

Abstract
Background
Hepatitis B virus (HBV) is accounting for over one million deaths annually due to immune-mediated chronic liver damage. Natural killer (NK) cells are abundant in the liver and contribute in HBV persistence. NK cytotoxic effects are controlled by signals from activating and inhibitory receptors. HBV may circumvent host antiviral immunity via the regulation of NK receptors and their ligands. We investigated the effect of viral replication and HBeAg mutations on NK mediators expression in the livers of chronic HBV (CHB) patients and in cell cultures.
Methods
HBV monomers bearing hotspot mutations in the basal core promoter and precore region were transfected into HepG2 cells using a plasmid-free assay. Serum viremia and liver HBV RNA were measured in 19 CHB patients. The expression of HBV RNA and of NKG2D ligands, B7H6, DNAX accessory molecule-1, lectin-like transcript 1 (LLT1), LFA-1 and TRAIL was measured in the livers of CHB patients and transfected cells.
Results
In general, high HBV replication in CHB patients and cell lines upregulated the mRNA of all NK cell ligands and particularly the inhibitory NK cell ligand, LLT1. The exception was the NKG2D ligand, MICA, that was significantly decreased in patients with high serum viremia and intrahepatic HBV RNA levels.
Conclusions
HBV replication has differential effects on NK cell ligands suggesting a potential escape mechanisms through up-regulation of LLT1 and down-regulation of MICA. A general trend towards upregulating NK cell ligands can be counteracted by decreasing MICA and hence weakening NK surveillance.
Keywords: Hepatitis B virus, NK cells, virus, LLT1, NKG2D, mutations, precore, viral replication
Introduction
Hepatitis B virus (HBV) is a hepatotropic DNA virus infecting humans for more than 1500 years [1]. To date, HBV is a leading cause of liver diseases worldwide, with more than 350 million people being persistently infected and at high risk developing liver failure, cirrhosis or hepatocellular carcinoma (HCC) [2,3]. An increased proportion of chronic disease is due to the emergence of viral variants that either abrogate or reduce HBeAg production [4]. One of the most critical mutations is the double A1762T/G1764A substitution in the basal core promoter (BCP) region and its appearance is usually followed by the development of the triple mutant A1762T/G1764A/C1766T [5]. The most common mutation that results in an HBeAg-negative phenotype is the G1896A variant of the precore region and is usually accompanied by a second G1899A mutation [6]. Variants with these mutations associate with more severe forms of liver damage and the development of HCC [7,8]. Studies on the effect of BCP mutations on viral replication remain controversial. Among other studies we have shown that they induce low viral transcription and replication (unpublished data) [9-11]. Precore mutated variants associate with high viral replication or have no effect on viral load (unpublished data) [11-13].
The outcome of HBV infection depends on the intensity and type of anti-viral immunity produced by the host. In acute infection the adaptive immune system mounts a strong, multi-specific response, but its effectiveness depends on the quality of the earlier innate immune response. NK cells are important effectors of the innate antiviral immunity. In addition to the direct killing of viral-infected cells without antigen-specific priming, they regulate the adaptive immune response by producing interferon (IFN)-γ, tumor necrosis factor (TNF)-α and immunoregulating cytokines. NK cells are enriched markedly in the liver, the site of HBV replication, implying that HBV has to evade NK cell-mediated immune responses to establish a persistent infection. In addition the high NK activity early in infection and during the incubation time suggests that they are key plays in viral clearance [14-17]. NK cells, however, can also negatively regulate specific antiviral immunity in CHB infection by contributing in the liver inflammation through TNF-related apoptosis-inducing ligand (TRAIL)- and Fas-mediated death [17,18] and by the direct killing of HBV-specific CD8+ T cells, which triggers the recruitment of inflammatory cells that amplify the hepatic damage [19,20].
The effector functions of NK cells are tightly regulated by the dynamic and coordinated balance of activating and inhibitory signals. In the present study, the NK cell receptors were selected on the basis of being known to control key cell functions. The main NK cell activating receptors studied in this respect include C-type lectin-like NK group 2D (NKG2D) receptor and immunoglobulin-like receptor NKp30. NKG2D is a potent activating receptor expressed in all NK cells, NKT cells and activated CD8+ T lymphocytes [21,22]. It binds to a family of ligands (NKG2DL) including the major histocompatibility complex class-I polypeptide-related chain (MIC) protein family that contains MICA and MICB; and the cytomegalovirus UL16-binding proteins (ULBP) family, which consists of five members, ULBP1-4 and RAET1G [23]. Another activating receptor involved in the direct regulation of NK cell-mediated recognition of cellular stress is the DNAX accessory molecule-1 (DNAM-1, CD226), which is constitutively expressed on NK cells, T cells, macrophages and a small subset of B cells [24]. Similar to NKG2D, DNAM-1 not only promotes adhesion and activation of NK cells and CTLs but also greatly enhances their cytotoxicity toward ligand-expressing targets [24]. Furthermore, lectin-like transcript 1 (LLT1, gene clec2d) is a type II transmembrane receptor that belongs to the C-type lectin like (CTL) superfamily of NK cell receptors. Six alternatively spliced transcripts of clec2d exist, with the isoform 1 (designed as LLT1) being the only one presented on the cell surface and hence the only isoform able to participate in the cell-to-cell transmission [25]. LLT1 is expressed mainly on activated lymphocytes, including NK, T cells and B cells, as well as on antigen presenting cells (APC) [26]. However, a decade ago, LLT1 was also identified as a physiological ligand of the NKR-P1 (CD161), expressed on all NK cells and subsets of T cells and represents the only member of the human NKR-P1 subfamily [27,28]. NK cell cytotoxicity and IFN-γ production is inhibited upon engagement of NKR-P1 on NK cell with LLT1 on target cell [27,28]. Upon viral infection, LLT1 expression is upregulated on surface of epithelial cells, while IFN-γ has been shown to increase LLT1 expression on B cells and APC [29]. Hence, LLT1-mediated signaling is thought to represent a system that regulates both innate and adaptive responses. For activated NK cells to achieve successful lysis, strong adhesion to target cells that is required is mediated by the β2 integrin LFA1 [30].
The objective of this study was to investigate the modulation of ligands for cell surface NK receptors in chronic HBV (CHB) patients and HepG2 cells transfected with BCP and precore mutants. Furthermore, we aimed to correlate patient’s viral replication and the differential viral capacity of hotspot mutants with the expression of NK receptors and their ligands at the RNA level.
Patients and methods
Patients
Nineteen patients (11 men and 8 women; mean age 44.5±12 years) with CHB were studied (Table 1). Tissue samples were obtained by percutaneous needle biopsy, snap-frozen and stored in liquid nitrogen. From all the individuals included in the study a serum sample was collected at the time of liver biopsy, stored at -80°C and HBV DNA levels were measured using real-time polymerase chain reaction (qPCR). Individuals with a history of excess alcohol, or positive for HCV, HDV or HIV were excluded. Informed consent was obtained from all the patients (The department of Hepatology St Mary’s Liver Tissue Bank, 06/Q0509/39). 1mg of liver tissue was processed using the gentleMACS Dissociator (Miltenyi Biotec SAS, Paris, France) prior to RNA extraction.
Table 1.
Clinical characteristics of the studied patients. Cirrhosis was measured with according to the Ishak scoring system and necro-inflammation with the Knodel system. Expression of liver HBV RNA was normalized with h-GAPDH and data represents the mean±SEM from 3 RT PCR experiments. Patients were divided into four groups according to their serum viremia and intrahepatic HBV RNA levels

Transient transfection experiments
The G1896A, G1899A, G1896A/G1899A, A1762T/G1764A and A1762T/G1764A/C1766T mutations were generated by splice extension mutagenesis using as a template a plasmid (p3.8II) containing a 1.2 genome length of HBV subtype adr (genotype C) [31] in a pBluescript II KS (+) background (Stratagene, CA, USA) (Table 2). Full-length HBV genomes were amplified from wild-type (WT) or mutant HBV genomes according to the method described by Günther, using the P1 sense (HBV positions: nt1821-1841) and P2 antisense primers (HBV positions: nt1823-1806) modified to contain the HindIII/SapI sites and the SacI/SapI sites, respectively (Table 2) [32]. PCRs were performed using the FastStart High Fidelity PCR System (Roche Applied Science, Germany) according to the manufacturer’s instructions. Each of the 3.2kb amplified HBV fragments were purified from agarose gels using the GeneJET Gel Extraction Kit (Thermo Scientific, Life Science Research, Lutterworth, UK) and were cloned into a pCRII TA-vector (Invitrogen, Paisley, UK). All constructs were sequenced to confirm the presence of the mutations and to ensure that no additional mutations were introduced in the process.
Table 2.
Primers used full-length HBV genome generation by splice extension mutagenesis

Linear HBV monomers were released by SapI restriction digestion (New England Biolabs, Beverly, MA), gel purified, and then used for transient transfection of HepG2 by lipofection. HepG2 cells were maintained in Dulbecco’s minimal essential medium supplemented with 10% fetal bovine serum, 2 mM L-Glutamine, 100 IU penicillin mL-1 and 100 μg streptomycin mL-1. Cells were seeded at a density of 4x105 cells in 60 mm diameter dishes (Corning, Flintshire, UK) and the following day were transfected with 500 ng HBV linear monomers using Lipofectamine Plus (Invitrogen) according to manufacturer’s instructions. The culture medium was changed 24 h after transfection, and cells were harvested at 48 and 72 h.
All transfections included 1 μg of reporter plasmid expressing a green fluorescent protein to monitor transfection efficiency by fluorescence-activated cell sorting analysis and ranged 43-50% while the empty pUC19 vector used as a negative control.
RNA quantification
Northern blot analysis
For Northern blot analysis, 10 μg of total RNA per sample was separated on a 1% formaldehyde-agarose gel and blotted onto a Zeta-Probe GT membrane (BioRad, Bio-Rad Laboratories, Hercules, CA). Radioactive probes were prepared by random priming, using either full-length HBV DNA or 18S cDNA template and 32P labeled αdCTP (Amersham). After hybridization the membrane was washed and exposed to X-Omat film (Kodak, Rochester, NY) at -80°C.
Relative quantification by real-time (RT) PCR
Total RNA was extracted from HepG2 cells 48 h after transfection with SapI -digested HBV DNA monomers and liver biopsies using Trizol reagent (Invitrogen) as recommended by the manufacturer. The RNA samples were treated with DNase (Qiagen, Crawley, UK) for 10 min at 25°C and were then purified using the RNeasy mini kit (Qiagen). RNA integrity was confirmed by agarose gel electrophoresis under UV, RNA concentrations were monitored by using the nanodrop and samples were stored at -80°C until further use.
Two μg RNA from each transfection and 500 ng RNA from each liver biopsy were reversed transcribed in 20 μL reactions using the RT2 First Strand kit (Qiagen). There were three biological replicates in each group for each transfection experiment and duplicates for each liver biopsy samples. The resulting cDNA samples were stored at -80°C until further use.
The relative levels of total HBV RNA transcripts concentrations were analyzed by real-time PCR with a TaqMan (Life technologies Paisley, UK) in a Light-Cycle (Roche Diagnostic, West Sussex, UK). Each PCR reaction was performed in a 75 μL reaction volume containing 5 μL cDNA, 0.5 mmol/L forward and reverse primers, 0.2 mmol/L 3’ FL-labeled probe, and 0.4mmol/L LC-labeled probe. The forward and reverse primers used were 5’-CTCGTGGTGGACTTCTCTC-3’and 5’-cagcaggatgaagaggaa-3’, and the probes were 5’-LC640-TGTCCTGGTTATCGCTGGATGTGTCT-PH-3’ and 5’-CAC TCACCAACCTCCTGTCCTCCAA-FL-3’. The h-GAPDH housekeeping gene Light Cycler Set (Roche DNA control kit, Roche Diagnostics) was used to normalize RNA expression.
For the detection of MICA, ULBP2, ULBP3, B7H6, CD226 (DNAM-1), CLEC2D (LLT1), ITGAL (LFA-1) and TNFSF10 (TRAIL) we performed RT PCR arrays with customized RT2 custom arrays containing pre-dispensed primer assays on a StepOne Plus RT PCR (AB Applied Biosystems, Thermo Scientific, Loughborough, UK) using RT2 SYBR Green/qPCR Master Mix (Qiagen). The custom-made array included two housekeeping genes (GAPDH and β-Actin) and three internal controls (human genomic DNA contamination control, reverse transcription control and positive PCR control). Each PCR reaction contained cDNA synthesized from 125 ng of total RNA. The thermocycler parameters were 95°C for 10 min, followed by 45 cycles of 95°C for 15 sec and 60°C for 1 min.
Relative changes in gene expression for both viral and cell RNA transcripts were calculated using ΔΔCt (threshold cycle) method. Threshold cycle numbers (Ct)2 above 35 were considered below detection level. The β-ACTIN housekeeping gene was used to normalize the RNA amounts. Results are expressed relative to β-ACTIN with NK markers as 2-ΔΔCT, where ΔΔCt = ΔCT-sample - ΔΔCT-control and where ΔCT = CT-target gene - ΔCT-ACT.
Statistical analysis
Two group comparisons of continuous variables were performed using the non-parametric Mann-Whitney test with two tailed values (GraphPrism 6). P values below 0.05 were considered statistical significant.
Results
Liver and serum viremia in CHB patients
We determined the expression of total HBV RNA in the liver of infected patients by RT PCR and compared this to the levels of serum viremia. It is well known that intrahepatic viremia does not correlate with serum viremia since the liver sampled cannot be representative. Indeed the levels of intrahepatic HBV RNA did not correlate with serum HBV viremia (r=0.34, P>0.1) (Fig. 1). We therefore divided the patients into four groups: 1) Low Serum Viremia patients, included those with serum viremia 1x101 – 1x104 viral copies/mL; 2) High Serum Viremia group, included patients with serum viremia 1x106 – 1x1010 viral copies/mL; 3) Low Liver HBV RNA patients, included those liver samples with 1x101 – 1x103 HBV RNA levels relative to β-ACTIN expression; and 4) High Liver HBV RNA samples with 1x104 – 1x105 HBV RNA levels (Table 1). Twelve of 19 patients were HBeAg-negative and HBeAb-positive; all patients had minimal inflammation levels; 1/19 patient was cirrhotic and 8/19 patients had elevated alanine aminotransferase (ALT) levels.
Figure 1.

Serum hepatitis B virus (HBV) DNA levels by qPCR and the relative expression of intrahepatic HBV RNA relative expression. Each dot represents a patient and data for HBV RNA relative expression is shown as the mean ± standard error of mean from 2 real-time polymerase chain reaction experiments
Expression of NK regulatory molecules
CHB patients. We examined the expression of the key NK cells ligands MICA, ULBP2, ULPB3, B7H6, the NK receptors DNAM-1, LFA-1, TRAIL and LLT1 in the livers of CHB patients (Fig. 2). In general, individuals with higher liver viral loads had higher expression of NK cell ligands and receptors. The exception to this was MICA, which was significantly downregulated in patients with high intrahepatic HBV RNA levels (P=0.02) and decreased in high viremic patients (P=0.05). We also compared the gene expression in 7 HBeAg(+) patients and 12 HBeAg(-) patients (Fig. 3). The expression of ULBP2, ULBP3, LLT1, LFA-1, and DNAM-1 was upregulated in HBeAg(+) patients, the majority of which had increased serum and liver HBV levels, while MICA was downregulated in this group of patients. Interestingly LLT1 is a ligand for the inhibitory receptor CD161, expressed on NK cells and intrahepatic T cells. Thus, this together with the down-regulation of MICA, may represent potential escape mechanisms for HBV, counteracting the trends observed for the other NKG2D ligands and also B7H6 the ligand for NKp30. Consistent with the lack of correlation between serum and liver HBV viremia, we observed no consistent trends of NK cell ligand expression and serum viremia.
Figure 2.
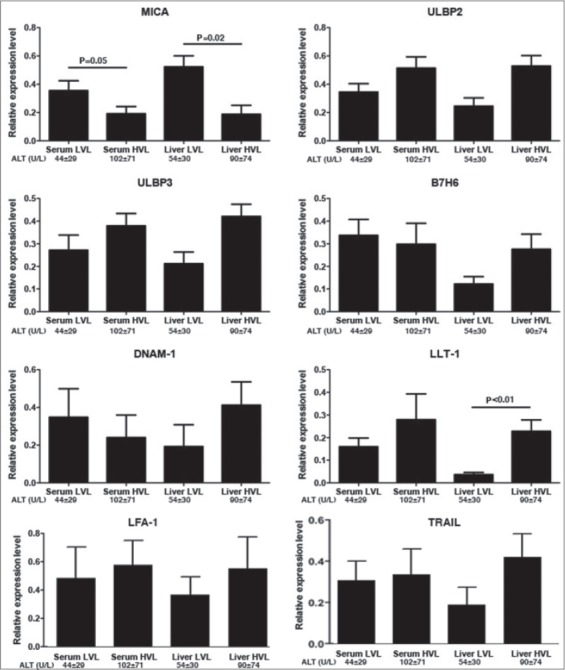
RNA levels of natural killer regulators in liver biopsies from chronic hepatitis B patients. Gene expression was normalized with β-ACTIN levels and data represents the mean ± standard error of mean (SEM) from 3 independent experiments of the relative gene expression between patients. The ALT levels are shown as the mean ± SEM LVL, low viral load; HVL, high viral load; ALT, alanine aminotransferases
Figure 3.

The RNA expression of natural killer regulators in liver biopsies from 7 HBeAg(-) and 12 HBeAg(+) chronic hepatitis B patients. Gene expression was normalized with β-ACTIN levels and data represents the mean ± standard error of mean (SEM) from 3 independent experiments of the relative gene expression between patients. The ALT levels are shown as the mean ± SEM LVL, low viral load; HVL, high viral load; HBeAg, HBV e antigen; HBeAb; ALT, alanine aminotransferases
HBV transfection experiments. We had previously constructed using site directed mutagenesis, the replication-competent plasmids bearing the BCP mutations 1762/64 and 1762/64/66 and the precore 1896, 1899 and 1896/1899 (unpublished data) (Table 2). Using the same HBV constructs and plasmid-free transfection system, we have previously demonstrated that compared to the WT and precore variants, BCP mutations result in lower viral replication capacity while the precore variants show similar replication fitness to the WT. BCP mutants consistently produced the decreased accumulation of all HBV RNA transcripts (Northern blotting) (Fig. 4); the viral replicative intermediates, including cccDNA (Southern blotting); and the culture HB surface antigen (HBsAg) levels (ELISA) (unpublished data).
Figure 4.
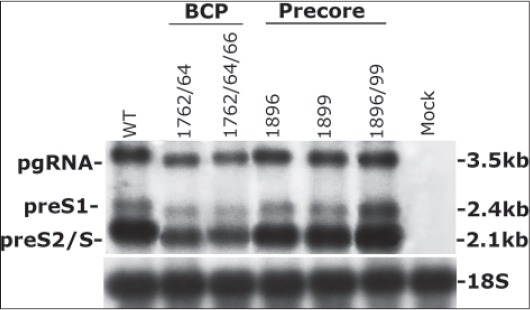
Northern blot analysis of hepatitis B virus (HBV) transcripts. 10 μg RNA were extracted from HepG2 cells after 48 h of transfection with 500 ng linear HBV DNA monomers carrying BCP or precore mutations pgRNA, pregenomic HBV mRNA; preS1 and preS2/S, pre-surface antigen S1 and S2/S mRNAs; BCP, basal core promoter
To investigate whether the differential replication of HBV induces changes in the expression of ligands for NK cell receptors on transfected HepG2 we performed RT qPCR (Fig. 5). Transfection of HepG2 cells induced the up-regulation of all NK cell ligands tested. MICA, ULBP3 and B7H6 transcript levels were not significantly altered after exposure to the WT and HBeAg mutant variants examined (Fig. 5). However we observed a weak trend towards an association of ULBP2 with viremia in that there were reduced levels of ULBP2 mRNA with the BCP mutants compared to WT and increased ULBP2 levels expression with the pre-core variants. However, the expression of LLT-1 in HepG2 transfected with the WT genome was significantly decreased compared to 1762/74 (P=0.01) and 1762/64/66 (P=0.04). Thus, LLT-1 correlated with the BCP mutations and their low replication efficiency. This is consistent with our findings on intrahepatic LLT1 RNA levels.
Figure 5.
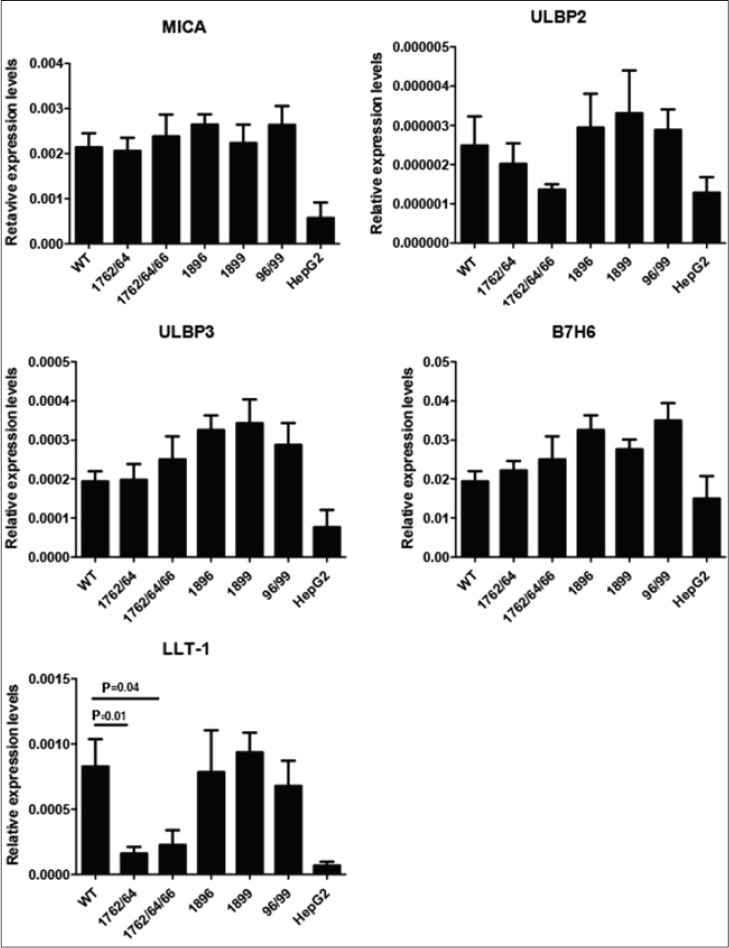
RNA expression of MICA, ULBP2, ULBP3 and B7H7 was assessed at 48 h post transfection by real-time polymerase chain reaction normalized to GAPDH or β-ACTIN expression. WT refers to wild-type virus, 1762/64 and 1762/64/66 are BCP variants and 1896, 1899 and 1896/99 are precore variants. Data is presented from 3 independent transfection experiments of HepG2 cells and values are expressed as mean ± standard error of mean
Discussion
Accumulating evidence suggest that activation of the NKG2D-ligand pathway contributes to the outcome of HBV infection. The unpredictable natural history of CHB infection makes it difficult to sample the immune correlates of viral replication activity longitudinally. The level of HBV DNA in serum is commonly used as a surrogate marker of HBV replication in the clinical setting but it does not reflect the levels of HBV in the liver [33]. To monitor HBV levels, in addition to serum viremia we assessed HBV RNA expression in the same liver sample that we determined NK ligand RNA expression. We found that MICA transcripts were decreased in highly viremic liver samples of CHB patients whereas there was a trend towards increased expression of ULBP2 and ULBP3 with HBV liver viremia. Thus, a general trend towards upregulating expression of NK cell ligands, could be counteracted by decreasing expression of MICA and hence weakening NK surveillance of the infected cells. These patients had minimal inflammation on their liver biopsies and therefore downregulation of MICA could be both harmful by inhibiting NK-induced viral clearance but also protective by preventing the infected hepatocytes from becoming targets for NK cell-mediated killing and hence preventing the development of liver injury. The NK markers B7H6, DNAM-1, LFA-1 and TRAIL were also slightly elevated in high viremic liver samples. Our results indicate that high HBV replication induces the upregulation of gene expression of these NK ligands. A recent study by Lunemann showed lower levels of NKG2A, DNAM-1 and TRAIL on the NK cells of CHB patients as compared to acute patients [34]. It has been previously shown that higher expression of NKG2D ligands on cancer cells is associated with down-regulation of NKG2D on NK cells related to soluble NKG2D ligand secretion [35,36]. We therefore propose that a similar process occurs in CHB.
In the course of CHB infection, HBeAg-negativity concurs with the emergence of the precore mutation 1896 and 1899 while the BCP mutation 1762/64 and 1762/64/66 reduces HBeAg production. We have previously showed that the BCP mutations result in lower viral replication capacity, whereas the major precore mutations have no effect on viral replication. The expression of all NKG2DL was increased when HepG2 cells were transfected by HBV suggesting that HBV induces NKG2DL upregulation. This finding is consistent with our observation that NK cell ligands, with the exception of MICA, are increased in HBV infection. However, consistent trends for the ligands for activating receptors were not observed between in vivo and in vitro assays nor between HBeAg(+) and HBeAg(-) patients, with the possible exception of ULBP2. In general, NKG2D is a system thought to be important for clearing viral infection, and especially in cytomegalovirus infection, in which the virus has developed multiple mechanisms to down-regulate different NKG2D ligands [37]. Consistent with previous studies, we found that MICA expression was reduced in the liver of CHB patients [38,39]. Transfection of HepG2 cells with all HBV constructs induced its expression suggesting that early in infection MICA is upregulated but in the persistent infection HBV weakens the immune surveillance of NK cells by inhibiting MICA expression.
Conversely, expression of LLT1 was increased in patients with high serum and intrahepatic viremia. Likewise, in HepG2 transfections we found low but detectable levels of LLT1 that were positively correlated with the differential viral replication of the HBeAg mutated variants. Upregulation of LLT1 and its subsequent engagement to their CD161 receptor expressed on NK cells have been shown to inhibit NK-mediated cytotoxity and IFN-γ production by T cells [27]. LLT1 expression has been reported to be absent in normal liver tissue but upregulated in HCC. Here we show for the first time that LLT1 is expressed in HBV infected liver and its levels associate with viral replication in HBV infection. Thus, HBV may circumvent the immunity of the host by inhibiting NK cytotoxicity and IFN-γ production via the upregulation of LLT1.
A detailed characterization of the molecular players that link the HBV stimuli to the regulation of NK cells will be critical to advance our knowledge on how HBV circumvents the host’s immunity. The manipulation of ligand expression can provide new insight on CHB immunopathogenesis and can lead to possibilities of developing effective treatment strategies.
Summary Box.
What is already known:
Natural killer (NK) cells play a key role in the outcome of hepatitis B virus (HBV) infection
HBV can circumvent host’s immunity by modulating the expression of NK receptors and their ligands
MICA is ligand of the activating NKG2D receptor and is important in clearing viral infection
LLT1 is expressed on NK cells and is a ligand of the NKR-P1 receptor of NK cells
What the new findings are:
The gene expression of ULBP2, ULPB3, B7H6, DNAM-1, LFA-1 and TRAIL was elevated in high HBV replication in vivo
HBV infection repressed MICA expression in the liver of chronic HBV patients whereas HBV transfection induced its expression, suggesting that early in infection MICA is upregulated
LLT1 expression was increased in patients with high serum and intrahepatic viremia
In HepG2 transfections LLT1 levels were positively correlated with the differential replication of HBeAg mutated variants
A general trend towards upregulating expression of NK ligands can be counteracted by decreasing expression of MICA and hence weakening NK surveillance of the infected cells
Acknowledgments
This study was supported by grants from the European Association of the study of Liver (EASL).
Biography
Imperial College, St. Mary’s Campus, London, UK; University Hospital of Messina, Messina, Italy; University of Nicosia Medical School, Nicosia, Cyprus; Henry Welcome Laboratories, Southampton General Hospital, Southampton, UK
Footnotes
Conflict of Interest: None
References
- 1.Zhou Y, Holmes EC. Bayesian estimates of the evolutionary rate and age of hepatitis B virus. J Mol Evol. 2007;65:197–205. doi: 10.1007/s00239-007-0054-1. [DOI] [PubMed] [Google Scholar]
- 2.Lupberger J, Hildt E. Hepatitis B virus-induced oncogenesis. World J Gastroenterol. 2007;13:74–81. doi: 10.3748/wjg.v13.i1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cassidy A, Mossman S, Olivieri A, De Ridder M, Leroux-Roels G. Hepatitis B vaccine effectiveness in the face of global HBV genotype diversity. Expert Rev Vaccines. 2011;10:1709–1715. doi: 10.1586/erv.11.151. [DOI] [PubMed] [Google Scholar]
- 4.Huang YH, Wu JC, Chang TT, et al. Association of core promoter/precore mutations and viral load in e antigen-negative chronic hepatitis B patients. J Viral Hepat. 2006;13:336–342. doi: 10.1111/j.1365-2893.2005.00688.x. [DOI] [PubMed] [Google Scholar]
- 5.Okamoto H, Tsuda F, Akahane Y, et al. Hepatitis B virus with mutations in the core promoter for an e antigen-negative phenotype in carriers with antibody to e antigen. J Virol. 1994;68:8102–8110. doi: 10.1128/jvi.68.12.8102-8110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carman WF, Jacyna MR, Hadziyannis S, et al. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet. 1989;2:588–591. doi: 10.1016/s0140-6736(89)90713-7. [DOI] [PubMed] [Google Scholar]
- 7.Baptista M, Kramvis A, Kew MC. High prevalence of 1762(T) 1764(A) mutations in the basic core promoter of hepatitis B virus isolated from black Africans with hepatocellular carcinoma compared with asymptomatic carriers. Hepatology. 1999;29:946–953. doi: 10.1002/hep.510290336. [DOI] [PubMed] [Google Scholar]
- 8.Li W, Chen G, Yu X, Shi Y, Peng M, Wei J. Accumulation of the mutations in basal core promoter of hepatitis B virus subgenotype C1 increase the risk of hepatocellular carcinoma in Southern China. Int J Clin Exp Pathol. 2013;6:1076–1085. [PMC free article] [PubMed] [Google Scholar]
- 9.Shi M, Zhang Y, Zhang J, Liu W, Xing L. Hepatitis B virus genotypes, precore mutations, and basal core promoter mutations in HBV-infected Chinese patients with persistently normal alanine aminotransferase and low serum HBV-DNA levels. Braz J Infect Dis. 2012;16:52–56. doi: 10.1016/s1413-8670(12)70274-x. [DOI] [PubMed] [Google Scholar]
- 10.Fang ZL, Sabin CA, Dong BQ, et al. The association of HBV core promoter double mutations (A1762T and G1764A) with viral load differs between HBeAg positive and anti-HBe positive individuals:a longitudinal analysis. J Hepatol. 2009;50:273–280. doi: 10.1016/j.jhep.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jammeh S, Tavner F, Watson R, Thomas HC, Karayiannis P. Effect of basal core promoter and pre-core mutations on hepatitis B virus replication. J Gen Virol. 2008;89:901–909. doi: 10.1099/vir.0.83468-0. [DOI] [PubMed] [Google Scholar]
- 12.Gunther S, Sommer G, Von Breunig F, et al. Amplification of full-length hepatitis B virus genomes from samples from patients with low levels of viremia:frequency and functional consequences of PCR-introduced mutations. J Clin Microbiol. 1998;36:531–538. doi: 10.1128/jcm.36.2.531-538.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parekh S, Zoulim F, Ahn SH, et al. Genome replication, virion secretion, and e antigen expression of naturally occurring hepatitis B virus core promoter mutants. J Virol. 2003;77:6601–6612. doi: 10.1128/JVI.77.12.6601-6612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Wang JJ, Gao S, et al. Decreased peripheral natural killer cells activity in the immune activated stage of chronic hepatitis B. PLoS One. 2014;9:e86927. doi: 10.1371/journal.pone.0086927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oliviero B, Varchetta S, Paudice E, et al. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology. 2009;137:1151.e1151–1160.e1157. doi: 10.1053/j.gastro.2009.05.047. [DOI] [PubMed] [Google Scholar]
- 16.Zhao J, Li Y, Jin L, et al. Natural killer cells are characterized by the concomitantly increased interferon-gamma and cytotoxicity in acute resolved hepatitis B patients. PLoS One. 2012;7:e49135. doi: 10.1371/journal.pone.0049135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zou Z, Xu D, Li B, et al. Compartmentalization and its implication for peripheral immunologically-competent cells to the liver in patients with HBV-related acute-on-chronic liver failure. Hepatol Res. 2009;39:1198–1207. doi: 10.1111/j.1872-034X.2009.00571.x. [DOI] [PubMed] [Google Scholar]
- 18.Dunn C, Brunetto M, Reynolds G, et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J Exp Med. 2007;204:667–680. doi: 10.1084/jem.20061287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bertoletti A, Maini MK. Protection or damage:a dual role for the virus-specific cytotoxic T lymphocyte response in hepatitis B and C infection? Curr Opin Microbiol. 2000;3:387–392. doi: 10.1016/s1369-5274(00)00109-0. [DOI] [PubMed] [Google Scholar]
- 20.Peppa D, Gill US, Reynolds G, et al. Up-regulation of a death receptor renders antiviral T cells susceptible to NK cell-mediated deletion. J Exp Med. 2013;210:99–114. doi: 10.1084/jem.20121172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2003;100:9452–9457. doi: 10.1073/pnas.1632807100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogasawara K, Hamerman JA, Ehrlich LR, et al. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity. 2004;20:757–767. doi: 10.1016/j.immuni.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Stern-Ginossar N, Mandelboim O. An integrated view of the regulation of NKG2D ligands. Immunology. 2009;128:1–6. doi: 10.1111/j.1365-2567.2009.03147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuchs A, Colonna M. The role of NK cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Semin Cancer Biol. 2006;16:359–366. doi: 10.1016/j.semcancer.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Germain C, Bihl F, Zahn S, et al. Characterization of alternatively spliced transcript variants of CLEC2D gene. J Biol Chem. 2010;285:36207–36215. doi: 10.1074/jbc.M110.179622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Germain C, Meier A, Jensen T, et al. Induction of lectin-like transcript 1 (LLT1) protein cell surface expression by pathogens and interferon-gamma contributes to modulate immune responses. J Biol Chem. 2011;286:37964–37975. doi: 10.1074/jbc.M111.285312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aldemir H, Prod’homme V, Dumaurier MJ, et al. Cutting edge:lectin-like transcript 1 is a ligand for the CD161 receptor. J Immunol. 2005;175:7791–7795. doi: 10.4049/jimmunol.175.12.7791. [DOI] [PubMed] [Google Scholar]
- 28.Rosen DB, Bettadapura J, Alsharifi M, Mathew PA, Warren HS, Lanier LL. Cutting edge:lectin-like transcript-1 is a ligand for the inhibitory human NKR-P1A receptor. J Immunol. 2005;175:7796–7799. doi: 10.4049/jimmunol.175.12.7796. [DOI] [PubMed] [Google Scholar]
- 29.Satkunanathan S, Kumar N, Bajorek M, Purbhoo MA, Culley FJ. Respiratory syncytial virus infection, TLR3 ligands, and proinflammatory cytokines induce CD161 ligand LLT1 expression on the respiratory epithelium. J Virol. 2014;88:2366–2373. doi: 10.1128/JVI.02789-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barber DF, Faure M, Long EO. LFA-1 contributes an early signal for NK cell cytotoxicity. J Immunol. 2004;173:3653–3659. doi: 10.4049/jimmunol.173.6.3653. [DOI] [PubMed] [Google Scholar]
- 31.Fu L, Cheng YC. Role of additional mutations outside the YMDD motif of hepatitis B virus polymerase in L(-)SddC (3TC) resistance. Biochem Pharmacol. 1998;55:1567–1572. doi: 10.1016/s0006-2952(98)00050-1. [DOI] [PubMed] [Google Scholar]
- 32.Gunther S, Li BC, Miska S, Kruger DH, Meisel H, Will H. A novel method for efficient amplification of whole hepatitis B virus genomes permits rapid functional analysis and reveals deletion mutants in immunosuppressed patients. J Virol. 1995;69:5437–5444. doi: 10.1128/jvi.69.9.5437-5444.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Werle-Lapostolle B, Bowden S, Locarnini S, et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126:1750–1758. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 34.Lunemann S, Malone FG, Hengst J, et al. Compromised function of natural killer cells in acute and chronic viral hepatitis. J Infect Dis. 2014;209:1362–1373. doi: 10.1093/infdis/jit561. [DOI] [PubMed] [Google Scholar]
- 35.Tong HV, Toan NL, Song LH, Bock CT, Kremsner PG, Velavan TP. Hepatitis B virus-induced hepatocellular carcinoma:functional roles of MICA variants. J Viral Hepat. 2013;20:687–698. doi: 10.1111/jvh.12089. [DOI] [PubMed] [Google Scholar]
- 36.Pollicino T, and Koumbi L. Role natural killer group 2D-ligand interactions in hepatitis B infection. World J Hepatol. 2015;7:819–824. doi: 10.4254/wjh.v7.i6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J Immunol. 2007;178:961–9. doi: 10.4049/jimmunol.178.2.961. [DOI] [PubMed] [Google Scholar]
- 38.Chen CJ, Yang HI, Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA. 2006;295:65–73. doi: 10.1001/jama.295.1.65. [DOI] [PubMed] [Google Scholar]
- 39.Tang KF, Chen M, Xie J, et al. Inhibition of hepatitis B virus replication by small interference RNA induces expression of MICA in HepG2.2.15 cells. Med Microbiol Immunol. 2009;198:27–32. doi: 10.1007/s00430-008-0101-6. [DOI] [PubMed] [Google Scholar]