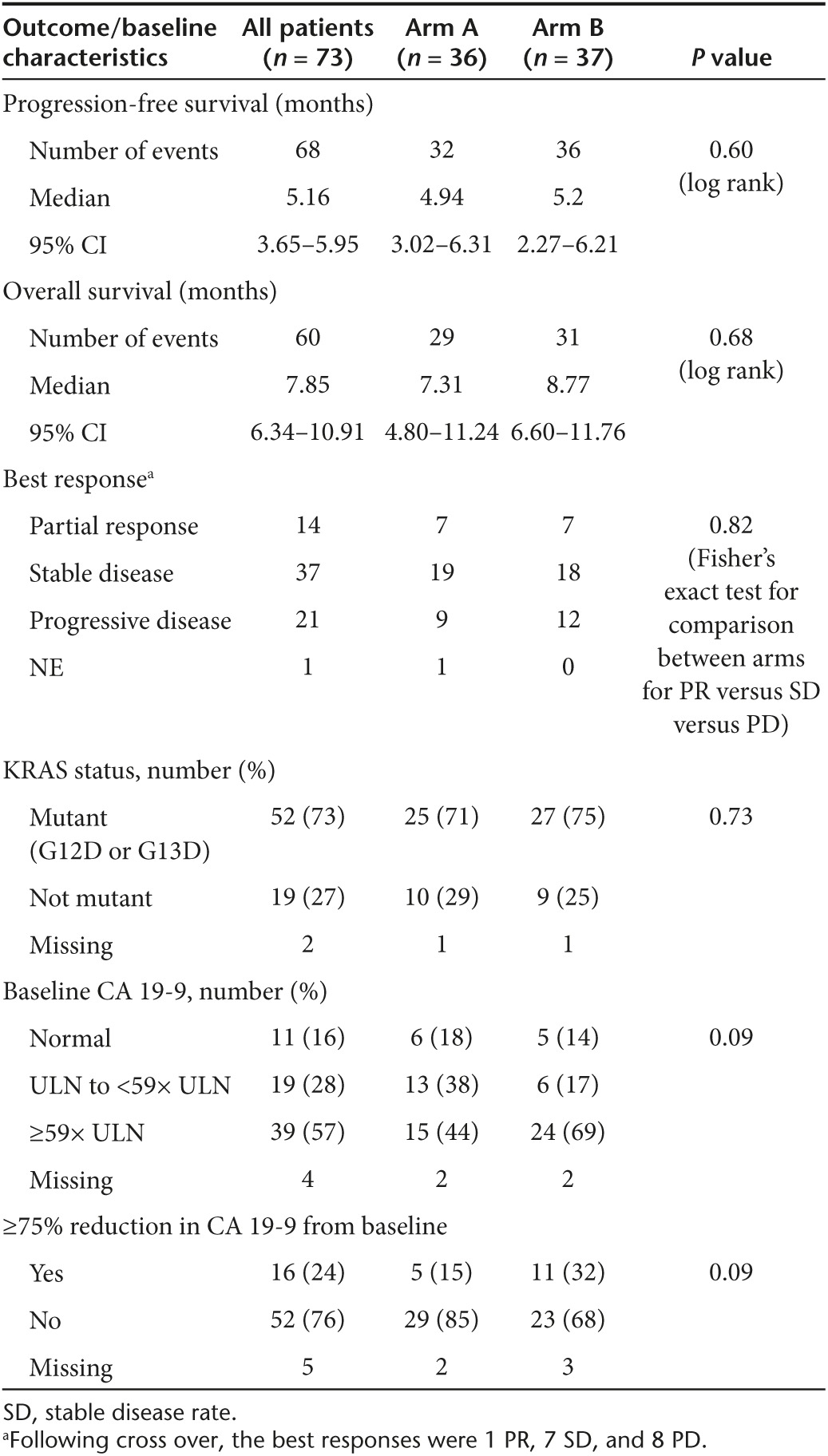
There were no differences in the distribution of best response to therapy (response versus stable disease versus disease progression/not evaluable) between the treatment arms (Table 3, P = 0.95). The partial response rate was 19% (95% CI: 11–30%) with seven responders in each arm. Stable disease was seen in 53% of patients in Arm A and in 49% of patients in Arm B (Table 3). The disease control rate (DCR) (PR and stable disease rate for 4+ months) was 56% in Arm A and 59% in Arm B. Seventeen patients crossed over from Arm B to Arm A and 16 received treatment. The median PFS after cross over was 1.7 months (95% CI: 1.4–2.6) and the median OS after cross over was 4.1 months (95% CI: 1.4–6.4).
KRAS mutational status did not predict response to pelareorep + carboplatin/paclitaxel
Tissue for KRAS mutational analysis was available for 71 patients. In concordance with previous literature, 73% of patients had a G12D or G13D KRAS mutation.4,5,6 There was no statistically significant difference in PFS in patients on Arm A or Arm B with tumors with KRAS mutations versus patients with tumors with wild-type KRAS (Table 3; Figure 2b) nor was there any difference in OS based on KRAS mutational status.
Systemic immune biomarkers, risk of progression, and response to therapy
Immunophenotypic analysis was conducted on plasma and peripheral blood mononuclear cells isolated from 70 patients who had evaluable pre- and post-treatment samples. Focusing on fold change after two cycles of treatment, we initially assessed the influence of markers on both shorter- (DCR) and longer-term (PFS) outcomes of disease control irrespective of treatment arm (see Supplementary Tables S3 and S4). Of the 32 cytokines and chemokines analyzed, changes in IL-6, IL-8, and VEGF-A were all negatively associated with DCR in the univariate setting (P = 0.026, P = 0.013, and P = 0.023, respectively). Study arm was not significantly associated with DCR, but when we formally adjusted for it in the model, only fold change in IL-6 and IL-8 remained negatively and significantly associated with DCR (P = 0.044 and P = 0.02, respectively). When analyzed in the context of PFS, increases in IL-8 (P = 0.0499), RANTES (P = 0.041), and fractalkine (P = 0.026) were significantly associated with higher hazard of progression, regardless of treatment arm (see Supplementary Table S3).
Since oncolytic viral therapy may impact the adaptive immune response and resulting antitumor immunity,10,11,12,13,14 potentially relevant immune cell subsets were analyzed by flow cytometry. Fold change in a limited number of phenotypically defined T-cell subsets was positively correlated with DCR in patients, regardless of treatment arm (see Supplementary Table S4). These included CD8+CD45RA+, a phenotype associated with naive or effector memory T cells,30 or T cells expressing both particular activating (CD4+CD137+, P = 0.11) or inhibitory (CD4+LAG3+, P = 0.04) receptors. In addition, increased fold change in NK-cells (CD3-CD56+) was associated with DCR (P = 0.046).
The relationship between B-cell subsets and clinical outcome was also examined in these patients. This analysis was particularly relevant as certain B-cell subsets may act as mediators of immune suppression in advanced cancer,31 or alternatively, in maintaining humoral immune responses against reovirus that might limit its therapeutic efficacy.32 This analysis revealed no significant association between fold change in the frequency of cells with an immunosuppressive, B regulatory cell phenotype (CD1d+CD24hiCD27+CD86+)31,33 with DCR or PFS (P = 0.34 and P = 0.87, respectively). However, fold change of mature, CD19+CD20+ B-cells was significantly associated with DCR (P = 0.02) (see Supplementary Table S4). No significant association between DCR and MDSC or other T-cell subsets expressing activation/inhibitory markers was evident (data not shown). Further, evaluation of neutralizing antireolysin antibody titers in plasma from patients on Arm A confirmed a robust humoral immune response at titers comparable to prior published studies using pelareorep alone (data not shown).
Pelareorep has distinct proinflammatory and immunomodulatory properties
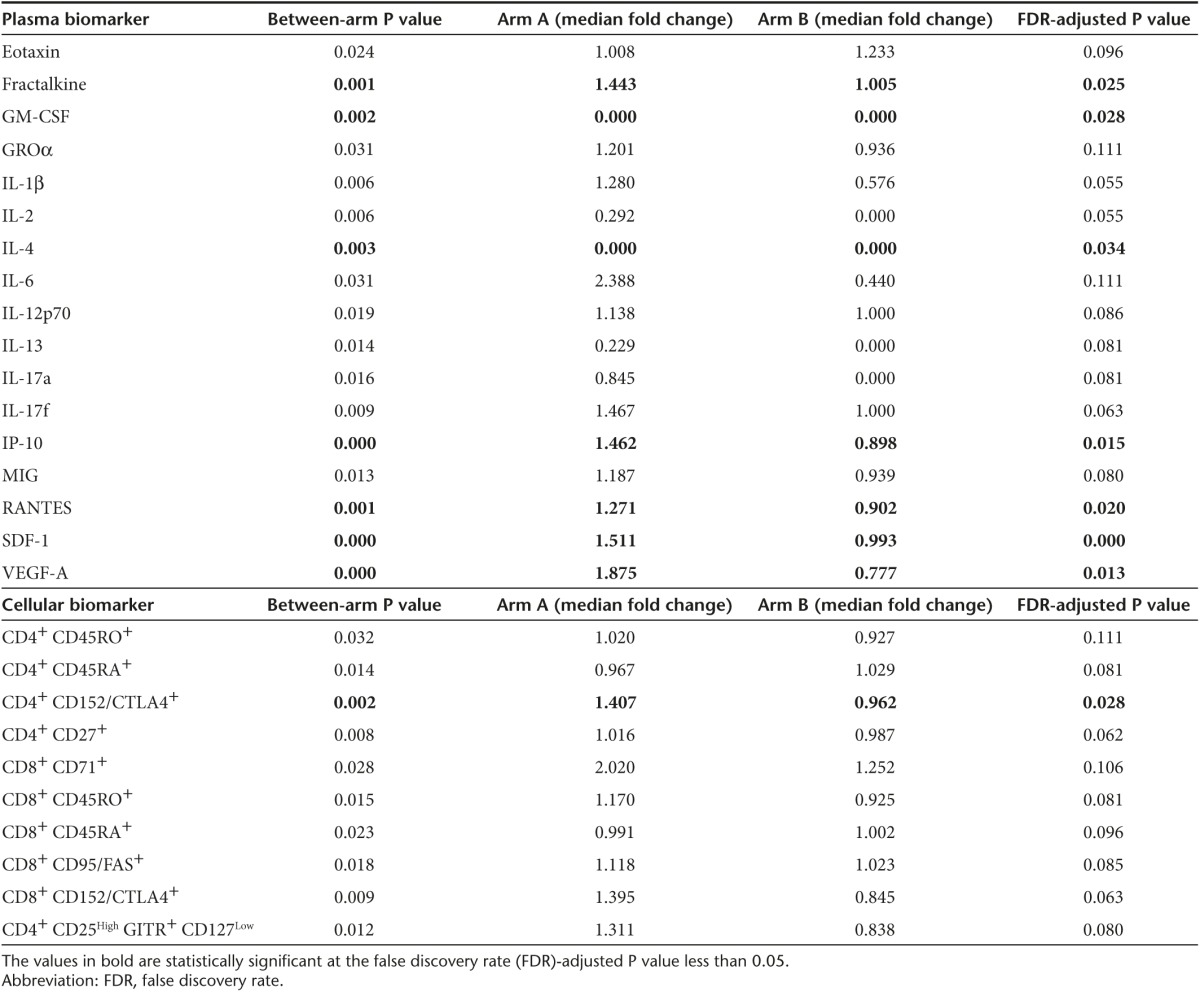
Differences in immune biomarkers between arms were also analyzed to better understand the impact of adding pelareorep to carboplatin/paclitaxel. The fold change in multiple cytokines (fractalkine, Gro-α, IL-1β, IL-2, IL-6, IL-12p70, IL-13, IL-17A, IL-17F, IP-10, MIG, RANTES, SDF-1, and VEGFA) was significantly greater in patients on Arm A as compared to Arm B, indicating that pelareorep exposure induces clear proinflammatory effects (Table 4, top). While changes in many of these markers were no longer significant after controlling for the false discovery rate, it is important to note that these immunological correlates were not a primary endpoint of the study, which was designed to be statistically powered to detect changes in PFS resulting from the addition of pelareorep to carboplatin/paclitaxel rather than to definitively identify relevant immunological predictors or markers of clinical benefit. Even with these limitations, after adjusting for false discovery rate, changes in fractalkine, IL-10, RANTES, SDF-1, and VEGF-A remained significantly greater in patients on Arm A versus Arm B, reiterating the apparent proinflammatory state induced following pelareorep treatment. Flow cytometric analysis also revealed significant increases in CD8+ T cells expressing the midearly activation markers CD71 and CD95, and in both CD4+ and CD8+ T cells expressing the maturation marker CD45RO in patients from Arm A (Table 4, bottom). In contrast, patients on Arm B had significant increases in CD4+ and CD8+ T cells expressing CD45RA, a marker of naive T cells (P = 0.014 and P = 0.023, respectively). Changes in the activation marker CD69 following treatment were not statistically significant, though the post-treatment time point examined (2 months following treatment initiation) may have been too late to detect changes in this very early T-cell activation marker. In contrast, this analysis also revealed an increase in an immunophenotypic signature indicative of immunosuppression for patients on Arm A as compared to Arm B. For example, these patients had significantly greater increases in Treg (P = 0.012), CD4+CTLA4+ (P = 0.002), CD8+CTLA4+ (P = 0.009) and CD8+TIM3+ (P = 0.047) T-cell subsets (Table 4). No significant differences in other immunophenotypic biomarkers were observed between arms, including MDSC, B regulatory cells, NK cells, CD4+TIM3+ or in any T-cell subsets expressing LAG3 or PD1 (see Supplementary Table S4 and data not shown).
Table 4. Significant immune biomarkers between arm.
Addition of pelareorep to carboplatin + paclitaxel did not worsen quality of life or toxicities
Finally, to determine the impact of the regimen on quality of life, Quality-adjusted Time without Symptoms of disease or Toxicity analysis was conducted (for methods see Supplemental Materials). Supplementary Table S5 shows the mean duration of health states by treatment arm. There was no statistically significant difference between Arm A and B for all of these health state durations (see Supplementary Tables S5 and S6; Supplementary Figure S3a,b). Data for the entire population are shown in Supplementary Tables S7 and S8 and Supplementary Figure S4).
Discussion
In this randomized phase 2 study, the addition of pelareorep to carboplatin and paclitaxel did not improve PFS compared to carboplatin and paclitaxel alone. Likewise, patients who progressed on Arm B and crossed over to Arm A did not experience a meaningful improvement in outcome upon addition of pelareorep. Pelareorep appeared to be safe to administer and was well tolerated. Apart from the occurrence of reversible lupus nephritis on the pelareorep arm, there was no significant difference in toxicity or Quality-adjusted Time without Symptoms of disease or Toxicity between the two arms.
We were particularly interested in studying pelareorep in PCA since KRAS mutations have been reported in 70–90% of PCA and the mechanism of pelareorep would suggest that it is more cytopathic in cells harboring a KRAS mutation.8,9 However, KRAS status was not associated with improvement of outcome with pelareorep in this study. We speculate that the target effect may have been neutralized in the context of other (unknown) mutations.
A number of immune biomarkers emerged across both treatment arms that were associated with improved DCR or PFS in this cohort. Among these were increased NK-cells or B-cells in patients with improved DCR. These interesting changes may reflect an improved reconstitution of circulating immune cells following administration of this taxane- and platinum-based treatment regimen. Such findings may be important in exploring future combination chemoimmunotherapy regimens using paclitaxel and carboplatin as a backbone in pancreas or other advanced cancers.
Our analysis also identified several phenotypic markers that are associated with immune suppression (including but not limited to IL-6, VEGF, and regulatory T cells) that were also increased in patients receiving pelareorep + carboplatin/paclitaxel as compared to chemotherapy alone. These data are consistent with prior preclinical studies showing that reovirus may promote immune suppression in tumor bearing mice.15 Alternatively, these markers could be upregulated as a consequence of viral infection, following stimulation of T cells via viral antigen. Regardless of the scenario, our analysis clearly indicates that pelareorep combined with chemotherapy was not a regimen sufficient to overcome the profound immunosuppression that is prevalent in patients with PCA.34,35 Patients on Arm A did have significantly increased expression of the checkpoint molecule CTLA4 on both CD4+ and CD8+ T-cell subsets. Thus, in addition to the role of these surface molecules as biomarkers of anergic or exhausted T-cells, they also represent actionable targets that could be combined therapeutically with pelareorep + carboplatin/paclitaxel.36 Indeed recent studies suggest that pelareorep may enhance the ability of antibodies targeting the programmed cell death protein 1 (PD1)/ programmed death ligand 1 (PD-L1) interaction to limit the growth of subcutaneous tumors in preclinical models.37 This study, in a murine melanoma model, reported that combination treatment with pelareorep + anti-PD-1 augmented antitumor T-cell interferon (IFN)γ secretion and NK cell killing of tumor cells. In turn this suggests that the addition of PD-1 or PD-L1 to pelareorep + carboplatin/paclitaxel has the potential to greatly increase the efficacy of this treatment regimen. Although we did not observe a significant difference in the percentage of CD8+ T-cells expressing PD1 between treatment arms, this may also remain a viable target that could be explored in animal models given the already elevated expression of this biomarker on T-cells from patients with advanced malignancy. Finally, it might also be beneficial to explore alternative chemotherapies to combine with immunotherapy and/or pelareorep, as these (e.g., gemcitabine + nab-paclitaxel, or FOLFIRINOX) may have alternative immunomodulatory properties. The combination of pelareorep and gemcitabine has been previously studied and gemcitabine was found to attenuate the neutralizing antireolysin antibody response.17
This study has a number of potential limitations to consider when interpreting immunologic data. First, although quite comprehensive, the data are derived from a single time point following treatment. Second, the changes in peripheral blood may not necessarily reflect that within the tumor microenvironment. Finally, it should be noted that these immunological analyses were not a primary endpoint of the study, but were carried out to better determine, mechanistically, why patients did not respond positively to the addition of pelareorep to carboplatin/paclitaxel. As such, this study was not statistically powered to be definitive with respect to the breadth of potentially relevant immunological biomarkers and correlates that we examined. Rather, these analyses identify immunological markers that will be the basis of future study. Despite these factors, this analysis has provided a robust and detailed phenotypic picture of immunologic changes in a population of patients who were treatment-naive at the start of this clinical trial. This is a particular strength, as it can disentangle both key immune biomarkers related to baseline immune function and those related to this regimen from immune changes that may occur as a consequence of prior aggressive myelo- or lymphoablative chemotherapy regimens.
As another interesting finding in this study, we show that carboplatin and paclitaxel appeared to perform quite well, with similar median PFS (5.2 months) and OS (8.8 months) on Arm B to that seen with gemcitabine and nab-paclitaxel-treated MPA patients in historical comparisons (PFS = 5.5 months, OS 8.5 months).3 We also recently performed a cost analysis of first-line pancreatic cancer regimens. Taking into consideration administration and toxicity costs, carboplatin and paclitaxel were over 5 times less expensive than nab-paclitaxel and gemcitabine.38 When the entire study population was analyzed for Quality-adjusted Time without Symptoms of disease or Toxicity, the majority of the mean PFS time was TWiST time (4.3 months) with good quality of life. We argue that our results warrant further investigation of carboplatin and paclitaxel as a potential first-line treatment in patients with PCA, particularly since the cost is significantly less than currently accepted standard therapies and our data suggest that it is efficacious.
Pelareorep is safe to administer in combination with carboplatin and paclitaxel, but does not increase PFS for MPA compared to carboplatin and paclitaxel alone. KRAS status did not influence response or survival outcomes following treatment with pelareorep and chemotherapy. Uniquely, outcomes with carboplatin and paclitaxel were historically comparable to standard gemcitabine and nab-paclitaxel, suggesting a more affordable taxane should be studied further in MPA. Immunologic studies suggest that the chemotherapy backbone improves the reconstitution of circulating immune cells with a more favorable outcome. Additionally, elevated expression of CTLA4 on T cells or expansion of Tregs may be key factors limiting the efficacy of oncolytic virotherapy in patients with metastatic pancreatic cancer. These findings may have implications for the design of future studies.
Materials and Methods
Study design and participants. In this phase 2, multicenter open-label randomized study, patients aged ≥18 years with histologically confirmed untreated MPA were recruited from four sites in the United States. Full eligibility criteria are provided in the protocol (see Supplemental Materials). The study was sponsored by the Cancer Therapy Evaluation Program of the National Cancer Institute (NCI). The protocol for this study was approved by Cancer Therapy Evaluation Program and the central and local institutional review boards. This study followed the protocols and standards of the Declaration of Helsinki, and patients gave written informed consent. The trial was registered at clinicaltrials.gov. (trial # NCT01280058).
Randomization and masking. Eligible patients were registered on study and randomized centrally at The Ohio State University. Patients were randomized in a 1:1 allocation to pelareorep carboplatin and paclitaxel (Arm A) or to carboplatin and paclitaxel (Arm B). The randomization method used a random permuted blocks approach with varying block sizes (Figure 1).
Procedures. Reolysin® (pelareorep, Reovirus Serotype3-Dearing Strain, NSC#729968, B-IND 13370, Oncolytics Biotech®, Calgary, AB, Canada) was supplied by NCI. Commercially available supplies of carboplatin and paclitaxel were used. All patients received intravenous infusions of paclitaxel on day 1 of each 21-day cycle at 175 mg/m2 over 3 hours followed by carboplatin at a dose area under the concentration–time curve of 5 mg/ml/minute over 30 minutes. On Arm A, pelareorep was administered after paclitaxel and carboplatin as a 60-minute intravenous infusion at a dose of 3 × 1010 tissue culture infectious dose 50 (TCID50)/day, on days 1–5 of each cycle (see Supplementary Figure S1, online only). Patients continued on therapy until they experienced disease progression, withdrew consent, or experienced intolerable toxicity that prevented further treatment. Patients in Arm B who had documented disease progression were allowed to cross over to Arm A.
Formalin-fixed paraffin-embedded tissue samples were collected at time of study entry. DNA was purified for KRAS mutational analysis.39 We evaluated plasma cytokines and chemokines as well as the immunophenotype of peripheral blood mononuclear cells on both arms at various time points; neutralizing antireovirus antibody titer levels were also assessed in Arm A patients32,40 (see Supplemental Materials for full correlative study methods).
Outcomes. The primary endpoint was PFS, defined as the time from study entry to date of documented progression and/or death. All patients who were randomized and began treatment were included in the evaluation of the primary endpoint. Patients who discontinued treatment and received an alternate therapy prior to disease progression were censored at that point. Alive and progression-free patients were censored at their last follow-up date. Secondary endpoints included objective response rate (ORR) by RECIST 1.1 criteria; DCR defined as PR or stable disease for at least 4 months; OS, defined as time from study entry to the time of death due to any cause. Correlative endpoints included immune studies and KRAS mutational analysis. Immune correlates were assessed at baseline and prior to the initiation of the third treatment cycle.
Statistical design. This randomized phase 2 study was designed to require 70 eligible and evaluable patients (35 per arm) to provide at least 90% power to detect an improvement in the median PFS from 3 to 5.5 months with the addition of pelareorep to carboplatin and paclitaxel. Based on historical data in this patient population, we hypothesized that the expected median PFS for carboplatin and paclitaxel was ~3 months. In this phase 2 setting with a focus on proof of concept, we assumed a one-sided Type I error rate of 0.2. SAS®9.3 and R3.1.2 for Windows statistical software were used.
Time-to-event outcomes (e.g., PFS, OS) were compared between arms using log-rank tests. PFS and OS distributions were evaluated using the methods of Kaplan and Meier, and univariate and multivariate Cox proportional hazards models were used to assess influence of factors or markers in relation to these survival endpoints. Patients who progressed on Arm B were allowed to cross over to Arm A. Primary PFS outcome analyses included those crossover patients in Arm B as they had already met the event of interest (disease progression). Differences in ORRs between treatment arms were assessed using Fisher's exact test. OS was evaluated and compared between the treatment arms using log-rank statistics and graphically using Kaplan and Meier methods. Differences in continuous markers between arms were assessed using Wilcoxon rank-sum tests.
Correlative endpoints were analyzed using descriptive statistics and summarized graphically. KRAS mutation status and immune markers were evaluated overall and by treatment arm. In addition, correlative markers were explored in relation to PFS, DCR, and ORR.
Toxicity was assessed using NCI CTCAE v4.0. All patients who received at least one dose of any of the therapeutic agents in a treatment arm were evaluable for toxicity and tolerability. Quality-adjusted Time without Symptoms of disease or Toxicity of treatment was calculated for each treatment arm and for the entire study population, using all grade 3 and 4 toxicities and the following grade 2 toxicities: nausea, fatigue, peripheral neuropathy, and diarrhea (see Supplemental Materials).
SUPPLEMENTARY MATERIAL Figure S1. Clinical trial schema. Figure S2. Overall survival. Figure S3. A and B. QTWiST analysis for Arm A (A) and Arm B (B). Figure S4. QTWiST analysis for both arms combined. Table S1. Univariate and multivariate analysis of PFS and OS by age. Table S2. Second-line regimens received following progression. Table S3. Relationship between plasma immune biomarkers as determined by bioplex analysis with disease control rate (DCR) or progression-free survival (PFS). Table S4. Relationship between cellular immune biomarkers as determined by flow cytometry with disease control rate (DCR) or progression-free survival (PFS). Table S5. Mean duration of health states by treatment arm. Table S6. Sensitivity analysis for QTWiST. Table S7. Mean duration of health states when all patients in the trial were included in the analysis. Table S8. Sensitivity analysis for all patients. Supplemental Materials • Clinical Protocol Pelareorep in Pancreatic Cancer • Methods for Correlative Studies
Acknowledgments
We would like to thank OSU CCC Analytical Cytometry Shared Resource and Clinical Trials Office for assistance with these studies. We are especially grateful to all patients who participated in this study. This study would not have been possible without the support of the following sources: the US National Cancer Institute's Cancer Therapy Evaluation Program (NCT01280058) and associated Phase 2 N01 program grant (HHSN261201100070C), an NIH postdoctoral training grant (5 T32 CA 90223-12) the William Hall Fund for Liver and Pancreatic Cancer Research, Oncolytics, and the Pelotonia Fellowship Program. Any opinions, findings, and conclusions expressed in this material are those of the authors and do not necessarily reflect those of the Pelotonia Fellowship Program. Anne Noonan (the Ohio State University) was a paid member of a data safety monitoring board for Helsinn. Matthew Farren (the Ohio State University) received salary support from the Pelotonia Research Foundation and an NIH training grant (5 T32 CA 90223-12). Susan Geyer (University of South Florida) reported no outside funding to disclose. Ying Huang (the Ohio State University) reported no outside funding to disclose. Sanaa Tahiri (the Ohio State University) reported no outside funding to disclose. Daniel Ahn (the Ohio State University) received salary support from an NIH training grant (1 T32 CA 165998-01). Sameh Mikhail (the Ohio State University) is a consultant/advisory board member for Bayer. Kristen Ciombor (the Ohio State University) received research support from Pfizer, Boston Biomedical, MedImmune, Onyx Pharmaceuticals, Bayer, Boehringer Ingelheim, and Bristol-Myers Squibb. Shubham Pant (Oklahoma University Cancer Institute) reported no outside funding to disclose. Santiago Aparo (Albert Einstein College of Medicine) reported no outside funding to disclose. Jennifer Sexton (the Ohio State University) received NIH/NCI salary support. John Marshall (Georgetown University) reported no outside funding to disclose. Thomas Mace (the Ohio State University) received NIH funding and institutional support. Christina Wu (the Ohio State University) served on a Bayer advisory board. Bassel El-Rayes (Emory University) received consulting fees (Merrimack, Genentech) research support (Bristol Myers Squibb, Boston Biomedical, Cleave Biosciences, SYNTA Pharmaceuticals, AVEO Pharmaceuticals, Taiho Oncology, Genentech, the Hoosier Cancer Research Network) and NIH research support. Cynthia Timmers (the Ohio State University) received research and salary support from Oncolytics and the NCI. James Zwiebel (US National Cancer Institute) had no funding to disclose. Gregory Lesinski (the Ohio State University) received institutional and divisional start-up funds (Ohio State University) NIH funding and independent grant support (Oncolytics, Karyopharm, Chirhoclin, the American College for Gastroenterology, Merck, and the Hoosier Cancer Research Network). Miguel Villalona-Calero (the Ohio State University) was the PI on the Phase 2 N01 program grant (HHSN261201100070C) under which this study was carried out. Tanios Bekaii-Saab (the Ohio State University) received research support from Oncolytics, the US National Cancer Institute, and the National Comprehensive Cancer Network. Greg Lesinski, Cynthia Timmers, and Tanios Bekaii-Saab received funding support from Oncolytics to support correlative laboratory research for this study; all other authors report no relevant potential conflicts of interest.
Supplementary Material
References
- Howlader N, N, A, Krapcho M, Garshell J, Miller D, Altekruse SF et al. (2014). SEER Cancer Statistics Review, 1975–2011. National Cancer Institute: Bethesda, MD. [Google Scholar]
- Conroy, T, Desseigne, F, Ychou, M, Bouché, O, Guimbaud, R, Bécouarn, Y et al.; Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup (2011). FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364: 1817–1825. [DOI] [PubMed] [Google Scholar]
- Von Hoff, DD, Ervin, T, Arena, FP, Chiorean, EG, Infante, J, Moore, M et al. (2013). Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369: 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almoguera, C, Shibata, D, Forrester, K, Martin, J, Arnheim, N and Perucho, M (1988). Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 53: 549–554. [DOI] [PubMed] [Google Scholar]
- Deramaudt, T and Rustgi, AK (2005). Mutant KRAS in the initiation of pancreatic cancer. Biochim Biophys Acta 1756: 97–101. [DOI] [PubMed] [Google Scholar]
- Jones, S, Zhang, X, Parsons, DW, Lin, JC, Leary, RJ, Angenendt, P et al. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321: 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty, R, Tran, H, Selvaggi, G, Hagerman, A, Thompson, B and Coffey, M (2015). The oncolytic virus, pelareorep, as a novel anticancer agent: a review. Invest New Drugs 33: 761–774. [DOI] [PubMed] [Google Scholar]
- Strong, JE, Coffey, MC, Tang, D, Sabinin, P and Lee, PW (1998). The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J 17: 3351–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos, JL (1989). ras oncogenes in human cancer: a review. Cancer Res 49: 4682–4689. [PubMed] [Google Scholar]
- Gujar, S, Dielschneider, R, Clements, D, Helson, E, Shmulevitz, M, Marcato, P et al. (2013). Multifaceted therapeutic targeting of ovarian peritoneal carcinomatosis through virus-induced immunomodulation. Mol Ther 21: 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujar, SA, Clements, D, Dielschneider, R, Helson, E, Marcato, P and Lee, PW (2014). Gemcitabine enhances the efficacy of reovirus-based oncotherapy through anti-tumour immunological mechanisms. Br J Cancer 110: 83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujar, SA, Marcato, P, Pan, D and Lee, PW (2010). Reovirus virotherapy overrides tumor antigen presentation evasion and promotes protective antitumor immunity. Mol Cancer Ther 9: 2924–2933. [DOI] [PubMed] [Google Scholar]
- Gujar, SA, Pan, DA, Marcato, P, Garant, KA and Lee, PW (2011). Oncolytic virus-initiated protective immunity against prostate cancer. Mol Ther 19: 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele, L, Errington, F, Prestwich, R, Ilett, E, Harrington, K, Pandha, H et al. (2011). Pro-inflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF-κB mediated and supports innate and adaptive anti-tumour immune priming. Mol Cancer 10: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements, DR, Sterea, AM, Kim, Y, Helson, E, Dean, CA, Nunokawa, A et al. (2015). Newly recruited CD11b+, GR-1+, Ly6C(high) myeloid cells augment tumor-associated immunosuppression immediately following the therapeutic administration of oncolytic reovirus. J Immunol 194: 4397–4412. [DOI] [PubMed] [Google Scholar]
- Gabrilovich, DI, Ostrand-Rosenberg, S and Bronte, V (2012). Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12: 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolkema, MP, Arkenau, HT, Harrington, K, Roxburgh, P, Morrison, R, Roulstone, V et al. (2011). A phase I study of the combination of intravenous reovirus type 3 Dearing and gemcitabine in patients with advanced cancer. Clin Cancer Res 17: 581–588. [DOI] [PubMed] [Google Scholar]
- Ma, WW and Hidalgo, M (2013). The winning formulation: the development of paclitaxel in pancreatic cancer. Clin Cancer Res 19: 5572–5579. [DOI] [PubMed] [Google Scholar]
- Emens, LA and Middleton, G (2015). The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res 3: 436–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apetoh, L, Ghiringhelli, F, Tesniere, A, Criollo, A, Ortiz, C, Lidereau, R et al. (2007). The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev 220: 47–59. [DOI] [PubMed] [Google Scholar]
- Apetoh, L, Ghiringhelli, F, Tesniere, A, Obeid, M, Ortiz, C, Criollo, A et al. (2007). Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 13: 1050–1059. [DOI] [PubMed] [Google Scholar]
- Lopes, G and Rocha Lima, CM (2005). Docetaxel in the management of advanced pancreatic cancer. Semin Oncol 32(2 suppl. 4): S10–S23. [DOI] [PubMed] [Google Scholar]
- Oettle, H, Arnold, D, Esser, M, Huhn, D and Riess, H (2000). Paclitaxel as weekly second-line therapy in patients with advanced pancreatic carcinoma. Anticancer Drugs 11: 635–638. [DOI] [PubMed] [Google Scholar]
- Whitehead, RP, Jacobson, J, Brown, TD, Taylor, SA, Weiss, GR and Macdonald, JS (1997). Phase II trial of paclitaxel and granulocyte colony-stimulating factor in patients with pancreatic carcinoma: a Southwest Oncology Group study. J Clin Oncol 15: 2414–2419. [DOI] [PubMed] [Google Scholar]
- Mikhail, S, Lustberg, MB, Ruppert, AS, Mortazavi, A, Monk, P, Kleiber, B et al. (2015). Biomodulation of capecitabine by paclitaxel and carboplatin in advanced solid tumors and adenocarcinoma of unknown primary. Cancer Chemother Pharmacol 76: 1005–1012. [DOI] [PubMed] [Google Scholar]
- Auerbach, M, Wampler, GL, Lokich, JJ, Fryer, D, Fryer, JG and Ahlgren, JD (1997). Treatment of advanced pancreatic carcinoma with a combination of protracted infusional 5-fluorouracil and weekly carboplatin: a Mid-Atlantic Oncology Program Study. Ann Oncol 8: 439–444. [DOI] [PubMed] [Google Scholar]
- Sei, S, Mussio, JK, Yang, QE, Nagashima, K, Parchment, RE, Coffey, MC et al. (2009). Synergistic antitumor activity of oncolytic reovirus and chemotherapeutic agents in non-small cell lung cancer cells. Mol Cancer 8: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadler, S, Yu, B, Lane, M, Klampfer, L, Sasazuki, T, Shirasawa, S, et al. (2004). 452 The oncolytic reovirus, Reolysin, augments the anticancer effects of cytotoxic agents in vitro against the ras-mutated human colon cancer cell line HCT116. Eur J Cancer Suppl 2: 135. [Google Scholar]
- Roulstone, V, Twigger, K, Zaidi, S, Pencavel, T, Kyula, JN, White, C et al. (2013). Synergistic cytotoxicity of oncolytic reovirus in combination with cisplatin–paclitaxel doublet chemotherapy. Gene Ther 20: 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann, D, Baars, PA, Rep, MH, Hooibrink, B, Kerkhof-Garde, SR, Klein, MR et al. (1997). Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med 186: 1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata, Y, Matsushita, T, Horikawa, M, Dilillo, DJ, Yanaba, K, Venturi, GM et al. (2011). Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 117: 530–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, CL, Twigger, KR, Vidal, L, De Bono, JS, Coffey, M, Heinemann, L et al. (2008). Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther 15: 911–920. [DOI] [PubMed] [Google Scholar]
- Blair, PA, Noreña, LY, Flores-Borja, F, Rawlings, DJ, Isenberg, DA, Ehrenstein, MR et al. (2010). CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity 32: 129–140. [DOI] [PubMed] [Google Scholar]
- Mace, TA, Ameen, Z, Collins, A, Wojcik, S, Mair, M, Young, GS et al. (2013). Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res 73: 3007–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnurr, M, Duewell, P, Bauer, C, Rothenfusser, S, Lauber, K, Endres, S et al. (2015). Strategies to relieve immunosuppression in pancreatic cancer. Immunotherapy 7: 363–376. [DOI] [PubMed] [Google Scholar]
- Mahoney, KM, Rennert, PD and Freeman, GJ (2015). Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 14: 561–584. [DOI] [PubMed] [Google Scholar]
- Rajani, K, Parrish, C, Kottke, T, Thompson, J, Zaidi, S, Ilett, L et al. (2016). Combination therapy with reovirus and anti-pd-1 blockade controls tumor growth through innate and adaptive immune responses. Mol Ther 24: 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DA, Krishna, K, Flowers C, El-Rayes BF, Bekaii-Saab TS, Noonan AM (2016). Cost description of chemotherapy regimens for the treatment of metastatic pancreas cancer (mPC). Med Oncol 33:48. [DOI] [PubMed] [Google Scholar]
- Kim, ST, Lim, do H, Jang, KT, Lim, T, Lee, J, Choi, YL c (2011). Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Mol Cancer Ther 10: 1993–1999. [DOI] [PubMed] [Google Scholar]
- Lesinski, GB, Kondadasula, SV, Crespin, T, Shen, L, Kendra, K, Walker, M et al. (2004). Multiparametric flow cytometric analysis of inter-patient variation in STAT1 phosphorylation following interferon Alfa immunotherapy. J Natl Cancer Inst 96: 1331–1342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.