

Abstract
The development of high efficiency perovskite solar cells has sparked a multitude of measurements on the optical properties of these materials. For the most studied methylammonium(MA)PbI3 perovskite, a large range (6–55 meV) of exciton binding energies has been reported by various experiments. The existence of excitons at room temperature is unclear. For the MAPbX3 perovskites we report on relativistic Bethe-Salpeter Equation calculations (GW-BSE). This method is capable to directly calculate excitonic properties from first-principles. At low temperatures it predicts exciton binding energies in agreement with the reported ‘large’ values. For MAPbI3, phonon modes present in this frequency range have a negligible contribution to the ionic screening. By calculating the polarization in time from finite temperature molecular dynamics, we show that at room temperature this does not change. We therefore exclude ionic screening as an explanation for the experimentally observed reduction of the exciton binding energy at room temperature and argue in favor of the formation of polarons.
In the last three years metal halide perovskites have come up as very promising solar cell materials1,2,3. Because of their relatively simple production procedure and high photovoltaic efficiency, they bear the potential of becoming competitive with current silicon based solar cells. The materials have an OMX3 perovskite structure (Organic(O), Metal(M), Halide(X)) and depending on the temperature up to three different crystal phases. The most frequently studied material is MAPbI3. At temperatures above 333 K, the lead and iodine atoms form a cubic perovskite structure enclosing a methylammonium (MA) molecule4. Combinations with the halogens Cl and Br can also be made and result in perovskite structures with different volumes and larger band gaps, not ideally suited for solar applications. According to early experimental measurements from the 1990s and 2000s, the MAPbX3 perovskites are semiconductors with optical band gaps (Δopt) ranging between ~1.6–3.1 eV5,6,7,8. The optical gap is slightly lower than the fundamental electronic band gap (Δ), because of the electron-hole (e-h) interaction present in the excited system. With the emergence of very efficient perovskite solar cells, the mechanism behind the material’s good energy conversion rate has become a focus of research. In this regard, one important issue is the relatively large exciton binding energies (Exb) reported for these materials, 6–55 meV for MAPbI35,7,9,10,11,12,13,14,15 and 76 meV7 for MAPbBr3. Intriguingly, many reported values are higher than kBT, which should make it difficult for electrons and holes to separate after excitation. It is then a mystery why these materials are so efficient in converting solar energy to power. As a solution to this puzzle, it has been proposed that ionic contributions from the PbI3 framework and the rotational freedom of MA molecules contribute to the screening properties, thereby reducing the exciton binding energy16,17,18,19. Alternative explanations invoke the formation of polarons, quasiparticles dressed by the ionic lattice that might lower the band gap19 below the excitonic onset. Furthermore, recent experiments indicate that temperature also plays a role. Y. Yamada et al.9 measured a reduction of Exb from ~30 meV at 13 K to ~6 meV at 300 K, and likewise, A. Miyata et al.10 measured a value of 16 ± 2 meV in the low temperature orthorhombic phase, but only a few meV at room temperature. Whether ionic screening does or does not affect Exb is under debate20. The large range of the reported Exb values indicate the need for a theoretical description. In this work we report about first principles calculations on these ionic systems and address excitons in their interplay with polar phonons.
The theoretical modeling of metal halide perovskites is extremely challenging as it involves the treatment of several subtle, but important effects that are difficult to compute accurately. The first issue is the lattice structure to consider. Temperature dependent crystal structures have been determined4, but uncertainties in the orientation of the organic part prevent an unequivocal structural resolution. The structural characteristics have been the subject of numerous studies based on different (Local Density, Generalized Gradient, van der Waals) DFT approximations21,22,23,24,25,26. For the cubic phase of MAPbI3, most calculations predict lattice constants in good to excellent agreement with experiment. However, differences in the orientation of the molecule and the resulting deformation of the unit cell have been reported. We address this issue here (i) by performing global search for minimum energy structures, (ii) calculating excitons for various unit cells, (iii) and finite temperature simulations. Second, the presence of heavy elements requires to consider relativistic effects including spin-orbit coupling (SOC)27,28. Furthermore, for a quantitative description of the electronic structure, it is essential to calculate many-body quasiparticle energies e.g. in the framework of the GW approximation27,29,30,31. Finally, to evaluate Exb and calculate accurate optical spectra, it is necessary to account for the e-h interaction. This can be done by the Bethe-Salpeter equation (BSE) following GW calculations31,32— a computationally exceedingly challenging endeavor if spin-orbit coupling is taken into account. It is therefore not astonishing that previous work has often given unsatisfactory results. Several quasiparticle GW calculations have been reported recently27,29,30,31, but a fully relativistic treatment including spin-orbit interaction was only performed in refs 27 and 33. Although, BSE calculations have been reported, these often neglect relativistic effects31,32 and report much too large binding energies. Even if relativistic effects are accounted for, the binding energies (Exb = 0.153 eV) are at least a factor 3 too large compared to any experimental values33. As we will show here, we can entirely resolve this issue when sampling the Brillouin zone with sufficient accuracy.
Computational method
The first-principles calculations use a plane-wave basis and the projector augmented wave (PAW) method34 as implemented in the vasp code35,36,37. For structure determination, the PBEsol (Perdew, Burke, Ernzerhof modified for solids)38 functional was used, if not otherwise noted. Cross checks were also performed using van der Waals corrected functionals, specifically, the PBE-D3 method of Grimme39 finding no relevant differences for the properties reported here. The MAPbX3 cubic perovskite unit cells (12 atoms per cell) were constructed starting from the cubic-phase of MAPbI3 determined by X-ray diffraction4 and seeking the global energy minimum by simulated annealing. To determine candidate structures, molecular dynamics simulations were performed with a linear decrease of the temperature from 800 K to 500 K in 50000 steps of 1.5 fs. Approximately every ~1000 steps a snapshot was taken and fully relaxed. This process was repeated from the lowest energy structure yet found. A unique global minimum was found for all considered materials (See Supplementary Materials). In the subsequent electronic structure calculations (GW and BSE), SOC was fully included, and for Pb the 5s25p65d10 orbitals were included in the valence27. Gaussian smearing with σ = 0.05 eV was used to broaden the one-electron levels. Many-body effects were accounted for by first calculating PBE orbitals, and then determining the quasiparticle energies and fundamental gaps in the GW0 approximation40,41. Here the one electron energies in G were iterated until the quasiparticle energies are converged, while keeping W0 fixed at the DFT-RPA level42. About 2100 empty bands on a 4 × 4 × 4 Γ-centered k-point grid and 128 points on the frequency grid are needed to obtain well converged band gaps.
To determine the optical properties, the Bethe-Salpeter equation for the polarizability43,44,45 was solved. The common Tamm-Dancoff approximation46, 32 occupied and unoccupied KS orbitals, the W0 of the preceding GW0 calculations, and 6 × 6 × 6 k-points centered on a low symmetry k-point were used.
To obtain k-point converged values for the exciton binding energy Exb at least 20 × 20 × 20 k-points are, however, required. These BSE calculations, were performed using only 2 (un)occupied orbitals and fitting W0 to a model dielectric function47 that depends parametrically on the macroscopic dielectric constant determined in the previous BSE calculations with few k-points. Since even GW calculations are prohibitive for so many k-points, we use PBE calculations and applied a scissor technique to raise the unoccupied KS eigenvalues (compare Fig. 1). At these dense k-point grids, the Exb becomes linearly dependent on the inverse of the total number of k-points48. The Exb values reported in this work are therefore obtained by linear extrapolation to obtain the limit of the infinitely dense k-point grid (See Supplementary Materials).
Figure 1. Fast band picture of excitons in MAPbX3.

Left: GW0 band structure of MAPbX3 in the pseudo-cubic phase, with X = I (red), Br (blue) and Cl (green) determined by Wannier interpolation64 from a calculation using 4 × 4 × 4 k-points. The band gaps at R are indicated in eV. The dashed lines in the background are the corresponding DFT+scissor band structures. Right: Zoom-in of the band structure (marked by the dotted lines) close to the R point. The radii of the circles represent the contribution of the e-h pair at that k-point (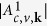 ) to the first exciton wave function.
) to the first exciton wave function.
The effect of different molecular orientations on the exciton binding energy have been assessed by BSE calculations on low energy configurations of the  FASnI3 and MAPbI3 super cells. In addition, we have constructed a
FASnI3 and MAPbI3 super cells. In addition, we have constructed a 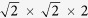 super cell for FASnI3. These structures were acquired by taking snap shots from Parallel Tempering Molecular Dynamics (PTMD) calculations at 300 K. The
super cell for FASnI3. These structures were acquired by taking snap shots from Parallel Tempering Molecular Dynamics (PTMD) calculations at 300 K. The  structures are the lowest energy configurations from the PTMD trajectory and were relaxed into their instantaneous ground state, while keeping the volume and cell shape fixed to the experiment. The
structures are the lowest energy configurations from the PTMD trajectory and were relaxed into their instantaneous ground state, while keeping the volume and cell shape fixed to the experiment. The 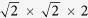 structure is a randomly picked configuration at 300 K from a separate PTMD calculation and was not relaxed. In the
structure is a randomly picked configuration at 300 K from a separate PTMD calculation and was not relaxed. In the  structures the molecular dipoles are orthogonally orientated w.r.t. each other and in the
structures the molecular dipoles are orthogonally orientated w.r.t. each other and in the 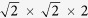 structures all the molecular dipoles have a different orientation. The same BSE calculation procedure was used as before, but the screening parameters and GW0 gap were not calculated; the values for the unit cell were used instead. This is a reasonable approximation, since calculation of the screening in the computationally more efficient random phase approximation shows little difference in electronic screening for different unit cells and different molecular orientations.
structures all the molecular dipoles have a different orientation. The same BSE calculation procedure was used as before, but the screening parameters and GW0 gap were not calculated; the values for the unit cell were used instead. This is a reasonable approximation, since calculation of the screening in the computationally more efficient random phase approximation shows little difference in electronic screening for different unit cells and different molecular orientations.
Details of the finite temperature dielectric function calculations are presented in the results section.
Results
In Fig. 1 (left), the calculated GW0 quasiparticle band structures of the three MAPbX3 structures are shown. The band gap at the R points is indicated and is in excellent agreement with experiment5,6,7,8. SOC shifts the band gap minimum to R′ making it slightly indirect30. The exciton wave function is expressed in an electron-hole product basis, 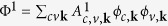 . The first eigenstate
. The first eigenstate 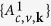 of the generalized BSE eigenvalue problem45 is visualized by plotting
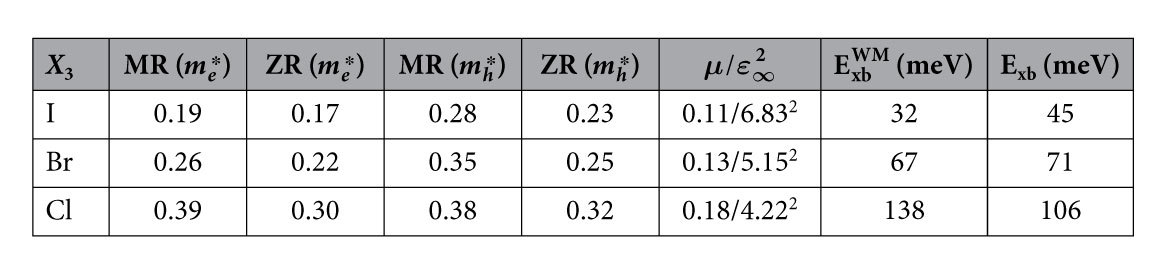
of the generalized BSE eigenvalue problem45 is visualized by plotting 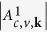 as a fat band structure. On the right hand side of Fig. 1 a zoom-in of the region close to R is made. It shows that the exciton is very localized in k-space, primarily consisting of states at the band extrema. Going from iodine to chlorine, the dispersion flattens (effective electron/hole masses increase), the band gap increases and, as a result, the extent of the exciton in k-space increases. The corresponding parameters are tabulated in Table 1. We have calculated the corresponding exciton binding energies also in the Wannier-Mott (WM) model for screened Coulomb interacting e-h pairs in parabolic bands:
as a fat band structure. On the right hand side of Fig. 1 a zoom-in of the region close to R is made. It shows that the exciton is very localized in k-space, primarily consisting of states at the band extrema. Going from iodine to chlorine, the dispersion flattens (effective electron/hole masses increase), the band gap increases and, as a result, the extent of the exciton in k-space increases. The corresponding parameters are tabulated in Table 1. We have calculated the corresponding exciton binding energies also in the Wannier-Mott (WM) model for screened Coulomb interacting e-h pairs in parabolic bands: 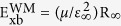 , with
, with 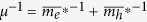 the effective mass of the e-h pair, ε∞ the high freq. dielectric constant and R∞ the Rydberg constant. Since we use the SOC split “Rashba-Dresselhaus”49 band structure in the BSE method, we can test the validity of the simple parabolic dispersion assumed in the WM model. We see (Table 1) that WM gives the correct order of the e-h interaction, however it results in a different ratio between I:Br and Br:Cl, which can not be trivially explained by small errors in μ or ε∞.
the effective mass of the e-h pair, ε∞ the high freq. dielectric constant and R∞ the Rydberg constant. Since we use the SOC split “Rashba-Dresselhaus”49 band structure in the BSE method, we can test the validity of the simple parabolic dispersion assumed in the WM model. We see (Table 1) that WM gives the correct order of the e-h interaction, however it results in a different ratio between I:Br and Br:Cl, which can not be trivially explained by small errors in μ or ε∞.
Table 1. Effective electron and hole masses (
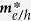 ) of the VBM and CBM in the MR and RZ directions from the GW
0+ SOC band structures, the ratio of the exciton effective mass (μ) over the high freq. dielectric constant squared (
) of the VBM and CBM in the MR and RZ directions from the GW
0+ SOC band structures, the ratio of the exciton effective mass (μ) over the high freq. dielectric constant squared (
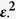 ), the Wannier-Mott -(
), the Wannier-Mott -(
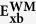 ) the BSE calculated (Exb) exciton binding energies.
) the BSE calculated (Exb) exciton binding energies.
An important question is, whether the ionic contributions to the screening can be disregarded in the BSE calculations. To explore this point, Fig. 2 shows the sum of the ionic and electronic contribution to the dielectric function at 0 K, ε(ω), with the ionic contribution calculated using density functional perturbation theory (DFPT)50,51,52. A sizable increase of the static dielectric constant (ε0) compared to the ‘ion-clamped’ high frequency dielectric constant (ε∞) is found. The increase comes from optically active phonon modes below 20 meV (see Im εp(ω)), clearly displaying the ionic nature of this material. However, the phonon modes present in the relevant energy window around Exb ≈ 45 meV (see inset) are practically not active.
Figure 2. Ionic and electronic contributions (εp, εe) to the dielectric function ε(ω) of MAPbI3.

The top/bottom figure shows the real/imaginary part of ε(ω). The solid red/dashed blue line are, respectively the results from the DFPT(T = 0 K)/MD(T = 300 K) method. The inset shows a zoom-in of Reεp(ω) close to the Exb of 45 meV.
Since the exciton binding seems to change with temperature, the second intriguing question is whether the screening changes at finite temperature. To explore this, we have developed a novel scheme to evaluate the dielectric ionic response at finite temperature that we briefly describe in this section (a more detailed description will be presented in a future work). The idea is inspired by methods usually used to determine the electronic contributions to the screening in time-dependent DFT53. Well equilibrated finite temperature ensembles are subjected to a short constant electric field in time Eδ(t) acting on the ions. The δ-pulse is a natural way of exciting all possible frequencies in the system. The force exerted by this field onto the ions is proportional to 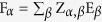 , where Zα,β are the Born effective charges evaluated by density functional perturbation theory, and α and β are Cartesian indices50. In the first time step, these forces are added, thereby exciting the ionic system. The issue is to find a suitable way to calculate the induced ionic polarization P(t) caused by the delta peak. Here, we calculate the induced polarization as δP(t) = (P+(t) − P−(t))/2, where P+(t) is the time evolution of the polarization for a positive delta peak Eδ(t), and P−(t) the time evolution of the polarization after a negative delta peak −Eδ(t). The evolving P(t) can in principle be evaluated using the Berry curvature54, but the Berry curvature often jumps discontinuously as the ions move. Hence, we evaluate the change of the polarization from the velocities vα(t) and the Born effective charges Zαβ(t) as
, where Zα,β are the Born effective charges evaluated by density functional perturbation theory, and α and β are Cartesian indices50. In the first time step, these forces are added, thereby exciting the ionic system. The issue is to find a suitable way to calculate the induced ionic polarization P(t) caused by the delta peak. Here, we calculate the induced polarization as δP(t) = (P+(t) − P−(t))/2, where P+(t) is the time evolution of the polarization for a positive delta peak Eδ(t), and P−(t) the time evolution of the polarization after a negative delta peak −Eδ(t). The evolving P(t) can in principle be evaluated using the Berry curvature54, but the Berry curvature often jumps discontinuously as the ions move. Hence, we evaluate the change of the polarization from the velocities vα(t) and the Born effective charges Zαβ(t) as 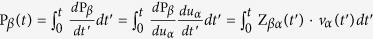 . The additional cost is small, since Z(t) varies very slowly and needs to be recalculated only about every 50 time steps. The Fourier transformation of δP(t) is directly related to the ionic polarizability55. We first tested this approach at T = 0 K and found exact agreement with perturbation theory. To obtain reasonably noise-free data at finite temperature, we use a 2 × 2 × 2 super cell and average over 80 starting configurations in order to converge the spectrum. After the δ-pulse, the system is allowed to evolve in the micro-canonical ensemble unperturbed for 3 ps, the short time somewhat limiting the spectral resolution. However, the 80 starting configurations were obtained by a taking independent snapshots every 0.7 ps from a well equilibrated 60 ps long finite temperature MD trajectory. Therefore, we expect most of the dynamics to be sampled. The PBE-D3 method of Grimme39 was used here (although PBEsol results are very similar) and the deuterium mass was used for the hydrogen atoms. This replacement only changes the hydrogen related modes above 100 meV and allows to increase the time step during the simulation.
. The additional cost is small, since Z(t) varies very slowly and needs to be recalculated only about every 50 time steps. The Fourier transformation of δP(t) is directly related to the ionic polarizability55. We first tested this approach at T = 0 K and found exact agreement with perturbation theory. To obtain reasonably noise-free data at finite temperature, we use a 2 × 2 × 2 super cell and average over 80 starting configurations in order to converge the spectrum. After the δ-pulse, the system is allowed to evolve in the micro-canonical ensemble unperturbed for 3 ps, the short time somewhat limiting the spectral resolution. However, the 80 starting configurations were obtained by a taking independent snapshots every 0.7 ps from a well equilibrated 60 ps long finite temperature MD trajectory. Therefore, we expect most of the dynamics to be sampled. The PBE-D3 method of Grimme39 was used here (although PBEsol results are very similar) and the deuterium mass was used for the hydrogen atoms. This replacement only changes the hydrogen related modes above 100 meV and allows to increase the time step during the simulation.
Clearly, the 300 K finite temperature polarizability (solid red line in Fig. 2) above 20 meV is very similar to the one at T = 0 K (dashed blue line in Fig. 2). The modes are at the same positions but broadened by fluctuations in the cage structure, as well as rotations of the molecules. Below 20 meV some differences are visible, however, in both methods the calculated ε0 is close to 30, in excellent agreement with the measured value of 28.856.
We now discuss the question whether ionic screening should be included in the calculation of the exciton binding energy. The values reported in Table 1 assume a fixed lattice, i.e. a vertical transition. It is well established from the Franck-Condon energy diagram that lattice relaxation in the excited state can only decrease the transition energy, i.e. thermal or adiabatic transition energies including relaxation are always below vertical transition energies57. Lattice relaxation therefore can only increase the exciton binding energy, since the fundamental gap minus the transition energy is defined as the exciton binding energy Exb. Whether relaxation needs to be included, i.e. whether the vertical or thermal transition energy is measured experimentally can be disputed, although common wisdom is that optical absorption virtually always measures vertical excitation energies. One argument is that the position of the (dominant) longitudinal optical phonon mode (ωLO) compared to the typical energy scales of the optical absorption determines whether ionic relaxation should be considered. In the effective mass approximation this leads to two extreme cases, 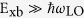 ,
,  and
and 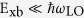 (ionic relaxation needs to be included)58,59. Since the dominant active phonon modes for MAPbI3 are all below 10 meV, we have
(ionic relaxation needs to be included)58,59. Since the dominant active phonon modes for MAPbI3 are all below 10 meV, we have 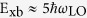 and the use of an εeff ≈ ε∞ ≈ 6.8 is entirely justified (see Fig. 2 for energies larger than 40 meV). The temperature independence of the ionic screening, furthermore, implies that the observed lowering of the exciton binding energy at elevated temperatures must have a different origin than changes in the ionic screening. In agreement with theory, recent room temperature time-resolved terahertz spectroscopy experiments, indicate a near constant screening ε(ω) = 5.5 in the frequency range of ω = 40–100 meV and Exb = 49 ± 3 meV60.
and the use of an εeff ≈ ε∞ ≈ 6.8 is entirely justified (see Fig. 2 for energies larger than 40 meV). The temperature independence of the ionic screening, furthermore, implies that the observed lowering of the exciton binding energy at elevated temperatures must have a different origin than changes in the ionic screening. In agreement with theory, recent room temperature time-resolved terahertz spectroscopy experiments, indicate a near constant screening ε(ω) = 5.5 in the frequency range of ω = 40–100 meV and Exb = 49 ± 3 meV60.
However, if the exciton binding energy Exb is not lowered by ionic screening, what mechanism then leads to carrier separation at higher temperatures? Our calculations also shed light on this. Individual electrons e− and holes h+ can be screened by the lattice, thereby forming ‘dressed’ quasiparticles (QP) known as polarons. Since the mesoscopic Wannier-Mott model was so precise, we again resort to a mesosocopic model, namely, Fröhlich’s theory for large polarons. In this model, polaron formation lowers the QP energy by 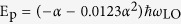 , with a coupling constant
, with a coupling constant 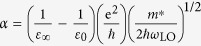 . Using the data from Table 1, a screening of ε0 = 30, ε∞ = 6, and
. Using the data from Table 1, a screening of ε0 = 30, ε∞ = 6, and 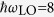 meV from Fig. 2 we obtain an α of 2.3/2.8. This lowers the QP energy of the electron and hole by 19 and 23 meV, respectively, and hence reduces the QP gap by 42 meV. This means that the charge separated polaronic state is only slightly less stable than the bound exciton. If we further recall that after excitation the electrons and holes are not yet close to the conduction or valence band edges, and that they are both individually scattered by lattice phonons loosing energy but possibly gaining momentum61, it is likely that they will rapidly separate in space and never reach their global groundstate, the bound exciton. Charge separation after optical excitation will be further eased by non-regularities in the electrostatic potential. And non-regularities exist aplenty in MAPbI3 at elevated temperatures: the polar MA molecules seem to prefer a short range ferroelectric order causing ferroelectric domains and a strong corrugation of the electrostatic potential17. A possible way to experimentally disentangle polaron formation and such molecular contributions and related corrugations in the potential is to perform control measurements on an OPbI3 perovskite with O cations that are non polar, for instance Cs.
meV from Fig. 2 we obtain an α of 2.3/2.8. This lowers the QP energy of the electron and hole by 19 and 23 meV, respectively, and hence reduces the QP gap by 42 meV. This means that the charge separated polaronic state is only slightly less stable than the bound exciton. If we further recall that after excitation the electrons and holes are not yet close to the conduction or valence band edges, and that they are both individually scattered by lattice phonons loosing energy but possibly gaining momentum61, it is likely that they will rapidly separate in space and never reach their global groundstate, the bound exciton. Charge separation after optical excitation will be further eased by non-regularities in the electrostatic potential. And non-regularities exist aplenty in MAPbI3 at elevated temperatures: the polar MA molecules seem to prefer a short range ferroelectric order causing ferroelectric domains and a strong corrugation of the electrostatic potential17. A possible way to experimentally disentangle polaron formation and such molecular contributions and related corrugations in the potential is to perform control measurements on an OPbI3 perovskite with O cations that are non polar, for instance Cs.
We like to comment briefly on the performance of a wider class of perovskites (OMX3). Specifically, we have replaced MA by formamidinium (FA) and Pb by Sn, thereby constructing twelve different perovskites. Their global lowest energy structure was calculated as before by simulated annealing and subsequent relaxation. FA is larger than MA and thereby changes the band gap of the perovskite. FASnI3 is a particular interesting candidate, previous work suggests that this is possibly a ferroelectric lead-free alternative for MAPbI349. In Fig. 3 we show the trend in the exciton binding energies w.r.t. the GW0 band gap. Clearly, the halogen species predominantly determines the gap. For each halogen, the strength of the exciton binding energy and the optical gap can be fine-tuned by varying the molecule or the metal atom. Nevertheless, only iodine based perovskites seem to posses sufficiently small band gaps and exciton binding energies to be suitable for solar cells. An overview of the band gaps calculated at the various level of theory and available experimental data has been presented in Table 2. Over the whole range a good agreement is found between the GW-BSE calculations on these small unit cell structures and experimentally observed band gaps. Small discrepancies can be caused by the unit cell approach taken in this work. For those structures, which have not yet been synthesized or for which the band gap has not yet been measured, we put these number forward as predictions.
Figure 3. Calculated exciton binding energies and GW0 band gaps of twelve metal halide perovskites (OMX3, {O = MA, FA, M = Pb, Sn, X = I, Br, Cl}).

Table 2. Calculated onset of optical absorption (Δopt = ΔGW − Exb), exciton binding energy (Exb), the GW0 (ΔGW), DFT (ΔDFT) and DFT without SOC (
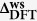 ) band gaps.
) band gaps.
Available experimental results are shown for comparison.
Discussion
The last point we need to consider is how different molecular orientations influence the results. In a real (super)structure the exciton wave function will span many unit cells with molecules that have different orientations. It has been suggested that the ordering and orientation of the molecules in the lattice aids the e-h dissociation process17,18,62. Both the MA and FA molecules have an intrinsic dipole moment and are only weakly bonded to the MX3 cage. It is known from Nuclear Magnetic Resonance Spectroscopy measurements that the MA molecules in MAPbX3 have the full rotational degree of freedom at room temperature and that reorientation is a fairly rapid process63. However, with a typical reorientation time in the pico-second time scale, it is the slowest screening mechanism present in the OMX3 perovskites. To asses the effect of different molecular orientations, BSE calculations have been performed on  and
and 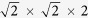 cells containing 2 and 4 molecules, respectively. For the
cells containing 2 and 4 molecules, respectively. For the  and the larger FASnI3 cell, the calculated exciton binding energies are 33 and 31 meV, respectively. Compared to the 31 meV predicted for the unit cell, the super cell approach does not give significantly different results for the exciton binding energy. The same holds for MAPbI3, where the
and the larger FASnI3 cell, the calculated exciton binding energies are 33 and 31 meV, respectively. Compared to the 31 meV predicted for the unit cell, the super cell approach does not give significantly different results for the exciton binding energy. The same holds for MAPbI3, where the  cell results in an exciton binding energy of 51 meV, which is only slightly larger than the 45 meV predicted for the unit cell.
cell results in an exciton binding energy of 51 meV, which is only slightly larger than the 45 meV predicted for the unit cell.
Conclusion
Accurate first principles calculations predict exciton binding energies of the order of 50, 70 and 110 meV for MAPbI3, MAPbBr3 and MAPbCl3, respectively. The agreement of the Wannier-Mott model with our high level calculations is good, provided that the model parameters are taken from accurate first principles calculations. The large exciton binding energy is clearly at variance with the observed high efficiency of solar cells, but in excellent agreement with most low temperature measurements. The much discussed ionic screening is almost temperature independent and substantially increases ε0 from around 6 to 30. However, the optically active modes are too slow (<10 meV) to effectively screen the excitons. For certain, we can rule out that a change of the ionic screening is responsible for the experimentally observed reduction of the exciton binding energy at room temperature. Instead, our calculations predict a different scenario: electrons and holes separate after optical excitation forming two individual polarons, lowering the fundamental gap by 42 meV. This scenario should now be carefully evaluated by experiments, and if validated, offers an intriguing option for the design of novel polaronic solar cell materials.
Additional Information
How to cite this article: Bokdam, M. et al. Role of Polar Phonons in the Photo Excited State of Metal Halide Perovskites. Sci. Rep. 6, 28618; doi: 10.1038/srep28618 (2016).
Supplementary Material
Acknowledgments
M.B., C.F. and D.D.S. acknowledge funding by the joint Austrian Science Fund - Indian Department of Science and Technology (DST) project INDOX (Grant No. I1490-N19). G.K. and C.F. acknowledge funding by Austrian Science Fund (FWF): F41 SFB ViCoM. The calculations were partly performed at the Vienna Scientific Cluster (VSC-3).
Footnotes
Author Contributions G.K., C.F., M.B and A.S. conceived the project; M.B. and G.K. proposed and implemented the new method to calculate the finite temperature dielectric function, and performed all DFT and GW-BSE calculations. D.D.S. and S.P. included insight from the experimental and theoretical standing of the perovskite field. All the authors contributed with discussions and writing of the manuscript.
References
- Green M. A., Ho-Baillie A. & Snaith H. J. The emergence of perovskite solar cells. Nature Phot. 8, 506–514 (2014). [Google Scholar]
- Grätzel M. The light and shade of perovskite solar cells. Nature Mater. 13, 838–842 (2014). [DOI] [PubMed] [Google Scholar]
- Kim H.-S., Im S. H. & Park N.-G. Organolead halide perovskite: New horizons in solar cell research. J. Phys. Chem. C 118, 5615–5625 (2014). [Google Scholar]
- Stoumpos C. C., Malliakas C. D. & Kanatzidis M. G. Semiconducting tin and lead iodide perovskites with organic cations: Phase transitions, high mobilities, and near-infrared photoluminescent properties. Inorg. Chem. 52, 9019–9038 (2013). [DOI] [PubMed] [Google Scholar]
- Hirasawa M., Ishihara T. & Goto T. Exciton features in 0-, 2-, and 3-dimensional networks of [pbi6]4- octahedra. J. Phys. Soc. Jpn. 63, 3870–3879 (1994). [Google Scholar]
- Papavassiliou G. & Koutselas I. Structural, optical and related properties of some natural three- and lower-dimensional semiconductor systems. Synthetic Metals 71, 1713–1714 (1995). [Google Scholar]
- Tanaka K. et al. Comparative study on the excitons in lead-halide-based perovskite-type crystals {CH3NH3PbBr3} {CH3NH3PbI3}. Sol. State Comm. 127, 619–623 (2003). [Google Scholar]
- Kitazawa N., Watanabe Y. & Nakamura Y. Optical properties of ch3nh3pbx3 (x = halogen) and their mixed-halide crystals. J. Mater. Sci. 37, 3585–3587 (2002). [Google Scholar]
- Yamada Y., Nakamura T., Endo M., Wakamiya A. & Kanemitsu Y. Photoelectronic responses in solution-processed perovskite ch3nh3pbi3 solar cells studied by photoluminescence and photoabsorption spectroscopy. IEEE J. Photovolt. 5, 401–405 (2015). [Google Scholar]
- Miyata A. et al. Direct measurement of the exciton binding energy and effective masses for charge carriers in organic-inorganic tri-halide perovskites. Nature Phys. 11, 582–587 (2015). [Google Scholar]
- Sun S. et al. The origin of high efficiency in low-temperature solution-processable bilayer organometal halide hybrid solar cells. Energy Environ. Sci. 7, 399–407 (2014). [Google Scholar]
- Saba M. et al. Correlated electronhole plasma in organometal perovskites. Nature Comm. 5, 5049 (2014). [DOI] [PubMed] [Google Scholar]
- Savenije T. J. et al. Thermally activated exciton dissociation and recombination control the carrier dynamics in organometal halide perovskite. J. Phys. Chem. Lett. 5, 2189–2194 (2014). [DOI] [PubMed] [Google Scholar]
- Zhang Q., Ha S. T., Liu X., Sum T. C. & Xiong Q. Room-temperature near-infrared high-q perovskite whispering-gallery planar nanolasers. Nano Lett. 14, 5995–6001 (2014). [DOI] [PubMed] [Google Scholar]
- D’Innocenzo V. et al. Excitons versus free charges in organo-lead tri-halide perovskites. Nature Comm. 5, 3586 (2014). [DOI] [PubMed] [Google Scholar]
- Huang L.-y. & Lambrecht W. R. L. Electronic band structure, phonons, and exciton binding energies of halide perovskites cssncl3, cssnbr3, and cssni3. Phys. Rev. B 88, 165203 (2013). [Google Scholar]
- Frost J. M. et al. Atomistic origins of high-performance in hybrid halide perovskite solar cells. Nano Lett. 14, 2584–2590 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even J., Pedesseau L. & Katan C. Analysis of multivalley and multibandgap absorption and enhancement of free carriers related to exciton screening in hybrid perovskites. J. Phys. Chem. C 118, 11566–11572 (2014). [Google Scholar]
- Menéndez-Proupin E., Beltrán Ros C. L. & Wahnn P. Non-hydrogenic excitons in perovskite ch3nh3pbi3. arXiv: 1507.00404v1 (2015).
- Filippetti A., Delugas P., Saba M. I. & Mattoni A. Entropy-suppressed ferroelectricity in hybrid lead-iodide perovskites. J. Phys. Chem. Lett. 6, 4909–4915 (2015). [DOI] [PubMed] [Google Scholar]
- Borriello I., Cantele G. & Ninno D. Ab initio investigation of hybrid organic-inorganic perovskites based on tin halides. Phys. Rev. B 77, 235214 (2008). [Google Scholar]
- Mosconi E., Amat A., Nazeeruddin M. K., Grätzel M. & De Angelis F. First-principles modeling of mixed halide organometal perovskites for photovoltaic applications. J. Phys. Chem. C 117, 13902–13913 (2013). [Google Scholar]
- Brivio F., Walker A. B. & Walsh A. Structural and electronic properties of hybrid perovskites for high-efficiency thin-film photovoltaics from first-principles. APL Materials 1, 042111 (2013). [Google Scholar]
- Feng J. & Xiao B. Crystal structures, optical properties, and effective mass tensors of ch3nh3pbx3 (x = i and br) phases predicted from hse06. J. Phys. Chem. Lett. 5, 1278–1282 (2014). [DOI] [PubMed] [Google Scholar]
- Egger D. A. & Kronik L. Role of dispersive interactions in determining structural properties of organicinorganic halide perovskites: Insights from first-principles calculations. J. Phys. Chem. Lett. 5, 2728–2733 (2014). [DOI] [PubMed] [Google Scholar]
- Motta C. et al. Revealing the role of organic cations in hybrid halide perovskite ch3nh3pbi3. Nature Comm. 6, 7026 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filip M. R. & Giustino F. gw quasiparticle band gap of the hybrid organic-inorganic perovskite ch3nh3pbi3: Effect of spin-orbit interaction, semicore electrons, and self-consistency. Phys. Rev. B 90, 245145 (2014). [Google Scholar]
- Menéndez-Proupin E., Palacios P., Wahnón P. & Conesa J. C. Self-consistent relativistic band structure of the CH3NH3PBI3 perovskite. Phys. Rev. B 90, 045207 (2014). [Google Scholar]
- Umari P., Mosconi E. & Angelis F. D. Relativistic gw calculations on ch3nh3pbi3 and ch3nh3sni3 perovskites for solar cell applications. Sci. Rep. 4, 4467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brivio F., Butler K. T., Walsh A. & van Schilfgaarde M. Relativistic quasiparticle self-consistent electronic structure of hybrid halide perovskite photovoltaic absorbers. Phys. Rev. B 89, 155204 (2014). [Google Scholar]
- Castelli I. E., Garca-Lastra J. M., Thygesen K. S. & Jacobsen K. W. Bandgap calculations and trends of organometal halide perovskites. APL Materials 2, 081514 (2014). [Google Scholar]
- Even J., Pedesseau L., Jancu J.-M. & Katan C. Dft and k.p modelling of the phase transitions of lead and tin halide perovskites for photovoltaic cells. Phys. Stat. Sol. RRL 8, 31–35 (2014). [Google Scholar]
- Ahmed T. et al. Optical properties of organometallic perovskite: An ab initio study using relativistic gw correction and bethe-salpeter equation. Europhys. Lett. 108, 67015 (2014). [Google Scholar]
- Blöchl P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994). [DOI] [PubMed] [Google Scholar]
- Kresse G. & Hafner J. Ab-initio molecular-dynamics for liquid-metals. Phys. Rev. B 47, 558–561 (1993). [DOI] [PubMed] [Google Scholar]
- Kresse G. & Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996). [DOI] [PubMed] [Google Scholar]
- Kresse G. & Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999). [Google Scholar]
- Perdew J. P. et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 136406 (2008). [DOI] [PubMed] [Google Scholar]
- Grimme S., Antony J., Ehrlich S. & Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (dft-d) for the 94 elements h-pu. J. Chem. Phys. 132, 154104 (2010). [DOI] [PubMed] [Google Scholar]
- Hedin L. New method for calculating the one-particle green’s function with application to the electron-gas problem. Phys. Rev. 139, A796–A823 (1965). [Google Scholar]
- Hybertsen M. S. & Louie S. G. Electron correlation in semiconductors and insulators: Band gaps and quasiparticle energies. Phys. Rev. B 34, 5390–5413 (1986). [DOI] [PubMed] [Google Scholar]
- Shishkin M. & Kresse G. Implementation and performance of the frequency-dependent gw method within the paw framework. Phys. Rev. B 74, 035101 (2006). [Google Scholar]
- Hanke W. & Sham L. J. Many-particle effects in the optical spectrum of a semiconductor. Phys. Rev. B 21, 4656–4673 (1980). [Google Scholar]
- Onida G., Reining L. & Rubio A. Electronic excitations: density-functional versus many-body green’s-function approaches. Rev. Mod. Phys. 74, 601–659 (2002). [Google Scholar]
- Sander T., Maggio E. & Kresse G. Beyond the tamm-dancoff approximation for extended systems using exact diagonalization. Phys. Rev. B 92, 045209 (2015). [Google Scholar]
- Dancoff S. M. Non-adiabatic meson theory of nuclear forces. Phys. Rev. 78, 382–385 (1950). [Google Scholar]
- Bechstedt F., Sole R. D., Cappellini G. & Reining L. An efficient method for calculating quasiparticle energies in semiconductors. Sol. State Comm. 84, 765–770 (1992). [Google Scholar]
-
Fuchs F., Rödl C., Schleife A. & Bechstedt F.
Efficient
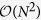 approach to solve the bethe-salpeter equation for excitonic bound states. Phys. Rev. B
78, 085103 (2008). [Google Scholar]
approach to solve the bethe-salpeter equation for excitonic bound states. Phys. Rev. B
78, 085103 (2008). [Google Scholar] - Stroppa A. et al. Tunable ferroelectric polarization and its interplay with spin-orbit coupling in tin iodide perovskites. Nature Comm. 5, 5900 (2014). [DOI] [PubMed] [Google Scholar]
- Wu X., Vanderbilt D. & Hamann D. R. Systematic treatment of displacements, strains, and electric fields in density-functional perturbation theory. Phys. Rev. B 72, 035105 (2005). [Google Scholar]
- Gajdoš M., Hummer K., Kresse G., Furthmüller J. & Bechstedt F. Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B 73, 045112 (2006). [Google Scholar]
- Pérez-Osorio M. A. et al. Vibrational properties of the organicinorganic halide perovskite ch3nh3pbi3 from theory and experiment: Factor group analysis, first-principles calculations, and low-temperature infrared spectra. J. Phys. Chem. C 119, 25703–25718 (2015). [Google Scholar]
- Walter M. et al. Time-dependent density-functional theory in the projector augmented-wave method. J. Chem. Phys. 128, 244101 (2008). [DOI] [PubMed] [Google Scholar]
- Resta R. Macroscopic polarization in crystalline dielectrics: the geometric phase approach. Rev. Mod. Phys. 66, 899–915 (1994). [Google Scholar]
- Thomas M., Brehm M., Fligg R., Vöhringer P. & Kirchner B. Computing vibrational spectra from ab initio molecular dynamics. Phys. Chem. Chem. Phys. 15, 6608–6622 (2013). [DOI] [PubMed] [Google Scholar]
- Poglitsch A. & Weber D. Dynamic disorder in methylammoniumtrihalogenoplumbates (ii) observed by millimeterwave spectroscopy. J. Chem. Phys. 87, 6373–6378 (1987). [Google Scholar]
- Freysoldt C. et al. First-principles calculations for point defects in solids. Rev. Mod. Phys. 86, 253–305. [Google Scholar]
- Bechstedt F., Seino K., Hahn P. H. & Schmidt W. G. Quasiparticle bands and optical spectra of highly ionic crystals: Aln and nacl. Phys. Rev. B 72, 245114 (2005). [Google Scholar]
- Bechstedt F. Many-Body Approach to Electronic Excitations 1 edn. (Springer-Verlag Berlin Heidelberg, 2015). [Google Scholar]
- Valverde-Chávez D. et al. Intrinsic femtosecond charge generation dynamics in single crystal ch3nh3pbi3. Energy Environ. Sci. 8, 3700–3707 (2015). [Google Scholar]
- Bernardi M., Vigil-Fowler D., Lischner J., Neaton J. B. & Louie S. G. Ab Initio study of hot carriers in the first picosecond after sunlight absorption in silicon. Phys. Rev. Lett. 112, 257402 (2014). [DOI] [PubMed] [Google Scholar]
- Ma J. & Wang L.-W. Nanoscale charge localization induced by random orientations of organic molecules in hybrid perovskite ch3nh3pbi3. Nano Lett. 15, 248–253 (2015). [DOI] [PubMed] [Google Scholar]
- Wasylishen R., Knop O. & Macdonald J. Cation rotation in methylammonium lead halides. Sol. State Comm. 56, 581–582 (1985). [Google Scholar]
- Mostofi A. A. et al. wannier90: A tool for obtaining maximally-localised wannier functions. Comp. Phys. Comm. 178, 685–699 (2008). [Google Scholar]
- Pang S. et al. Nh2chnh2pbi3: An alternative organolead iodide perovskite sensitizer for mesoscopic solar cells. Chem. Mat. 26, 1485–1491 (2014). [Google Scholar]
- Eperon G. E. et al. Formamidinium lead trihalide: a broadly tunable perovskite for efficient planar heterojunction solar cells. Energy Environ. Sci. 7, 982–988 (2014). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.