

Abstract
Bone marrow mesenchymal stem cells (BMSCs) exhibit an increased propensity toward adipocyte differentiation accompanied by a reduction in osteogenesis in osteoporotic bone marrow. However, limited knowledge is available concerning the role of long non-coding RNAs (lncRNAs) in the differentiation of BMSCs into adipocytes. In this study, we demonstrated that lncRNA H19 and microRNA-675 (miR-675) derived from H19 were significantly downregulated in BMSCs that were differentiating into adipocytes. Overexpression of H19 and miR-675 inhibited adipogenesis, while knockdown of their endogenous expression accelerated adipogenic differentiation. Mechanistically, we found that miR-675 targeted the 3′ untranslated regions of the histone deacetylase (HDAC) 4–6 transcripts and resulted in deregulation of HDACs 4–6, essential molecules in adipogenesis. In turn, trichostatin A, an HDAC inhibitor, significantly reduced CCCTC-binding factor (CTCF) occupancy in the imprinting control region upstream of the H19 gene locus and subsequently downregulated the expression of H19. These results show that the CTCF/H19/miR-675/HDAC regulatory pathway plays an important role in the commitment of BMSCs into adipocytes.
Marrow fat accumulation is characteristic of aging, estrogen deficiency, chemotherapy, glucocorticoid therapy, and radiotherapy1, and it is frequently associated with morbidity in osteoporosis2. Bone marrow fat cells and bone cells share a common progenitor, and they arise from bone marrow mesenchymal stem cells (BMSCs)3. An inverse relationship exists between marrow fat production and bone formation4. One possible cause of fat deposition is the aberrant commitment of BMSCs to the adipocyte lineage5,6. Thus, further understanding of the molecular mechanisms that initiate the differentiation of BMSCs into adipocytes may lead to the development of therapies that prevent excessive bone marrow adipogenesis and deficient bone formation.
Non-protein-coding RNAs (ncRNAs) have emerged as important epigenetic regulators in biological control and pathology7. A class of small ncRNAs has been identified as important regulators in the adipogenic differentiation of BMSCs. For example, miR-188 regulates the age-related adipogenic differentiation of BMSCs and increases bone marrow adiposity8; overexpression of miR-29a protects against glucocorticoid-induced fatty marrow and bone loss by inhibiting histone deacetylase 4 (HDAC 4)9; and miR-320 promotes the lineage-specific commitment of BMSCs to the adipocyte lineage by directly targeting RUNX210. In contrast, the global expression patterns and functional contributions of long non-coding RNAs (lncRNAs), tentatively defined as ncRNAs >200 nucleotides (nt) in length11,12, in BMSC adipogenic differentiation are still largely unknown.
H19 is a paternally-imprinted gene that does not encode a protein, but rather a 2.3-kb ncRNA13. It harbors an miRNA-containing hairpin and generates miR-675 in a classic Drosha and Dicer splicing-dependent manner14. Recent studies have highlighted the role of H19 in embryonic placental growth and cellular differentiation15,16,17. However, its functions in the adipogenic differentiation of BMSCs remain unclear. Here, we revealed that H19 and its derivative miR-675 inhibited adipogenesis in BMSCs by reducing the expression of class II HDACs 4, 5, and 6, essential molecules in adipogenesis. We also found that HDAC inhibition reduced H19 expression by decreasing the CCCTC-binding factor (CTCF) occupancy on the imprinting control region (ICR) of H19. These results provide better understanding of lncRNA regulator involved in the commitment of BMSCs to adipocytes.
Results
H19 and miR-675 expression is downregulated during adipocyte differentiation
The dynamic expression profiles of H19 and miR-675 were detected in BMSCs after their induction to the adipogenic lineage. Quantitative reverse-transcription (qRT-PCR) showed that H19 expression was significantly reduced after adipogenic induction and decreased by 80% on day 11 (Fig. 1A). The expression of miR-675, derived from H19, showed a similar trend and decreased by 50% after adipocyte differentiation (Fig. 1A). The mRNA and protein expression of the genes associated with adipogenic differentiation, peroxisome proliferator-activated receptor-γ (PPARγ), CCAAT-enhancer-binding proteins-α (C/EBPα), and fatty-acid binding protein 4 (FABP4), was significantly upregulated after adipogenesis (Fig. 1B,C).
Figure 1. Expression profiles of lncRNA H19 and H19-derived miR-675 during adipogenic differentiation of BMSCs.

(A) Left: relative expression of H19 during differentiation as determined by qRT-PCR analysis. Right: relative miR-675 transcript levels at the indicated time points during differentiation. (B) Relative mRNA expression levels of the adipogenic markers PPARγ, C/EBPα, and FABP4 at the indicated time points, as in (A). RNA expression at the time points was normalized to that on day 0. (C) Western blot analysis (left) and quantification (right) of protein expression of PPARγ, C/EBPα, FABP4, and the internal control GAPDH at the indicated time points, as in (A). Results are presented as mean ± SD (*P < 0.05, **P < 0.01 compared to BMSCs without adipocyte induction).
Overexpression of H19 and miR-675 inhibits adipocyte differentiation
LncRNA H19 was located both in nucleus and cytoplasm, and it was enriched in the cytoplasmic fraction (Supplementary Figure S1A). To determine whether H19 and miR-675 directly affect adipocyte differentiation, lentiviruses were used to overexpress H19 and miR-675 in BMSCs. The efficiency of lentiviral transduction was >90% and the expression of H19 and miR-675 was significantly upregulated by >8-fold (Supplementary Figure S1B), as described previously15. After induction to the adipogenic lineage, H19 and miR-675 significantly inhibited intracellular lipid accumulation as indicated by Oil red O staining (Fig. 2A). The mRNA and protein expression of the adipocyte-specific factors PPARγ, C/EBPα, and FABP4 was significantly reduced by the overexpression of H19 and miR-675 (Fig. 2B,C). To determine whether H19 acts on adipogenesis via miR-675, we designed a mutant H19 that carried mutation in the sequences of miR-675. The inhibition of adipocyte differentiation induced by H19 was abrogated through mutation of miR-675, as indicated by Oil red O staining and the expression of adipocyte-specific genes (Fig. 2A–C).
Figure 2. H19 and miR-675 inhibited adipogenic differentiation of BMSCs.
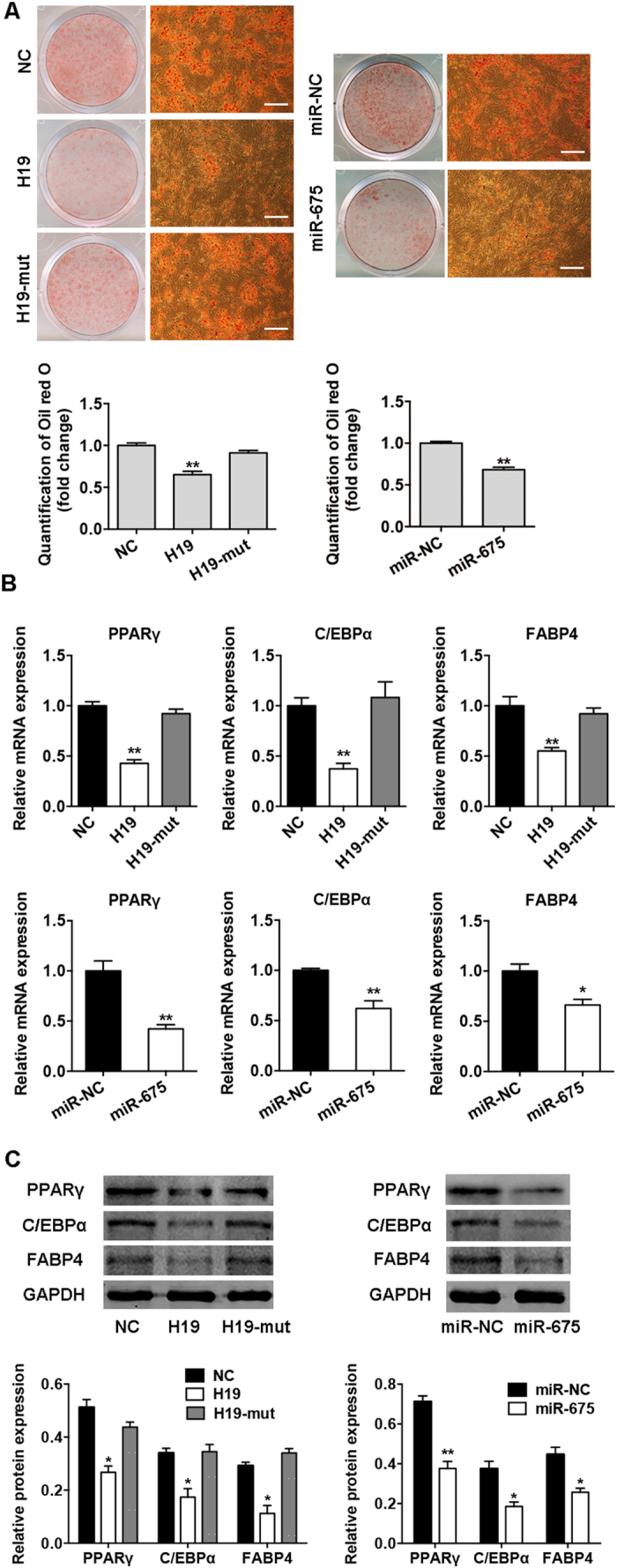
(A) Images of Oil red O staining in BMSCs transfected with H19, H19-mut, miR-675, or their control vectors (NC, miR-NC) on day 10 of differentiation. Scale bar, 500 μm. Histograms show quantification of Oil red O staining by spectrophotometry (normalized to control groups). (B) Relative mRNA expression of the adipogenic factors PPARγ, C/EBPα, and FABP4 measured by qRT-PCR on day 10 of adipogenic induction in BMSCs transfected with H19, H19-mut, miR-675, or their control vectors. (C) Western blot analysis of PPARγ, C/EBPα, FABP4, and GAPDH on day 10 of adipogenic induction in BMSCs transfected with H19, H19-mut, miR-675, or their control vectors. Histograms show quantification of the band intensities. Results are presented as mean ± SD (*P < 0.05, **P < 0.01).
Knockdown of H19 and miR-675 promotes adipocyte differentiation
To further confirm the effects of H19 and miR-675 on adipocyte differentiation, we knocked down the endogenous H19 and miR-675 in BMSCs using lentivirus transfection. To control for potential off-target shRNA effects, we used two different shRNA sequences targeting H19. The expression of H19 and miR-675 was significantly reduced by ~70% (Supplementary Figure S1B), as described previously15. The inhibition of H19 and miR-675 promoted adipocyte formation as shown by Oil red O staining (Fig. 3A), and knockdown of H19 and miR-675 also significantly upregulated the mRNA and protein expression of the adipogenic marker genes PPARγ, C/EBPα, and FABP4 (Fig. 3B,C).
Figure 3. Knockdown of H19 and miR-675 promoted adipogenic differentiation of BMSCs.
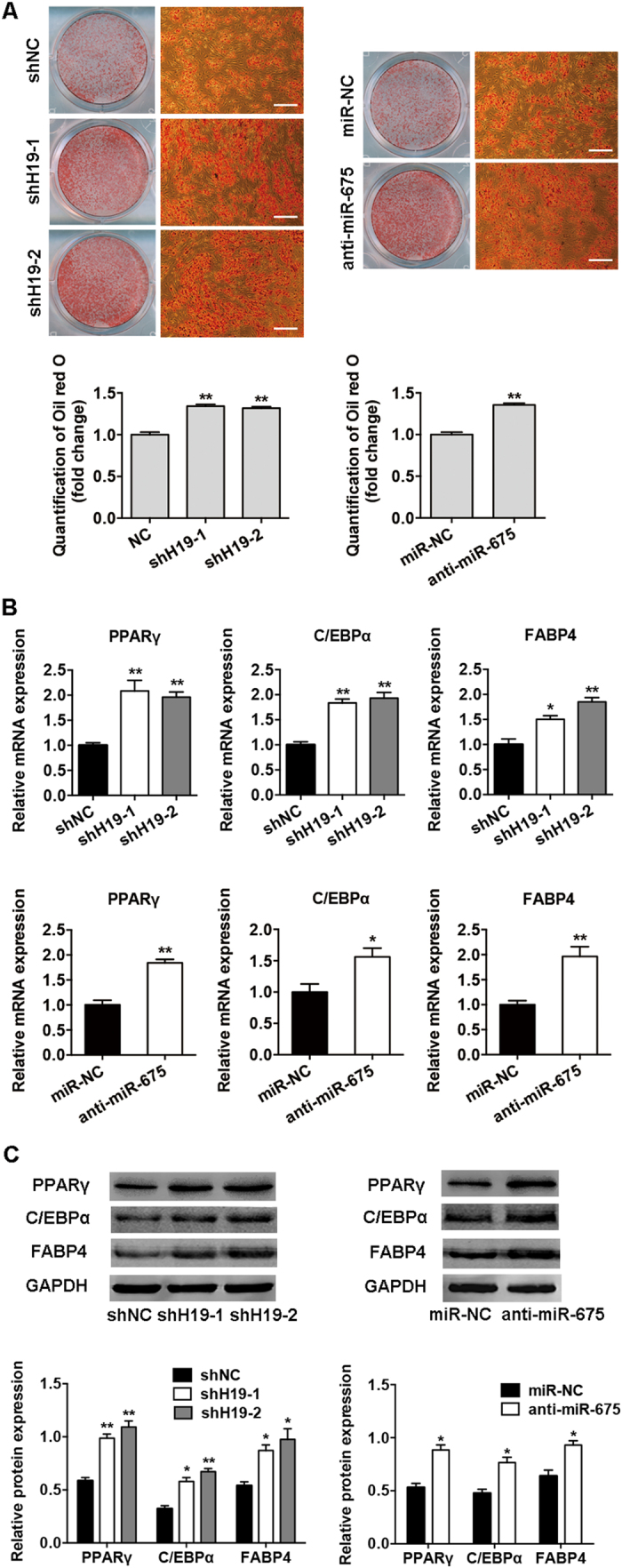
(A) Images of Oil red O staining in BMSCs transfected with shH19, anti-miR-675, or their scrambled vectors (shNC, miR-NC) on day 10 of differentiation. Scale bar, 500 μm. Histograms show quantification of Oil red O staining by spectrophotometry (normalized to control groups). (B) Relative mRNA expression of the adipogenic genes PPARγ, C/EBPα, and FABP4 on day 10 of differentiation in BMSCs transfected with shH19, anti-miR-675, or their scrambled vectors. (C) Western blot analysis (upper) and quantification (down) of protein expression of PPARγ, C/EBPα, FABP4, and GAPDH on day 10 of differentiation in BMSCs transfected with shH19, anti-miR-675, or their scrambled vectors. Data are presented as mean ± SD (*P < 0.05, **P < 0.01).
HDACs 4, 5, and 6 are directly targeted by miR-675
HDACs respond to signals that regulate a broad and complex array of physiological processes, including adipocyte differentiation and metabolism18. Our previous study showed that H19 and miR-675 reduce the expression of HDACs 4 and 515, but the mechanism remains unclear. So, we further assessed the expression of HDACs 1–6 in BMSCs overexpressing miR-675. Compared with the negative control, miR-675 substantially reduced the mRNA and protein levels of class II HDACs 4, 5, and 6, and slightly inhibited the expression of HDAC 1 (by ~20%), whereas no effect on the expression of HDACs 2 and 3 was found (Fig. 4A,B). Consistently, the ectopic overexpression of H19 downregulated the expression of HDACs 4, 5, and 6 in BMSCs, and the repression was relieved through mutation of miR-675 sequences (Fig. 4C).
Figure 4. H19 and miR-675 downregulated the expression of HDACs 4, 5, and 6.

(A) Quantification of mRNA expression of HDACs 1–6 measured by qRT-PCR in BMSCs overexpressing miR-675 relative to the miRNA control group (miR-NC). (B) Western blot analysis of HDACs 1–6 and GAPDH in BMSCs overexpressing miR-675 or miR-NC. Histograms show quantification of the band intensities. (C) Western blot analysis of HDACs 4–6 and GAPDH in BMSCs overexpressing H19, H19-mut, and the control vector (NC). Histograms show quantification of the band intensities. Results are presented as mean ± SD (*P < 0.05, **P < 0.01).
We then performed bioinformatic predictions of miRNA targets using RNA22 software19. The transcripts of HDACs 4–6 were found to contain several putative miR-675-binding sites (Supplementary Tables S1–S3). Among these, miR-675 possessed the maximum likelihood of binding to the 3′ untranslated region (UTR) of HDAC 4 (4183–4206 nt) (ΔG = −22.4 kcal/mol), the 3′UTR of HDAC 5 (4443–4465 nt) (ΔG = −19.1 kcal/mol), and the 3′UTR of HDAC 6 (3832–3853 nt) (ΔG = −19.1 kcal/mol) (Fig. 5A). Thus, we chose these target sites and separately ligated them to a luciferase reporter (Fig. 5B) to determine whether miR-675 directly targets these sites. The ectopic overexpression of miR-675 significantly inhibited luciferase activity in the HDAC 4, 5, and 6 constructs (Fig. 5C). Mutation of the miR-675-binding site abolished the inhibitory effect of miR-675 on the HDAC 4, 5, and 6 reporter activity (Fig. 5C).
Figure 5. miR-675 directly targeted the 3′ untranslated regions (UTRs) of HDAC 4, 5, and 6 transcriptions.

(A) Schematic diagrams of the wild-type and mutant binding sites of miR-675 located in the 3′UTRs of HDAC 4, 5, and 6 transcriptions. (B) Schematic showing the constructed luciferase reporter system containing the binding sites of miR-675. (C) Luciferase activity of 293T cells co-transfected with 100 nM miR-675 mimic or miRNA mimic control (mimic NC) and the luciferase constructs carrying the 3′UTR of HDAC 4, 5, or 6. Results are shown as mean ± SD (**P < 0.01).
Knockdown of HDACs 4, 5, and 6 inhibits adipocyte differentiation
To determine the role of HDACs 4, 5, and 6 in adipogenesis, we first measured their expression patterns during adipocyte differentiation. The expression of HDACs 4–6 was gradually upregulated with the highest expression on day 11 (Fig. 6A,B), which was inversely correlated with H19 expression during adipogenesis. We then used specific small-interfering RNAs (siRNAs) to suppress their endogenous expression. The specific siRNAs were transfected into BMSCs in growth medium, which was changed to adipogenic-differentiation medium on day 1. On day 3, the siRNAs were transfected again, and the cells were harvested on day 7. Successful knockdown of HDACs 4, 5, and 6 (Fig. 6D) inhibited the adipocyte differentiation of BMSCs as indicated by Oil red O staining (Fig. 6C), and the mRNA and protein expression of adipogenic genes PPARγ, C/EBPα, and FABP4 was also significantly downregulated (Fig. 6E,F).
Figure 6. Knockdown of HDACs 4–6 by RNA interference suppressed the adipogenic differentiation of BMSCs.

(A) Relative mRNA expression levels of HDACs 4–6 at the indicated time points during adipocyte differentiation. (B) Western blot analysis of HDACs 4–6 and GAPDH at the indicated time points during adipogenesis. Histograms show quantification of the band intensities. RNA and protein expression at each time point was compared to the group without adipocyte induction. (C) Images of Oil red O staining in BMSCs transfected with specific siRNAs on day 7 of adipogenic differentiation. Scale bar, 500 μm. Histograms show quantification of Oil red O staining normalized to the siNC group. (D) Quantification of mRNA expression of HDACs 4–6 measured by qRT-PCR after transfection with siHDAC4-6 or siNC. (E) Relative mRNA expression of the adipocyte-specific genes PPARγ, C/EBPα, and FABP4 measured by qRT-PCR on day 7 of adipogenic induction in BMSCs transfected with specific siRNAs. (F) Western blot analysis of PPARγ, C/EBPα, FABP4, and GAPDH on day 7 of adipogenic induction in BMSCs transfected with specific siRNAs. Histograms show quantification of the band intensities. Data are shown as mean ± SD (*P < 0.05, **P < 0.01).
Trichostatin A (TSA) reduces CTCF occupancy in the H19 imprinting control region (ICR) and downregulates H19 expression
H19 is an imprinted gene, and its expression is regulated by chromatin structure and epigenetic mechanisms20,21. Thus, we sought to determine the epigenetic effect of HDACs on the expression of H19 and miR-675. We treated BMSCs with TSA (400 nM), an HDAC inhibitor, for 3, 7, and 14 days. This treatment significantly suppressed H19 expression in a time-dependent manner. The expression of H19 was significantly reduced after 3 days, while further treatment caused further reduction and H19 expression was suppressed to a low level after 7 days (Fig. 7A). The expression of miR-675 was also downregulated after TSA treatment, and displayed a pattern similar to H19 expression (Fig. 7A).
Figure 7. HDAC inhibition reduced the binding of CTCF in the H19 imprinting control region (ICR).

(A) Left: relative expression of H19, as determined by qRT-PCR analysis, in BMSCs with or without TSA (400 nM) for 3, 7, and 14 days. Right: relative miR-675 expression in BMSCs with or without TSA (400 nM) for 3, 7, and 14 days. (B) Left: schematic of the H19 gene locus. Right: ChIP assay of CTCF occupancy in the H19 ICR. Soluble chromatin from BMSCs with or without TSA was immunoprecipitated with CTCF or IgG antibodies, and the immunoprecipitated DNA was analyzed by qRT-PCR. (C) Left: relative expression of H19, as determined by qRT-PCR analysis, in BMSCs transfected with siHDAC4-6 or siNC. Right: ChIP assay of CTCF occupancy in the H19 ICR from BMSCs transfected with siHDAC4-6 or siNC. Data are shown as mean ± SD (*P < 0.05, **P < 0.01, compared with non-treated or siNC group).
H19 expression is controlled by CTCF binding to the ICR upstream of the H19 gene. The CTCF protein promotes enhancer function at the H19 promoter (Fig. 7B)22,23. To determine whether TSA treatment leads to changes in the CTCF-binding status at the H19 ICR, we analyzed the levels of CTCF occupancy in this region using chromatin immunoprecipitation (ChIP). Indeed, endogenous CTCF directly interacted with the H19 ICR in BMSCs. And following TSA treatment, the occupancy of CTCF protein in the H19 ICR was reduced (Fig. 7B).
TSA inhibits the enzymatic HDAC activity without significant change of their expression (Supplementary Figure S2). To confirm the role of HDACs in H19 transcription, we applied siRNA gene knockdown studies and found that knockdown of HDACs 4–6 resulted in a phenotype similar to that observed after TSA treatment. H19 expression was reduced by HDAC 4–6 knockdown, and CTCF occupancy in H19 ICR was also moderately reduced with significant change in HDAC6 knockdown group (Fig. 7C).
Discussion
In this study, we demonstrated that lncRNA H19 and H19-derived miR-675 were significantly downregulated in BMSCs that were differentiating into adipocytes. Overexpression of H19 and miR-675 in BMSCs inhibited adipogenesis, while knockdown of their expression accelerated adipogenic differentiation. These phenomena indicate that H19 plays key roles in the process of BMSC adipogenesis. A few lncRNAs, such as ADINR24, PU.1-as25, and NEAT126, have been found to directly participate in the genetic control of adipogenic differentiation in adipose-derived stem cells. However, the stem cells from different tissues differ in their metabolic activity and ability to differentiate27,28. The roles of lncRNAs in BMSC adipogenesis are still largely unknown. It has been reported that when the balance between adipogenesis and osteogenesis is disturbed and BMSCs tend to differentiate into adipocytes rather than osteoblasts, marrow fat progressively accumulates and bone loss occurs4,5,6. Several studies have identified regulators of the switch between osteogenesis and adipogenesis in BMSCs, such as miR-1888, Maf 29, Ezh2, and Kdm6a30. Here, we demonstrated that H19 plays inhibitory roles in adipogenesis, while our previous study showed that H19 promotes osteoblast differentiation15. Thus, H19 seems to function in the switch of osteoblast and adipocyte differentiation of BMSCs. In a preclinical mouse model, BMSC-specific inhibition of miR-188 by intra-bone marrow injection of aptamer-antagomiR-188 increases bone formation and decreases bone marrow fat accumulation8. Our studies indicating a role of lncRNA H19 in the shift from adipogenesis to osteogenesis in BMSCs may also provides a potential target for treating fatty marrow and bone loss.
LncRNAs exert their functions via diverse mechanisms, including co-transcriptional regulation, modulation of gene expression, scaffolding of nuclear or cytoplasmic complexes, and pairing with other RNAs11. Another means by which lncRNAs acquire functionality is by acting as precursors of miRNAs31 or as sinks for pools of active miRNAs32 to regulate transcripts targeted by that set of miRNAs. Several previous studies have shown that miR-675 confers functionality on H1914,16,17,33. Our results addressing the inhibitory effect of miR-675 on adipogenic differentiation and lost of function in H19-mut group suggest that miR-675 is at least partially responsible for the inhibition of adipogenesis induced by H19. However, as lncRNAs act via diverse mechanisms, H19 may interact with other molecules involved in the complex biology of adipogenic differentiation. In this study, we used PPARγ, FABP4, and C/EBPα as adipocyte markers, all of which were significantly upregulated during adipocyte differentiation. However, a previous study has showed that FABP4 is induced by PPARγ but negatively regulates PPARγ activity in macrophages and adipocytes34. There is no consensus on the interrelationship between FABP4 and PPARγ, and our results were consistent with some previous studies concerning adipocyte differentiation24,35,36,37. The discrepancy of FABP4 expression in previous studies may be attributable to the different cells and tissues.
We found that miR-675 bound directly to the 3′UTRs of class II HDACs 4–6 and downregulated their mRNA and protein levels, while minor effects were found on the expression of class I HDACs 1, 2, and 3. The expression of HDACs 4–6 was inversely correlated with H19 and miR-675 expression during adipocyte differentiation. The downregulation of class II HDACs 4, 5, and 6 inhibited the adipogenic differentiation of BMSCs. Consistent with our results, previous studies have demonstrated the essential role of class II HDACs in adipocyte differentiation. Growing evidence shows that class I is directly involved in regulation of cell growth and apoptosis, whereas class II members regulate differentiation processes38. Treatment of mesenchymal stem cells with pan-HDAC inhibitors, class II-specific inhibitors, or specific siRNAs targeting HDACs, attenuates adipogenesis and reduces the expression of adipocyte markers following the induction of differentiation38,39,40,41,42. H19 and miR-675 inhibited the adipogenic differentiation of BMSCs at least partially through the downregulation of HDACs.
HDAC inhibition reduced CTCF enrichment in the H19 ICR and downregulated H19 expression. H19 has been reported to be regulated by chromatin structure and epigenetic mechanisms, including DNA methylation, CTCF insulator, and enhancer activity20,21. Correct positioning of nucleosomes within the ICR is required for CTCF stably binding, which promotes enhancer function at the H19 promoter22,23. TSA treatment reduced the occupancy of the CTCF protein in the H19 ICR and abolished the boundary activity of the ICR, thereby downregulating H19 expression. There is evidence that regional changes in acetylation within the promoter of H19 occur after TSA treatment, and H19 expression is significantly reduced43. And there is also evidence that HDACs from nuclear extracts are bound by the CTCF zinc-finger domain, and CTCF may function by associating with the HDAC complex44. From these results, it is not clear whether the reduced CTCF occupancy is causally related to the histone acetylation status of H19 promoter, but it implies the role of HDACs in this lncRNA transcription. Also, knockdown of HDACs 4–6 resulted in the similar but weaker phenotype compared to TSA treatment, indicating that other histone or non-histone acetylation may be involved. The precise molecular mechanism that underlies the regulation of specific histone variations needs further investigation.
In conclusion, H19 and H19-derived miR-675 inhibits the adipocyte differentiation of BMSCs through the epigenetic modulation of HDACs; miR-675 directly targets HDACs 4, 5, and 6; and the inhibition of HDACs reduces the levels of CTCF occupancy in the H19 ICR and reduces H19 expression (Fig. 8). Further research may elucidate whether H19 and miR-675 modulate the shift of cell lineage commitment of BMSCs in vivo and provide a potential therapeutic target for bone marrow adiposity.
Figure 8. Schematic of pathways involved in the inhibition of BMSC adipogenic differentiation by H19 and miR-675.

H19-derived miR-675 directly targeted HDACs 4, 5, and 6. TSA, an HDAC inhibitor, reduced the levels of CTCF occupancy in the H19 imprinting control region and reduced H19 expression.
Methods
Cell cultures and adipocyte differentiation
Primary BMSC lines from three donors were obtained from ScienCell (San Diego, CA, USA) and cultured at sub-confluent density in growth medium consisting of α-minimum essential medium supplemented with 10% fetal bovine serum and 1% antibiotics. All cell-based in vitro experiments were repeated in triplicate. For the adipocyte differentiation experiment, cells were allowed to become confluent for 1 day, and then cultured in standard growth medium supplemented with 50 nM insulin (Sigma-Aldrich, Saint Louis, MO, USA), 100 nM dexamethasone (Sigma-Aldrich), 0.5 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich), and 200 μM indomethacin (Sigma-Aldrich). The adipogenic medium was changed every 2 days, and cells were harvested at the indicated times. The 293T cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum and 1% antibiotics.
Lentivirus infection
Recombinant lentiviruses harboring full-length H19, mutant H19 (H19-mut), or H19-targeting sequences (shH19-1 and shH19-2) were constructed as described previously15. Site-directed mutagenesis of the H19 sequences was performed using the Site-Directed Mutagenesis Kit (SBS Genetech, Beijing, China). It carried mutation in the sequences of miR-675, as follows: TGG TGC GGA GAG GGC CCA CAG TG was changed to TCC ACG CGA GAG GGC CCA CAG TG. A vacant lentiviral vector (NC) and a scrambled non-targeting vector (shNC) were used as control group. Recombinant lentiviruses harboring miR-675 or miR-675 inhibitor sequences (anti-miR-675) and the control vector (miR-NC) were obtained from Integrated Biotech Solutions Co. (Shanghai, China). Transfection of the BMSCs was performed by exposing them to dilutions of the viral supernatant in the presence of polybrene (5 μg/ml) for 24 h.
Oil red O staining
The cells were washed with phosphate-buffered saline (PBS) and fixed in 10% formalin for 30 min. The cells were then rinsed with 60% isopropanol. Oil red O (0.3%, Sigma-Aldrich) was added and incubated for 10 min with gentle agitation. After staining, the cells were washed with distilled water to remove unbound dye, visualized by light microscopy, and photographed. For quantitative assessment, the Oil red O was eluted by 100% isopropanol and quantified by spectrophotometric absorbance at 520 nm against a blank (100% isopropanol).
RNA oligoribonucleotides and chemicals
A chemically-modified double-stranded miR-675 mimic and the corresponding miRNA mimic control (mimic NC) were obtained from RiboBio Co. (Guangzhou, China). siRNAs targeting HDAC 4, 5, and 6 transcripts (siHDAC4–6) and the corresponding siRNA control (siNC) were from Integrated Biotech Solutions Co. The sequences are listed in Supplementary Table S4. TSA (Sigma-Aldrich) was dissolved in dimethyl sulfoxide.
Transient transfection
Cells at 70–80% confluence were transfected with plasmids, miRNA mimics, or siRNAs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions as described previously45.
Reporter vectors
Predicted miR-675 target genes and their target binding sites were investigated using RNA22 software19. The reporter vectors were constructed by Integrated Biotech Solutions Co. Briefly, the 3′UTRs of HDAC 4, 5, and 6 mRNA, containing the predicted miR-675 binding sites, were synthesized and cloned into a modified version of pcDNA3.1(+) containing a firefly luciferase reporter gene (a gift from Professor Brigid L.M. Hogan, Duke University, Durham, NC, USA)46, at a position downstream of the luciferase reporter gene as described previously45. Site-directed mutagenesis of selected putative seeding-sequence regions was performed using the Site-Directed Mutagenesis Kit from SBS Genetech. All constructs were confirmed by DNA sequencing.
Dual luciferase reporter assay
Luciferase assays were performed as described previously45. Briefly, 293T cells grown in 48-well plates were transfected with 100 nM miR-675 mimic or control, 40 ng luciferase reporter, and 4 ng pRL-TK, a plasmid expressing Renilla luciferase (Promega, Madison, WI, USA) using Lipofectamine 2000 (Invitrogen). The Renilla and firefly luciferase activities were measured 24 h after transfection using the Dual Luciferase Reporter Assay System (Promega). All luciferase values were normalized to those of Renilla luciferase and expressed as fold-induction relative to the basal activity.
RNA isolation and qRT-PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions and then reverse-transcribed into cDNA using the cDNA Reverse Transcription Kit from Applied Biosystems (Foster City, CA, USA). qRT-PCR was conducted using SYBR Green Master Mix on the ABI Prism 7500 real-time PCR System (Applied Biosystems) as described45. The following thermal settings were used: 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The primers used for H19, miR-675, HDACs 1–6, PPARγ, C/EBPα, FABP4, U6 (internal control for miRNAs), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, internal control for mRNAs and lncRNAs) are listed in Supplementary Table S4. The data were analyzed using the 2−ΔΔCt relative expression method as described previously45.
Cell fractionation
Cytoplasmic and nuclear RNAs were fractionated using a Nuclei Isolation Kit (KeyGEN, Nanjing, China). Briefly, cells were harvested in lysis buffer, treated with Reagent A, incubated on ice for 15 min, followed by centrifugation at 4 °C. The pellet was then resuspended in lysis buffer followed by centrifugation. The supernatant was transferred to a new tube as the cytoplasmic fraction; the pellet was resuspended in Medium Buffer A and then added to a new tube with Medium Buffer B, followed by centrifugation at 4 °C. The supernatant was saved as the cytoplasmic fraction. The pellet was used as the nuclear fraction. RNA was extracted from both fractions using TRIzol.
Western blot analysis
Western blot analysis was performed as described previously45. Briefly, cells were harvested, washed with PBS, and lysed in RIPA buffer. Proteins were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Primary antibodies against PPARγ (Cell Signaling Technology, Beverly, MA, USA), HDACs 1–6 (Cell Signaling Technology), C/EBPα (HuaxingBio Science, Beijing, China), FABP4 (HuaxingBio Science), and GAPDH (Abcam, Cambridge, UK) were diluted 1:1,000. The intensities of the bands obtained by Western blot analysis were quantified using ImageJ software (http://rsb.info.nih.gov/ij/). The background was subtracted, and the signal of each target band was normalized to that of the GAPDH band.
ChIP assay
ChIP assays were performed using the EZ-Magna ChIP assay kit (Merck Millipore, Darmstadt, Germany) according to the manufacturer’s instructions. Briefly, cells were washed with PBS and cross-linked with 1% formaldehyde for 10 min. Chromatin was sonicated on ice to generate chromatin fragments of 500–2000 bp. Then the DNA-protein complexes were isolated using antibodies against CTCF (Cell Signaling Technology) or isotype IgG (Cell Signaling Technology). The protein/DNA complexes were then eluted and reverse cross-linked. Input control DNA or immunoprecipitated DNA was quantified by qRT-PCR, using SimpleChIP Human H19/IGF2 ICR Primers (Cell Signaling Technology). Relative enrichment was calculated as the amount of amplified DNA normalized to the input and relative to values obtained after IgG immunoprecipitation.
Statistical analysis
Statistical analyses were performed using SPSS version 16.0 (SPSS, Chicago, IL, USA). All data are expressed as mean ± standard deviation (SD). Differences between groups were analyzed using Student’s t-test. In cases of multiple-group testing, one-way analysis of variance was conducted. A two-tailed value of P < 0.05 was considered statistically significant.
Additional Information
How to cite this article: Huang, Y. et al. Long Non-coding RNA H19 Inhibits Adipocyte Differentiation of Bone Marrow Mesenchymal Stem Cells through Epigenetic Modulation of Histone Deacetylases. Sci. Rep. 6, 28897; doi: 10.1038/srep28897 (2016).
Supplementary Material
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81402235) and from the Peking University School of Stomatology Fund for Talented Young Investigators (PKUSS20140104).
Footnotes
Author Contributions Y.H. performed experiments and wrote the manuscript. Y.Z., C.J. and X.L. contributed to data collection and analysis. L.J. and W.L. supervised the work and edited the manuscript. All authors approved the final version of the manuscript.
References
- Georgiou K. R., Hui S. K. & Xian C. J. Regulatory pathways associated with bone loss and bone marrow adiposity caused by aging, chemotherapy, glucocorticoid therapy and radiotherapy. American journal of stem cells 1, 205–224 (2012). [PMC free article] [PubMed] [Google Scholar]
- Yeung D. K. et al. Osteoporosis is associated with increased marrow fat content and decreased marrow fat unsaturation: a proton MR spectroscopy study. Journal of magnetic resonance imaging 22, 279–285 (2005). [DOI] [PubMed] [Google Scholar]
- Pittenger M. F. et al. Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 (1999). [DOI] [PubMed] [Google Scholar]
- Greco E. A., Lenzi A. & Migliaccio S. The obesity of bone. Therapeutic advances in endocrinology and metabolism 6, 273–286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerman E. J., Teng K., Lipschitz D. A. & Lecka-Czernik B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging cell 3, 379–389 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pino A. M., Rosen C. J. & Rodriguez J. P. In osteoporosis, differentiation of mesenchymal stem cells (MSCs) improves bone marrow adipogenesis. Biological research 45, 279–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick J. S. RNA regulation: a new genetics? Nature reviews. Genetics 5, 316–323 (2004). [DOI] [PubMed] [Google Scholar]
- Li C. J. et al. MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. The Journal of clinical investigation 125, 1509–1522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J. Y. et al. MicroRNA-29a mitigates glucocorticoid induction of bone loss and fatty marrow by rescuing Runx2 acetylation. Bone 81, 80–88 (2015). [DOI] [PubMed] [Google Scholar]
- Hamam D. et al. microRNA-320/RUNX2 axis regulates adipocytic differentiation of human mesenchymal (skeletal) stem cells. Cell death & disease 5, e1499 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I. & Bartel D. P. lincRNAs: genomics, evolution, and mechanisms. Cell 154, 26–46 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista P. J. & Chang H. Y. Long noncoding RNAs: cellular address codes in development and disease. Cell 152, 1298–1307 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. & Tycko B. Monoallelic expression of the human H19 gene. Nature genetics 1, 40–44 (1992). [DOI] [PubMed] [Google Scholar]
- Cai X. & Cullen B. R. The imprinted H19 noncoding RNA is a primary microRNA precursor. Rna 13, 313–316 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Zheng Y., Jia L. & Li W. Long Noncoding RNA H19 Promotes Osteoblast Differentiation Via TGF-beta1/Smad3/HDAC Signaling Pathway by Deriving miR-675. Stem cells 33, 3481–3492 (2015). [DOI] [PubMed] [Google Scholar]
- Dey B. K., Pfeifer K. & Dutta A. The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes & development 28, 491–501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keniry A. et al. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nature cell biology 14, 659–665 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Peng J. & Jiang S. Role of histone acetyltransferases and histone deacetylases in adipocyte differentiation and adipogenesis. European journal of cell biology 93, 170–177 (2014). [DOI] [PubMed] [Google Scholar]
- Miranda K. C. et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 126, 1203–1217 (2006). [DOI] [PubMed] [Google Scholar]
- Sasaki H., Ishihara K. & Kato R. Mechanisms of Igf2/H19 imprinting: DNA methylation, chromatin and long-distance gene regulation. Journal of biochemistry 127, 711–715 (2000). [DOI] [PubMed] [Google Scholar]
- Gabory A., Jammes H. & Dandolo L. The H19 locus: role of an imprinted non-coding RNA in growth and development. BioEssays : news and reviews in molecular, cellular and developmental biology 32, 473–480 (2010). [DOI] [PubMed] [Google Scholar]
- Hark A. T. et al. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405, 486–489 (2000). [DOI] [PubMed] [Google Scholar]
- Bell A. C. & Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405, 482–485 (2000). [DOI] [PubMed] [Google Scholar]
- Xiao T. et al. Long Noncoding RNA ADINR Regulates Adipogenesis by Transcriptionally Activating C/EBPalpha. Stem cell reports 5, 856–865 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang W. J. et al. Knockdown of PU.1 AS lncRNA inhibits adipogenesis through enhancing PU.1 mRNA translation. Journal of cellular biochemistry 114, 2500–2512 (2013). [DOI] [PubMed] [Google Scholar]
- Cooper D. R. et al. Long Non-Coding RNA NEAT1 Associates with SRp40 to Temporally Regulate PPARgamma2 Splicing during Adipogenesis in 3T3-L1 Cells. Genes 5, 1050–1063 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco P., Robey P. G. & Simmons P. J. Mesenchymal stem cells: revisiting history, concepts, and assays. Cell stem cell 2, 313–319 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachon-Pena G. et al. Stromal stem cells from adipose tissue and bone marrow of age-matched female donors display distinct immunophenotypic profiles. Journal of cellular physiology 226, 843–851 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K. et al. Maf promotes osteoblast differentiation in mice by mediating the age-related switch in mesenchymal cell differentiation. The Journal of clinical investigation 120, 3455–3465 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemming S. et al. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem cells 32, 802–815 (2014). [DOI] [PubMed] [Google Scholar]
- Wilusz J. E., Sunwoo H. & Spector D. L. Long noncoding RNAs: functional surprises from the RNA world. Genes & development 23, 1494–1504 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmena L., Poliseno L., Tay Y., Kats L. & Pandolfi P. P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146, 353–358 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W. P. et al. Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis 31, 350–358 (2010). [DOI] [PubMed] [Google Scholar]
- Garin-Shkolnik T., Rudich A., Hotamisligil G. S. & Rubinstein M. FABP4 attenuates PPARgamma and adipogenesis and is inversely correlated with PPARgamma in adipose tissues. Diabetes 63, 900–911 (2014). [DOI] [PubMed] [Google Scholar]
- Kim Y. J., Hwang S. J., Bae Y. C. & Jung J. S. MiR-21 regulates adipogenic differentiation through the modulation of TGF-beta signaling in mesenchymal stem cells derived from human adipose tissue. Stem cells 27, 3093–3102 (2009). [DOI] [PubMed] [Google Scholar]
- Shi C. et al. miR-148a is Associated with Obesity and Modulates Adipocyte Differentiation of Mesenchymal Stem Cells through Wnt Signaling. Scientific reports 5, 9930 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. et al. miR-140-5p regulates adipocyte differentiation by targeting transforming growth factor-beta signaling. Scientific reports 5, 18118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebbioso A. et al. HDACs class II-selective inhibition alters nuclear receptor-dependent differentiation. Journal of molecular endocrinology 45, 219–228 (2010). [DOI] [PubMed] [Google Scholar]
- Lee S. et al. Histone deacetylase inhibitors decrease proliferation potential and multilineage differentiation capability of human mesenchymal stem cells. Cell proliferation 42, 711–720 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weems J. & Olson A. L. Class II histone deacetylases limit GLUT4 gene expression during adipocyte differentiation. The Journal of biological chemistry 286, 460–468 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weems J. C., Griesel B. A. & Olson A. L. Class II histone deacetylases downregulate GLUT4 transcription in response to increased cAMP signaling in cultured adipocytes and fasting mice. Diabetes 61, 1404–1414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. et al. Upregulation of miR-22 promotes osteogenic differentiation and inhibits adipogenic differentiation of human adipose tissue-derived mesenchymal stem cells by repressing HDAC6 protein expression. Stem cells and development 21, 2531–2540 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean V., O’Neill L., Sado T., Turner B. & Ferguson-Smith A. Relationship between DNA methylation, histone H4 acetylation and gene expression in the mouse imprinted Igf2-H19 domain. FEBS letters 488, 165–169 (2001). [DOI] [PubMed] [Google Scholar]
- Lutz M. et al. Transcriptional repression by the insulator protein CTCF involves histone deacetylases. Nucleic acids research 28, 1707–1713 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L. F. et al. Expression, regulation and roles of miR-26a and MEG3 in tongue squamous cell carcinoma. International journal of cancer. Journal international du cancer 135, 2282–2293 (2014). [DOI] [PubMed] [Google Scholar]
- Lu Y., Thomson J. M., Wong H. Y., Hammond S. M. & Hogan B. L. Transgenic over-expression of the microRNA miR-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Developmental biology 310, 442–453 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.