

Abstract
Leukemic stem cells (LSCs) have been proven to drive leukemia initiation, progression and relapse, and are increasingly being used as a critical target for therapeutic intervention. As an essential feature in LSCs, reactive oxygen species (ROS) homeostasis has been extensively exploited in the past decade for targeting LSCs in acute myeloid leukemia (AML). Most, if not all, agents that show therapeutic benefits are able to alter redox status by inducing ROS, which confers selectivity in eradicating AML stem cells but sparing normal counterparts. In this review, we provide the comprehensive update of ROS-generating agents in the context of their impacts on our understanding of the pathogenesis of AML and its therapy. We anticipate that further characterizing these ROS agents will help us combat against AML in the coming era of LSC-targeting strategy.
Keywords: Acute myeloid leukemia (AML), reactive oxygen species (ROS), leukemic stem cells (LSCs), targeting therapy
Introduction
Acute myeloid leukemia (AML) is a malignant disorder, consisting of a heterogeneous group of clonal hematopoietic stem cell neoplasms. AML is characterized by aberrant accumulation of immature myelocytes in the bone marrow, which interferes with the production of normal blood cells. The last four decades have seen great advances in treatment-optimization and supportive care. However, long-term effective cure has been by far undefined (1). After being treated with chemotherapy, most patients can achieve complete remission (CR), but only about 20-25% patients remain long-term disease-free survival (DFS) (obtained from the NCI’s SEER database) (2). It becomes even worse for elderly patients (>60 years); the figure drops downs to 10-15% only. It is therefore not surprising that the death rate can reach as high as 50% for young adult patients and 90% for elderly AML patients (3). Low DFS is largely explained by refractoriness to chemotherapeutic strategies and relapse after CR, both of which become the main bottlenecks in current AML therapy.
The high frequency of drug resistance and relapse after conventional chemotherapy is a result of failure of targeting leukemic stem cells (LSCs). Accumulating evidences have supported that LSCs are key drivers of leukemia initiation, progression and relapse. Similar to normal hematopoietic stem cells (HSCs), LSCs exhibit stem cell-like characteristics such as the capacity for self-renewal, multi-potent differentiation potential and relative quiescence (4). Higher expression of multi-drug resistance (MDR) proteins renders LSCs resistant to conventional chemotherapeutic agents. The persistence of surviving LSCs is responsible for the high frequency of relapse (5-8). Undoubtedly, successful therapeutic strategies should also eliminate these LSCs. It is therefore crucial that therapies be developed targeting the quiescent and drug-resistant LSCs. Despite the similarities shared by LSCs and HSCs, LSCs often possess several unique features. To name but a few, they include: (I) the abnormal expression of CD markers (e.g., CD44, CD47, CD96, and CD123) (9-12); (II) constitutive activation of nuclear factor kappa B (NF-κB) (13); (III) active Wnt/β-catenin signaling (14); (IV) elevated levels of interferon regulatory factor-1 (IRF-1) and death-associated protein kinase (DAPK) (15); and (V) the sensitiveness to reactive oxygen species (ROS) homeostasis (16-18). Thankfully, these characteristics may provide important hints for designing LSC-targeted therapy.
In this review, we mainly focus on ROS and their roles in AML therapies, especially in targeting AML stem cells. ROS are a heterogeneous group of small molecules, including super-oxide, hydrogen peroxide, singlet oxygen, ozone, hypo-halous acids and organic peroxides (19-21). Due to highly chemically reactive nature, it is much easier for ROS to damage DNA, proteins and lipids than molecular oxygen does. In hematopoietic cells, ROS are largely generated in the mitochondria, through nicotinamide adenine dinucleotide phosphate (NADPH) oxidases and other ROS-related metabolic pathways (e.g., polyamine metabolism, cytochrome P450 and xanthine oxidase) (22). In hematological malignancies, oxidative stress overload (due to increased ROS) has been identified in several other myeloid neoplasms in addition to AML, thus representing a universal feature in myeloid diseases (23,24). Several studies have reported the association between increased levels of oxidative stress and AML relapse, indicating that ROS over-production may be a critical factor in AML initiation and progression. More importantly, ROS may be an optimal therapeutic target for AML-stem cells therapy. This is evident in our recent report and many others; ROS-generating agents can selectively eradicate AML-stem cells via modulating ROS production (25-34).
Pathologic features of AML stem cells
LSCs account for a rare sub-population of leukemia cells, but are the real instigator of currently poor prognosis in AML therapies. There is a hot debate on their origin, but a consensus being reached is that LSCs do not come from single source; they can originate either from long-term HSCs (LT-HSCs) and short-term HSCs (ST-HSCs), or multi-potent progenitors (MPPs), or even more differentiated progenitors [i.e., common myeloid progenitors (CMP) and granulocyte/macrophage progenitors (GMP)] (35). Similar to normal HSCs, LSCs exhibit stem cell-like characteristics such as the capacity for unlimited self-renewal, impaired hematopoietic differentiation and the ability of generating heterogeneous leukemia cells that comprise leukemia. However, the signaling pathways controlling “stemness” in LSCs are not exactly the same as those in HSCs. These pathways are disturbed in LSCs. For example, self-renewal associated pathways [B lymphoma Mo-MLV insertion region 1 homolog (Bmi-1), telomerase and signal transducer and activator of transcription 5 (STAT5)] (36-39) and developmental pathways [Notch, Wnt/β-catenin and sonic hedgehog (Shh)] (14,40,41) are aberrantly activated. Other miscellaneous pathways related to apoptosis, differentiation and drug resistance [NF-κB, fms-like tyrosine kinase-3 (FLT3), phosphoinositide 3-kinase (PI3K) /AKT/mammalian target of rapamycin (mTOR) and P-glycoprotein (P-gp)] are constitutively activated (8,13,42-45). Recent studies have also shown unique epigenetic pathways requisite for maintaining AML-stem cells self-renewal, including highly expressed histone H3 lysine 36 dimethyl-specific demethylase KDM2b/JHDM1b (46). In addition to these pathways, LSCs also have specific immunophenotypic markers, including CD117, CD82, CD47, CD96, CD32, CD33, CD25, Cyclin-A1, H-Y antigens, C-type lectin-like molecule-1 (CLL-1), T cell immunoglobulin mucin-3 (TIM3) and CD44 (10,11,47-56). The greatest difference is their unique location in bone marrow. Most LSCs are concentrated within the osteoblast-rich area. This area is of low oxygen pressure and is highly immune to cytotoxicity induced by chemotherapy. Under low-oxygen condition, AML-stem cells are arrested at stable quiescent cycling stage, and over-express a higher P-gp, MDR-related protein 1 (MRP), breast cancer resistance protein (BRCP) and lung-resistance protein (LRP) (8). The quiescence and higher MDR expression confer drug-efflux capabilities on LSCs to escape from chemotherapies.
ROS homeostasis
The generation source of ROS
Intracellular ROS can be produced exogenously and endogenously. The exogenous sources of ROS include smoke, air pollutants, ultraviolet radiation, γ-irradiation, and many chemicals able of generating ROS. The endogenous sources of ROS are much diverse. It can come from mitochondria, a family of NADPH oxidases (NOXs), and ROS-related metabolisms (57). The mitochondrial respiratory system is believed to be the major intracellular source of ROS. Upon pro-oxidant stimuli, ROS (e.g., O2·–, HO·) are generated by accompanying unstable mitochondrial membrane potential (Δѱ), the electron delivery through the mitochondrial respiratory complexes and proton gradient establishment across the inner mitochondrial membrane. These metabolisms are usually catalyzed by cytochrome P450 enzymes, polyamine and amino acid oxidases, xanthine oxidase, lipoxygenases, cyclooxygenases, flavoenzyme ERO1 in the endoplasmic reticulum (ER), flavin-dependent demethylase, and nitric oxide synthases, to name but a few (58-60). Other sources include free iron or copper ions, haem groups, and metal storage proteins responsible of converting O2·– and/or H2O2 to OH·.
The scavenger system of ROS
Cells have evolved an elaborate defense system to scavenge excess ROS so as to avoid oxidative stress damage. This scavenger system is important to maintain ROS homeostasis, especially for hematopoiesis (61). To reach oxidation-reduction (redox) equilibrium, a variety of cellular antioxidant enzymes and non-enzymatic molecules can be utilized. The enzymatic antioxidant enzymes include uperoxide dismutases, catalases, glutathione peroxidases, glutathione reductase, thioredoxins, thioredoxin reductases, methionine sulphoxide reductases, peroxiredoxins and peroxynitrite reductases. The non-enzymatic molecules include ascorbate, pryuvate, α-ketoglutarate (α-KG) and oxaloacetate; all these can directly react with ROS for which is kept at a homeostatic level.
The influent roles of ROS in HSCs
It has long been recognized that ROS play a critical role in balancing self-renewal and myeloid differentiation of HSCs. Studies using animal models has demonstrated that coordinated regulation of ROS levels is of great importance for HCS quiescence (62,63). Mice lacking the ataxia telangiectasia mutated (ATM) can develop bone marrow failure that is correlated with increased levels of ROS (64). The impairment of HSCs in ATM-/- mice is due to the aberrant activation of p38 MAPK signaling pathway caused by increased ROS (65). The forkhead O (FoxO) family of transcription factors is important to control ROS levels of quiescence HSCs compartment (66). Mice with conditional loss of FoxO1, FoxO3, and FoxO4 suffer from decreased number of HSCs, impaired capabilities of hematopoietic reconstitution in competitive and noncompetitive recipient mice. However, increased levels of ROS induce differentiation of HSCs into myeloid progenitors but in a manner independent of FoxO activity (67,68). The findings above in mice are also validated in fruit flies: the accumulation of ROS is an important factor triggering myeloid progenitor differentiation from HSCs (67). Mechanistic studies reveal that increased levels of ROS induce HSCs to exit from the niche for the differentiation via suppressing the expression of N-cadherin and reducing HSCs-osteoblast adhesion (63). However, abnormal robust ROS accumulation is closely associated with HSC senescence (69). ROS-induced HSC senescence is regulated by signaling pathways including FoxOs, ATM, mTOR, TSC1, Bmi1 and AKT. Other pathways such as p53-p21 and p16-Rb activation can also rapidly induce ROS increase and then trigger HSC senescence.
The emerging roles of ROS in AML
Cancer cells rely on glycolysis but not aerobic mitochondrial respiration for energy supply, and tend to live in the lower pressure of oxygen environment than their normal counterparts. Low level of intracellular ROS is even common when it comes to cancer stem cells (CSCs), regardless of their tissue origins (e.g., breast, gallbladder, liver, lymphoma and so forth) (70-75). It is likely a metabolic hallmark of CSCs. The knowledge about the association between ROS levels and cancer (and CSCs) is largely learnt from the studies on AML. First, some genetic abnormalities in AML are directly relevant to ROS metabolism. One of the most common genetic abnormalities is Ras [including 10% of neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS) mutation, 2% of v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutation and rare v-Ha-ras Harvey rat sarcoma viral oncogene homolog (HRAS) mutation], leading to constitutive activation of Ras signaling. CD34+ human hematopoietic progenitor cells with RAS mutations are capable of inducing ROS production (68). Genome-wide sequencing studies have identified 7% isocitrate dehydrogenase 1 (IDH1) and 8% isocitrate dehydrogenase 2 (IDH2) somatic mutations in AML patients. These phenotypic mutations impair the normal enzymatic activity of IDH1/2, leading to decreased production of NAD(P)H, α-KG and glutathione, and increased production of oncometabolite 2-hydroxyglutarate (2-HG). These genetic abnormalities can disturb the balance between pro-oxidative and anti-oxidative states, which eventually increases the ROS production (76,77). Recent published data demonstrate that active FLT3 signaling (over 30% AML patients with FLT3 mutation) is also closely related to increased ROS production (24,78,79). Second, AML-stem cells tend to have a lower metabolic activity (thanks to their quiescent nature), thus a lower ROS production than the AML cells as a whole. Through delicate functional assays, Lagadinou et al. demonstrate that the majority of AML-stem cells are enriched in the ROS-low population. Moreover, this study further shows that these ROS-low cells overexpress BCL-2, and are more sensitive than ROS-high counterparts to cell death induced by the BCL-2 inhibitor. The BCL-2 is an apoptosis antagonist, characterizing the apoptotic-resistance feature of AML-stem cells (31,80). Thanks to the ROS difference, different responses to BCL-2 inhibitors can be used for AML therapies.
Agents selectively induce AML-stem cells cell death via ROS
In this section, we provide a detailed review of ROS-generating agents that show potentials to target primary AML-stem cells (summarized in Table 1). ROS play a decisive role in eradicating AML-stem cells. Though the mechanisms of how ROS are produced by these agents remain unclear, increasing evidence suggest that impairment of the scavenge system might play the central role in ROS generation. This is exemplified by (I) 3-deazaneplanocin A (DZNep) up-regulates thioredoxin-binding protein 2 (TXNIP) to inhibit thioredoxin activity, which in turn causes ROS production (33); (II) treatment with ABT-263 causes decreased levels of reduced glutathione (GSH) in AML-stem cells (31); (III) antioxidants such as N-acetyl-l-cysteine (NAC), vitamin C or α-tocopherol can protect AML-stem cells from targeting (25,32,34); (IV) NF-κB, a key transcription factor for the redox balance, is inhibited by most AML-stem cells targeting agents (26,27,29,30,34). Besides ROS induction, other mechanisms mainly include NF-κB inactivation, p53 activation, FLT3 inhibition, Wnt inhibition, PKC inhibition and membrane disruption. Integration of ROS with other key stemness/survival signaling pathways brings powerful toxic efficacy on AML-stem cells (Figure 1). Until now, 11 agents have been verified in AML-stem cell targeting capabilities.
Table 1. Agents favoring oxidative stress for targeting AML-stem cells.
| Targeting agents | Molecular structure | Involved mechanisms | Ref |
|---|---|---|---|
| Niclosamide | 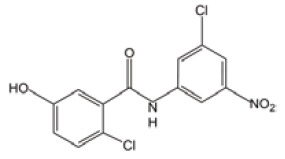 |
ROS induction | (30) |
| NF-κB inactivation | |||
| ABT-737 | 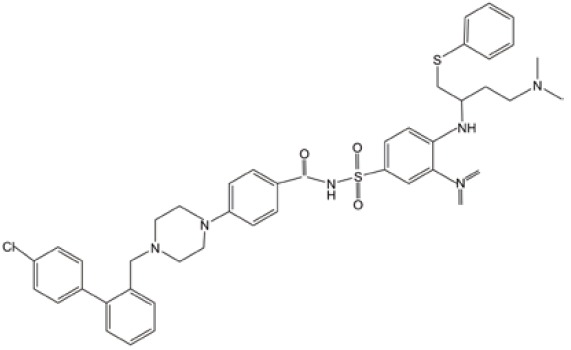 |
BCL-2 inhibition | (31) |
| ROS-induction | |||
| GSH depletion | |||
| ABT-263 | 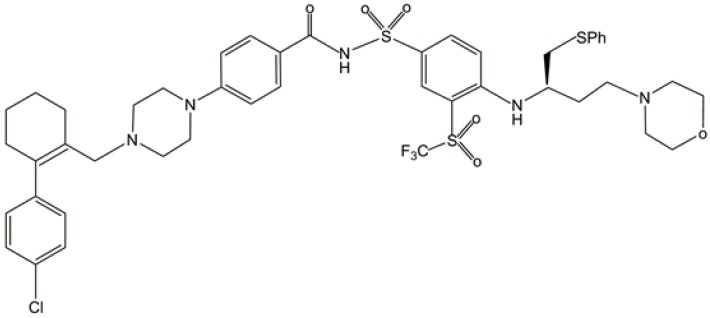 |
OXPHOS inhibition | (31) |
| ATP depletion | |||
| Glycolysis inhibition | |||
| Celastrol | 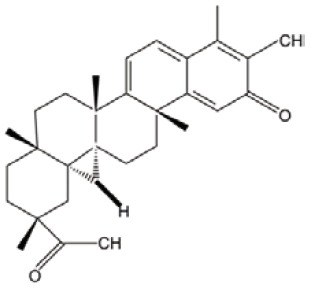 |
ROS induction | (29) |
| NF-κB inactivation | |||
| HNE |  |
ROS induction | (29) |
| NF-κB inactivation | |||
| DZNep |  |
ROS induction | (33) |
| ER stress | |||
| Mefloquine |  |
ROS induction | (32) |
| Lysosome disruption | |||
| PTL |  |
ROS induction | (26) |
| p53 activation | |||
| NF-κB inactivation | |||
| DMAPT | 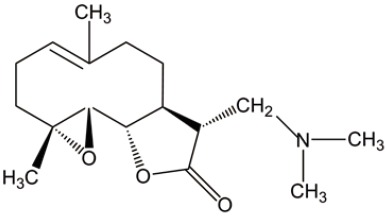 |
ROS induction | (27) |
| p53 activation | |||
| NF-κB inactivation | |||
| Fenretinide | 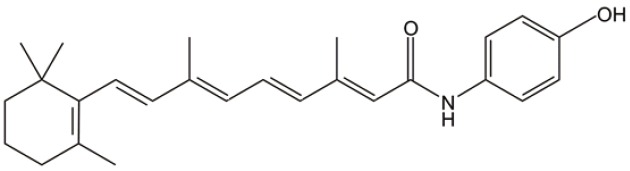 |
ROS induction | (34) |
| NF-κB inactivation | |||
| Wnt inhibtion | |||
| TDZD-8 | 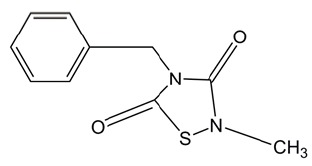 |
ROS induction | (25) |
| Membrane disruption | |||
| PKC inhibition | |||
| FLT3 inhibition |
AML, acute myeloid leukemia; HNE, 4-hydroxy-2-nonenal; DZNep, 3-deazaneplanocin A; PTL, parthenolide; DMAPT, dimethylaminoparthenolide; TDZD-8, 4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione; ROS, reactive oxygen species; NF-κB, nuclear factor kappa B; GSH, glutathione; ER, endoplasmic reticulum; FLT3, fms-like tyrosine kinase-3.
Figure 1.
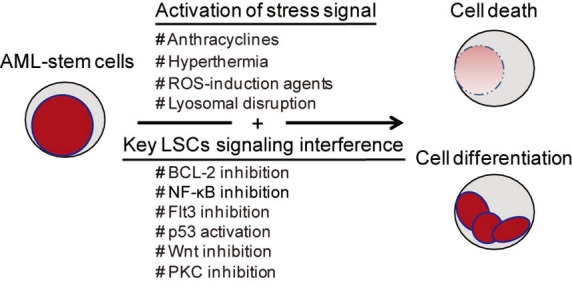
Therapeutic strategies for targeting leukemia stem cells in AML. AML, acute myeloid leukemia; ROS, reactive oxygen species; NF-κB, nuclear factor kappa B; FLT3, fms-like tyrosine kinase-3.
Bcl-2 inhibitors
Small molecule BCL-2 inhibitors, ABT-737 and ABT-263, have been shown to selectively kill ROS-low AML-stem cells (31). In vivo experiments reveal that BCL-2 inhibitors severely impair mitochondrial oxidative phosphorylation, rapidly deplete intracellular ATP production, and induce a robust generation of mitochondrial ROS. More importantly, there are no cytotoxic effects observed on ROS-high AML cells (including higher proliferate rate AML cells and AML blast cells). BCL-2 inhibitors have entered a phase I clinical trial for evaluating the clinical efficacy in AML/ myelodysplastic syndrome (MDS) treatment. In this trial, the small-molecule pan-BCL-2 inhibitor, called ‘obatoclax mesylate’, was prescribed to 44 patients with refractory leukemia and myelodysplasia to assess its safety and optimal dose. One AML patient with mixed lineage leukemia t (9;11) rearrangement achieved a CR that lasted for 8 months. Three of 14 MDS patients showed hematologic improvement for less red blood cells or platelet transfusion dependence (81). This clinical trial comes to the conclusion that obatoclax mesylate is a well-tolerated pan-BCL-2 inhibitor with early efficiency, calling for next-phase trial for its eventual clinical use in targeting AML stem cells.
Parthenolide (PTL) and dimethylaminoparthenolide (DMAPT)
PTL is a naturally occurring small molecule that has been evaluated for in vitro and in vivo efficacy on AML progenitor and stem cell populations. However, low soluble feature makes its pharmacologic potential less attractive. Instead, DMAPT, a dimethylamino analog of PTL, demonstrates 1,000-fold greater solubility in water than PTL. Both of DMAPT and PTL show similar efficacy on AML-stem cells through the similar molecular mechanisms, including the elevated ROS, p53 activation and NF-κB inactivation. Canine xenograft experiments, an equivalent to phase I clinical trials, show that the pharmacologic properties of DMAPT are superior to PTL (26,27). These data call for further studies on clinical safety and translational efficacy of DMAPT/PTL in AML treatment.
Fenretinide
Fenretinide is a well-tolerated vitamin A derivative that lacks a carboxyl functional group likely necessary for retinoid receptor activity. Our most recent study has shown that it is capable of eradicating LSCs but not normal hematopoietic progenitor/stem cells, at physiologically achievable concentrations (5 µM). Fenretinide-induced AML-stem cells death is associated with the rapid generation of ROS, induction of genes responsible for stress responses and apoptosis, and repression of genes involved in NF-κB and Wnt signaling (34). Though there is no clinical trial ongoing for AML treatment, fenretinide has been verified its safety and low toxicity in phase II-III clinical trials for solid tumors such as small cell lung cancer, breast cancer and prostate cancer (82-85). Moreover, via bioinformatics analysis we have observed that fenretinide down-regulated genes are significantly correlated with genes related to a poor prognosis/relpase of AML. We anticipate that fenretinide is a potent AML-stem cells targeting candidate in the treatment of AML.
Niclosamide
Niclosamide has antihelminthic activity specific to most tapeworms but not other worms (e.g., pinworms and roundworms), and has been approved for use by Food and Drug Administration (FDA) (30). Mechanistic studies have shown that it is able to inhibit the transcription and DNA binding of NF-kappaB, and block the tumor necrosis factor-induced signaling pathway. Interestingly, nicloamide also increases increases the levels of ROS in AML cells (especially in CD34+CD38– subpopulation), kills AML progenitor/stem cells and inhibits their AML repopulation capability in xenograft recipients. When used in combination with frontline chemotherapeutic regimens such as cytarabine, etoposide and daunorubicin, nicloamide has a synergistic efficacy. These combinations suggest multi-layer anti-leukemia effects, and deserve immediate studies to clarify chemo-sensitive potential and inhibition of AML repopulation in nude mice.
Celastrol and 4-hydroxy-2-nonenal (HNE)
Both agents can effectively eradicate AML at the bulk, progenitor, and stem cell level. They were first identified through an in silico screen method, called gene expression-based high-throughput screening (GE-HTS) and the Connectivity Map (29).The concept is based on gene expression signature reversal strategy for screening newly AML-stem cells targeting agents. In-depth experiments suggest that two agents share mechanisms including ROS induction and NF-κB inactivation. No clinical trials have been reported, and so there is no data for their safety and efficacy in AML treatment.
4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione (TDZD-8)
TDZD-8 is a non-ATP competitive inhibitor of GSK-3β. It is widely studied as a cytoprotective agent to treat septic and nonseptic shock, lung injury, arthritis, spinal cord injury, colitis and Alzheimer disease. Guzman et al. explores another functional facet of TDZD-8, namely selectively killing AML-stem/progenitor cells. Cellular and molecular studies indicate that its mode-of-action is correlated with rapid loss of membrane integrity, depletion of free thiols/rapid ROS generation and inhibition of both the PKC and FLT3 signaling pathways. It should be noted that TDZD-8 induces cell-death effects not only on AML, but also chronic myeloid leukemia (CML) and acute lymphoblastic leukemia (ALL) (25).
3-deazaneplanocin A (DZNep)
DZNep is a histone methyltransferase inhibitor that targets AML leukemia cells and shows certain capabilities on AML-stem/progenitor cells (33). It can disrupt polycomb-repressive complex 2 (PRC2), deplete enhancer of zeste homolog 2 (EZH2), reactivate TXNIP, inhibit thioredoxin activity, increase ROS, and then lead to AML cell apoptosis. A large body of knowledge has been accumulated for epigenetics-based treatment in AML. Compared to other epigenetic modulators, DZNep is unique in treating AML through a ROS-generation mechanism.
Mefloquine
Most recently, Sukhai et al. an antimalarial agent called ‘mefloquine’ for its capacity in targeting newly AML-stem cells (32). This study reveals a previously unappreciated mechanism for AML-stem cells killing. Mefloquine is able to disrupt lysosome integrity releasing hydrolases, lipases, proteases and cathepsins. This disruption subsequently increases the levels of ROS and triggers death of AML cells and stem cells in a caspase-independent manner. Mefloquine is widely used for malaria therapy and chemoprevention, its clinical safety has been already characterized in large cohorts of clinical trials. Its AML targeting efficacy will be next for evaluation.
Agents selectively induce AML-stem cells differentiation via ROS
Differentiation therapy achieves remarkable success in the acute promyelocytic leukemia (APL) therapy. Whether this therapeutic strategy can be extended to non-APL subtypes of AML, especially in targeting LSCs, still remains undefined. A recent study carried by Callens et al. suggests that inducing LSC differentiation by ROS-modulating agents is likely also be feasible (86,87). First, they find that iron deprivation therapy can induce generation of ROS in a time and dose-dependent manner. The iron deprivation is established using iron chelator deferasirox (DFX), deferoxamine (DFO), or transferrin receptor-specific antibody (e.g., TfR1 antibody and mAb A24). Second, leukemia cells (including primary AML cells from various subtypes) are promoted into mature monocytic differentiation. They also design paralleled experiments to reinforce the role of iron chelating treatments in ROS production, and to clarify the cause-effect between iron chelating treatments-induced ROS and monocytic differentiation of AML blast cells. Third, microarray analysis shows that iron chelating treatments-induced expression profiling is similar to that induced by Vitamin D3, a well-known inducer of HL60 cells monocytic differentiation. Last but not least, a refractory AML/MDS patient receiving oral DFX (1 g/d) and 25-hydroxycholecalciferol (4,000 IU/d) shows a decrease of AML blast cells and an increase of mature differentiated monocytes. Together, iron chelating—induced ROS generation and differentiation in AML, especially in refractory AML/MDS patients (likely due to the persistence of LSCs), opens up a possibility of using ROS-dependent LSC differentiation.
Concluding remarks and future challenges
The past 40 years have seen much progress in understanding biology and treatment of AML, as highlighted by, HSCs transplantation, sophiscated risk-stratification and great success of APL treatment. However, it seems to be hard for AML to be curable based on the current front-line therapies; they are limited by the inability in eradicating LSCs. Novel therapeutic regimens are required to target LSCs but not normal counterparts are required. The ROS-modulating agents are ideal because LSCs are much more susceptible to ROS than normal HSCs. In this review, we have given a relatively complete overview of agents that rely on ROS in part or in whole to potentially treat AML. Despite their promises, there are still many challenges that need to be addressed in the future:
From our current understanding, both pro-oxidant (inducing oxidative stress) and antioxidant (reducing oxidative) environments are beneficial to cancer therapy. In terms of AML (especially AML stem cells), pro-oxidants seem to be much preferred over antioxidants. However, we are still ignorant of how to fine-tune balance between pro-oxidant and antioxidant to achieve the maximum therapeutic proficiency.
We are aware of the fact that LSCs are more sensitive to ROS than normal HSCs, but do not know exactly why. There must be some built-in links between physiologic and pathophysiologic levels of ROS. It is critical for evaluating any ROS-generating agent and determining its optimal dosage in eradicating LSCs but sparing normal counterparts.
ROS has multiple endogenous sources, and identifying these sources is always useful for designing efficient ROS-targeting agents. It is unclear which species of ROS is specific to leukemic cells (especially AML-stem cells). This clarification will greatly improve the efficiency of ROS-targeting AML therapy.
It should be clear that ROS is not the only mechanism of targeting LSCs. It is integrated into functional networks. Unfortunately, how ROS cooperates with other functional pathways to specially eradicating LSCs is little known.
Increased levels of ROS can also promote cell proliferation, cell survival and drug resistance. It remains to test whether ROS-generation agents will induce secondary resistance.
The iron chelation therapy represents a new ROS-based strategy of inducing AML-stem cells differentiation. It is interesting to know whether this therapy is reproducible for sub-populations of AML stem cells.
Acknowledgements
Funding: This work was supported in part by Ministry of Science and Technology Grants of China (No. 2012AA02A211) and National Natural Science Foundation Grants of China (No. 81270625, No. 31301041, No. 81300401 and No. 31171257).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood 2005;106:1154-63. [DOI] [PubMed] [Google Scholar]
- 2.Howlader N, Noone AM, Krapcho M, et al. eds. SEER Cancer Statistics Review, 1975-2010, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2010/, based on November 2012 SEER data submission, posted to the SEER web site, April 2013.
- 3.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013;63:11-30. [DOI] [PubMed] [Google Scholar]
- 4.Huntly BJ, Gilliland DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer 2005;5:311-21. [DOI] [PubMed] [Google Scholar]
- 5.Wulf GG, Wang RY, Kuehnle I, et al. A leukemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood 2001;98:1166-73. [DOI] [PubMed] [Google Scholar]
- 6.Sikic BI. Multidrug resistance and stem cells in acute myeloid leukemia. Clin Cancer Res 2006;12:3231-2. [DOI] [PubMed] [Google Scholar]
- 7.Feuring-Buske M, Hogge DE. Hoechst 33342 efflux identifies a subpopulation of cytogenetically normal CD34(+)CD38(-) progenitor cells from patients with acute myeloid leukemia. Blood 2001;97:3882-9. [DOI] [PubMed] [Google Scholar]
- 8.de Figueiredo-Pontes LL, Pintao MC, Oliveira LC, et al. Determination of P-glycoprotein, MDR-related protein 1, breast cancer resistance protein, and lung-resistance protein expression in leukemic stem cells of acute myeloid leukemia. Cytometry B Clin Cytom 2008;74:163-8. [DOI] [PubMed] [Google Scholar]
- 9.Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000;14:1777-84. [DOI] [PubMed] [Google Scholar]
- 10.Jin L, Hope KJ, Zhai Q, et al. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med 2006;12:1167-74. [DOI] [PubMed] [Google Scholar]
- 11.Hosen N, Park CY, Tatsumi N, et al. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A 2007;104:11008-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Majeti R, Chao MP, Alizadeh AA, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009;138:286-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guzman ML, Neering SJ, Upchurch D, et al. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 2001;98:2301-7. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Krivtsov AV, Sinha AU, et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010;327:1650-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guzman ML, Upchurch D, Grimes B, et al. Expression of tumor-suppressor genes interferon regulatory factor 1 and death-associated protein kinase in primitive acute myelogenous leukemia cells. Blood 2001;97:2177-9. [DOI] [PubMed] [Google Scholar]
- 16.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 2009;8:579-91. [DOI] [PubMed] [Google Scholar]
- 17.Tothova Z, Gilliland DG. FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell 2007;1:140-52. [DOI] [PubMed] [Google Scholar]
- 18.Abdel-Wahab O, Levine RL. Metabolism and the leukemic stem cell. J Exp Med 2010;207:677-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hole PS, Darley RL, Tonks A. Do reactive oxygen species play a role in myeloid leukemias? Blood 2011;117:5816-26. [DOI] [PubMed] [Google Scholar]
- 20.Valko M, Leibfritz D, Moncol J, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007;39:44-84. [DOI] [PubMed] [Google Scholar]
- 21.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol 2003;15:247-54. [DOI] [PubMed] [Google Scholar]
- 22.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009;417:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farquhar MJ, Bowen DT. Oxidative stress and the myelodysplastic syndromes. Int J Hematol 2003;77:342-350. [DOI] [PubMed] [Google Scholar]
- 24.Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett 2008;270:1-9. [DOI] [PubMed] [Google Scholar]
- 25.Guzman ML, Li X, Corbett CA, et al. Rapid and selective death of leukemia stem and progenitor cells induced by the compound 4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione (TDZD-8). Blood 2007;110:4436-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guzman ML, Rossi RM, Karnischky L, et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 2005;105:4163-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guzman ML, Rossi RM, Neelakantan S, et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood 2007;110:4427-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guzman ML, Swiderski CF, Howard DS, et al. Preferential induction of apoptosis for primary human leukemic stem cells. Proc Natl Acad Sci U S A 2002;99:16220-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hassane DC, Guzman ML, Corbett C, et al. Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood 2008;111:5654-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin Y, Lu Z, Ding K, et al. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer Res 2010;70:2516-27. [DOI] [PubMed] [Google Scholar]
- 31.Lagadinou ED, Sach A, Callahan K, et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell stem cell 2013;12:329-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sukhai MA, Prabha S, Hurren R, et al. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. J Clin Invest 2013;123:315-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou J, Bi C, Cheong LL, et al. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood 2011;118:2830-9. [DOI] [PubMed] [Google Scholar]
- 34.Zhang H, Mi JQ, Fang H, et al. Preferential eradication of acute myelogenous leukemia stem cells by fenretinide. Proc Natl Acad Sci U S A 2013;110:5606-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goardon N, Marchi E, Atzberger A, et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer cell 2011;19:138-152. [DOI] [PubMed] [Google Scholar]
- 36.Brümmendorf TH, Balabanov S. Telomere length dynamics in normal hematopoiesis and in disease states characterized by increased stem cell turnover. Leukemia 2006;20:1706-16. [DOI] [PubMed] [Google Scholar]
- 37.Hiyama E, Hiyama K. Telomere and telomerase in stem cells. Br J Cancer 2007;96:1020-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003;423:255-60. [DOI] [PubMed] [Google Scholar]
- 39.Tam WF, Hähnel PS, Schüler A, et al. STAT5 is crucial to maintain leukemic stem cells in acute myelogenous leukemias induced by MOZ-TIF2. Cancer Res 2013;73:373-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lobry C, Ntziachristos P, Ndiaye-Lobry D, et al. Notch pathway activation targets AML-initiating cell homeostasis and differentiation. J Exp Med 2013;210:301-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009;458:776-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding Q, Gu R, Liang J, et al. PI-103 sensitizes acute myeloid leukemia stem cells to daunorubicin-induced cytotoxicity. Med Oncol 2013;30:395. [DOI] [PubMed] [Google Scholar]
- 43.Hong Z, Xiao M, Yang Y, et al. Arsenic disulfide synergizes with the phosphoinositide 3-kinase inhibitor PI-103 to eradicate acute myeloid leukemia stem cells by inducing differentiation. Carcinogenesis 2011;32:1550-8. [DOI] [PubMed] [Google Scholar]
- 44.Levis M, Murphy KM, Pham R, et al. Internal tandem duplications of the FLT3 gene are present in leukemia stem cells. Blood 2005;106:673-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao S, Konopleva M, Cabreira-Hansen M, et al. Inhibition of phosphatidylinositol 3-kinase dephosphorylates BAD and promotes apoptosis in myeloid leukemias. Leukemia 2004;18:267-75. [DOI] [PubMed] [Google Scholar]
- 46.He J, Nguyen AT, Zhang Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood 2011;117:3869-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blair A, Hogge DE, Sutherland HJ. Most acute myeloid leukemia progenitor cells with long-term proliferative ability in vitro and in vivo have the phenotype CD34(+)/CD71(-)/HLA-DR. Blood 1998;92:4325-35. [PubMed] [Google Scholar]
- 48.Chao MP, Alizadeh AA, Tang C, et al. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res 2011;71:1374-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kikushige Y, Akashi K. TIM-3 as a therapeutic target for malignant stem cells in acute myelogenous leukemia. Ann N Y Acad Sci 2012;1266:118-23. [DOI] [PubMed] [Google Scholar]
- 50.Nishioka C, Ikezoe T, Furihata M, et al. CD34+/CD38– acute myelogenous leukemia cells aberrantly express CD82 which regulates adhesion and survival of leukemia stem cells. Int J Cancer 2013;132:2006-19. [DOI] [PubMed] [Google Scholar]
- 51.Ochsenreither S, Majeti R, Schmitt T, et al. Cyclin-A1 represents a new immunogenic targetable antigen expressed in acute myeloid leukemia stem cells with characteristics of a cancer-testis antigen. Blood 2012;119:5492-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosinski KV, Fujii N, Mito JK, et al. DDX3Y encodes a class I MHC-restricted H-Y antigen that is expressed in leukemic stem cells. Blood 2008;111:4817-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sachlos E, Risueno RM, Laronde S, et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 2012;149:1284-97. [DOI] [PubMed] [Google Scholar]
- 54.Scolnik MP, Morilla R, de Bracco MM, et al. CD34 and CD117 are overexpressed in AML and may be valuable to detect minimal residual disease. Leuk Res 2002;26:615-9. [DOI] [PubMed] [Google Scholar]
- 55.van Rhenen A, van Dongen GA, Kelder A, et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007;110:2659-66. [DOI] [PubMed] [Google Scholar]
- 56.Walter RB, Appelbaum FR, Estey EH, et al. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012;119:6198-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245-313. [DOI] [PubMed] [Google Scholar]
- 58.Mehraein-Ghomi F, Basu HS, Church DR, et al. Androgen receptor requires JunD as a coactivator to switch on an oxidative stress generation pathway in prostate cancer cells. Cancer Res 2010;70:4560-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radic Biol Med 2002;33:774-97. [DOI] [PubMed] [Google Scholar]
- 60.Bernhardt R. Cytochrome P450. structure, function, and generation of reactive oxygen species. Rev Physiol Biochem Pharmacol 1996;127:137-221. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y, Du Y, Le W, et al. Redox control of the survival of healthy and diseased cells. Antioxid Redox Signal 2011;15:2867-908. [DOI] [PubMed] [Google Scholar]
- 62.Chen C, Liu Y, Liu R, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med 2008;205:2397-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007;110:3056-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ito K, Hirao A, Arai F, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004;431:997-1002. [DOI] [PubMed] [Google Scholar]
- 65.Ito K, Hirao A, Arai F, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 2006;12:446-51. [DOI] [PubMed] [Google Scholar]
- 66.Tothova Z, Kollipara R, Huntly BJ, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007;128:325-39. [DOI] [PubMed] [Google Scholar]
- 67.Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 2009;461:537-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hole PS, Pearn L, Tonks AJ, et al. Ras-induced reactive oxygen species promote growth factor-independent proliferation in human CD34+ hematopoietic progenitor cells. Blood 2010;115:1238-46. [DOI] [PubMed] [Google Scholar]
- 69.Yahata T, Takanashi T, Muguruma Y, et al. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood 2011;118:2941-50. [DOI] [PubMed] [Google Scholar]
- 70.Shi X, Zhang Y, Zheng J, Pan J: Reactive oxygen species in cancer stem cells. Antioxid Redox Signal 2012;16:1215-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009;458:780-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li XX, Dong Y, Wang W, et al. Emodin as an effective agent in targeting cancer stem-like side population cells of gallbladder carcinoma. Stem Cells Dev 2013;22:554-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ikeda J, Mamat S, Tian T, Wang Y, Luo W, Rahadiani N, Aozasa K, Morii E: Reactive oxygen species and aldehyde dehydrogenase activity in Hodgkin lymphoma cells. Lab Invest 2012;92:606-14. [DOI] [PubMed] [Google Scholar]
- 74.Achuthan S, Santhoshkumar TR, Prabhakar J, et al. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J Biol Chem 2011;286:37813-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haraguchi N, Ishii H, Mimori K, et al. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest 2010;120:3326-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462:739-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Corcoran A, Cotter TG. FLT3-driven redox-modulation of Ezrin regulates leukaemic cell migration. Free Radic Res 2013;47:20-34. [DOI] [PubMed] [Google Scholar]
- 79.Sallmyr A, Fan J, Datta K, et al. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood 2008;111:3173-82. [DOI] [PubMed] [Google Scholar]
- 80.Hogdal LJ, Letai A. BCL-2 Inhibition: Stemming the Tide of Myeloid Malignancies. Cell stem cell 2013;12:269-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schimmer AD, O’Brien S, Kantarjian H, et al. A phase I study of the pan bcl-2 family inhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clin Cancer Res 2008;14:8295-301. [DOI] [PubMed] [Google Scholar]
- 82.Puduvalli VK, Yung WK, Hess KR, et al. Phase II study of fenretinide (NSC 374551) in adults with recurrent malignant gliomas: A North American Brain Tumor Consortium study. J Clin Oncol 2004;22:4282-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sabichi AL, Lerner SP, Atkinson EN, et al. Phase III prevention trial of fenretinide in patients with resected non-muscle-invasive bladder cancer. Clin Cancer Res 2008;14:224-229. [DOI] [PubMed] [Google Scholar]
- 84.Schneider BJ, Worden FP, Gadgeel SM, et al. Phase II trial of fenretinide (NSC 374551) in patients with recurrent small cell lung cancer. Invest New Drugs 2009;27:571-8. [DOI] [PubMed] [Google Scholar]
- 85.Veronesi U, Mariani L, Decensi A, et al. Fifteen-year results of a randomized phase III trial of fenretinide to prevent second breast cancer. Ann Oncol 2006;17:1065-71. [DOI] [PubMed] [Google Scholar]
- 86.Callens C, Coulon S, Naudin J, et al. Targeting iron homeostasis induces cellular differentiation and synergizes with differentiating agents in acute myeloid leukemia. J Exp Med 2010;207:731-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chaston TB, Watts RN, Yuan J, et al. Potent antitumor activity of novel iron chelators derived from di-2-pyridylketone isonicotinoyl hydrazone involves fenton-derived free radical generation. Clin Cancer Res 2004;10:7365-74. [DOI] [PubMed] [Google Scholar]