

Abstract
Aside from the CTNNB1 and adenomatous polyposis coli (APC) mutations, the genetic profile of pediatric aggressive fibromatosis (AF) has remained poorly characterized. The aim of this study was to shed more light on the mutational spectrum of pediatric AF, comparing it with its adult counterpart, with a view to identifying biomarkers for use as prognostic factors or new potential therapeutic targets. CTNNB1,APC,AKT1,BRAF TP53, and RET Sanger sequencing and next‐generation sequencing (NGS) with the 50‐gene Ion AmpliSeq Cancer Hotspot Panel v2 were performed on formalin‐fixed samples from 28 pediatric and 33 adult AFs. The prognostic value of CTNNB1,AKT1, and BRAF mutations in pediatric AF patients was investigated. Recurrence‐free survival (RFS) curves were estimated with the Kaplan–Meier method and statistical comparisons were drawn using the log‐rank test. In addition to the CTNNB1 mutation (64%), pediatric AF showed AKT1 (31%), BRAF (19%), and TP53 (9%) mutations, whereas only the CTNNB1 mutation was found in adult AF. The polymorphism Q472H VEGFR was identified in both pediatric (56%) and adult (40%) AF. Our results indicate that the mutational spectrum of pediatric AF is more complex than that of adult AF, with multiple gene mutations involving not only CTNNB1 but also AKT1 and BRAF. This intriguing finding may have clinical implications and warrants further investigations.
Keywords: AKT1; BRAF; CTNNB1, TP53; pediatric aggressive fibromatosis
Introduction
Aggressive fibromatosis (AF), also known as desmoid‐type fibromatosis, is a rare, locally aggressive nonmetastasizing fibroblastic proliferation with an incidence of 0.2–0.4 per 100,000 cases/year. It mainly affects young to middle‐aged adults, but children are also sometimes involved 1. Its growth may be fairly slow, lasting over several years and it can appear nearly anywhere in the body 2.
The pathogenesis of AF is not completely understood, although many factors can be implicated in its inception and progression, such as a genetic predisposition (familial adenomatous polyposis [FAP] and Gardner syndrome) 3.
The biological and clinical patterns of AF in children are generally considered the same as in adults and treatment recommendations are usually similar. Nowadays, a stepwise approach is generally adopted, involving “wait‐and‐see” and medical options, and local treatments that include nonmutilating surgery in selected cases 4, 5, 6, 7, 8, 9, 10.
As in adults, AF in children may occur sporadically or in association with FAP; in both settings, an activation of the Wnt/beta‐catenin signaling has been documented 11. Consistently, more than 60% of sporadic cases of pediatric AF show nuclear beta‐catenin expression and mutations of its gene CTNNB1, whereas syndromic pediatric AF exhibit adenomatous polyposis coli (APC) germline mutations 12.
The CTNNB1 somatic mutations seen in sporadic pediatric AF are clustered in two codons and are represented exclusively by three different types of single‐nucleotide substitution leading to the amino acid changes T41A, S45F, and S45P. Intriguingly, Bo et al. 12. found that the S45F mutation occurred mainly in recurrent pediatric cases, whereas the T41A mutation was most frequently seen in primary‐onset cases, suggesting that S45F mutations may represent a risk factor for recurrence, as in surgically treated adult AF 13.
Aside from the CTNNB1 and APC mutations, the genetic profile of pediatric AF has been poorly characterized. A few molecular analyses have been performed on adult AF using comparative genomic hybridization or high‐density single‐nucleotide polymorphism array, and shown loss of 5q (including the APC locus), 6q and 8p23, but such studies are completely lacking in pediatric AF 14, 15.
As in many other soft tissue sarcomas, it remains to be seen whether AF has the same biological background and the same clinical behavior when it occurs in children as opposed to adults.
The aim of this study was to gain some insight on the mutational spectrum of pediatric AF, comparing it with its adult counterpart, in an effort to identify potential biomarkers that might be used as prognostic factors or therapeutic targets.
Material and Methods
Patients and samples
Twenty‐eight primary pediatric AFs treated at the IRCCS Istituto Nazionale dei Tumori in Milan between 1990 and 2011 were considered in this study. The characteristics of the patients and their disease are described in Table 1. Eleven patients underwent biopsy, 13 nonradical surgery, and four radical surgery. Among patients underwent biopsy, four received chemotherapy (low‐dose chemotherapy, i.e., methotrexate and vinblastine in two patients and methotrexate and vinorelbine in three cases) immediately after diagnosis, while five patients received chemotherapy (methotrexate and vinorelbine in four cases and vincristine + actinomycin D + cyclophosphamide in one case) after evidence of progression.
Table 1.
Clinical, CTNNB1, and adenomatous polyposis coli Sanger sequencing data in pediatric sporadic aggressive fibromatosis
| N. | Sex/age | Type | Site | CTNNB1 | APC MCR |
|---|---|---|---|---|---|
| 1 | f/15 | Sporadic | Proximal lower limb | T41A | nd |
| 2 | m/3 | Sporadic | H&N | T41A | nd |
| 3 | f/12 | Sporadic | Proximal lower limb | T41A | nd |
| 4 | f/18 | Sporadic | Trunk + Abdomen | T41A | nd |
| 5 | m/2 | Sporadic | H&N | T41A | nd |
| 6 | f/6 | Sporadic | H&N | T41A | nd |
| 7 | f/0.2 | Sporadic | H&N | T41A | nd |
| 8 | m/18 | Sporadic | Trunk | T41A + S45F | nd |
| 9 | m/2 | Sporadic | Trunk + Distal upper limb | S45F | nd |
| 10 | m/9 | Sporadic | H&N | S45F | nd |
| 11 | m/18 | Sporadic | Distal lower limb | S45F | nd |
| 12 | f/12 | Sporadic | Proximal upper limb | S45F | nd |
| 13 | f/18 | Sporadic | Abdominal wall | S45F | nd |
| 14 | m/18 | Sporadic | Distal lower limb | S45P | nd |
| 15 | f/12 | Sporadic | Distal lower limb | S45P | nd |
| 16 | m/4 | Sporadic | Proximal lower limb | S45P | nd |
| 17 | f/18 | Sporadic | Proximal lower limb | S45P | nd |
| 18 | f/18 | Sporadic | Distal lower limb | S45P | nd |
| 19 | m/14 | Gardner syndrome | Abdominal wall | wt | wt |
| 20 | f/15 | Sporadic | Proximal lower limb | wt | wt |
| 21 | f/15 | Gardner syndrome | Abdomen | wt | wt |
| 22 | f/13 | Sporadic | Trunk | wt | E1544K |
| 23 | m/5 | Sporadic | Distal upper limb | wt | wt |
| 24 | m/7 | Sporadic | Trunk | wt | wt |
| 25 | f/9 | Sporadic | Distal upper limb | wt | wt |
| 26 | f/14 | Sporadic | Distal lower limb | wt | na |
| 27 | f/14 | Sporadic | Proximal lower limb | wt | na |
| 28 | 3 months | Sporadic | H&N | wt | wt |
f, female; m, male; wt, wild type; nd, not done; na, not assessable; H&N, head and neck; MCR, mutated cluster region.
The study sample was compared with a group of 33 surgically treated patients with sporadic adult AF, selected on the grounds of their known CTNNB1 mutational status, whose characteristics are detailed in Table 2.
Table 2.
Clinical, next‐generation sequencing (NGS), and Sanger sequencing data in adult aggressive fibromatosis
| N. | Sex/age (years) | Type | Site | CTNNB1Sanger | NGS | ||
|---|---|---|---|---|---|---|---|
| 1 | f/47 | Sporadic | Abdominal wall | T41A | CTNNB1 T41A | ||
| 2 | f/60 | Sporadic | Proximal upper limb | T41A | CTNNB1 T41A | ||
| 3 | m/27 | Sporadic | Proximal lower limb | T41A | CTNNB1 T41A | ||
| 4 | f/32 | Sporadic | Abdominal wall | S45F | CTNNB1 S45F | ||
| 5 | m/46 | Sporadic | Trunk | S45F | CTNNB1 S45F | ||
| 6 | f/36 | Sporadic | Abdominal wall | S45P | CTNNB1 S45P | ||
| 7 | f/35 | Sporadic | Chest | wt | No mutation | ||
| 8 | f/60 | Sporadic | Peritoneum | wt | No mutation | ||
| 9 | f/35 | Sporadic | Abdominal wall | wt | No mutation | ||
| 10 | f/65 | Sporadic | Proximal lower limb | wt | CTNNB1 T41A | ||
| Sanger sequencing | |||||||
| AKT1 | BRAF | TP53 | |||||
| 11 | m | Sporadic | Chest wall | T41A | wt | wt | wt |
| 12 | f/32 | Sporadic | Shoulder | T41A | wt | wt | wt |
| 13 | f/43 | Sporadic | Abdomen | T41A | wt | wt | wt |
| 14 | m/34 | Sporadic | Abdomen | T41A | wt | wt | wt |
| 15 | f/24 | Sporadic | Abdominal wall | T41A | wt | wt | wt |
| 16 | f/31 | Sporadic | Abdominal wall | T41A | wt | wt | wt |
| 17 | m/31 | Sporadic | Trunk | T41A | wt | wt | wt |
| 18 | f/49 | Sporadic | Mesentery | T41A | wt | wt | wt |
| 19 | f/28 | Sporadic | Abdomen | T41A | wt | wt | wt |
| 20 | f/31 | Sporadic | Abdominal wall | S45F | wt | wt | wt |
| 21 | f/42 | Sporadic | Proximal lower limb | S45F | wt | wt | wt |
| 22 | f/35 | Sporadic | Abdominal wall | S45F | wt | wt | wt |
| 23 | m/50 | Sporadic | Proximal lower limb | S45F | wt | wt | wt |
| 24 | f/41 | Sporadic | Abdominal wall | S45F | wt | wt | wt |
| 25 | f/31 | Sporadic | Abdominal wall | S45P | wt | wt | wt |
| 26 | f/37 | Sporadic | Abdominal wall | S45P | wt | wt | wt |
| 27 | f/34 | Sporadic | Abdominal wall | S45P | wt | wt | wt |
| 28 | m/32 | Sporadic | Abdomen | wt | wt | wt | wt |
| 29 | m/61 | Sporadic | Trunk | wt | wt | wt | wt |
| 30 | f/70 | Sporadic | Chest wall | wt | wt | wt | wt |
| 31 | f/36 | Sporadic | Proximal lower limb | wt | wt | wt | wt |
| 32 | f/39 | Sporadic | Abdominal wall | wt | wt | wt | wt |
| 33 | m/36 | Sporadic | Abdomen‐retroperitoneum | wt | wt | wt | wt |
| 70% | 0 | 0 | 0 | ||||
f, female; m, male; wt, wild type.
DNA extraction
Genomic DNA was extracted from 5 μm sections cut from formalin‐fixed and paraffin‐embedded (FFPE) tumor samples with the GeneRead DNA FFPE kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions.
Sanger sequencing
PCR was performed using specific primers for CTNNB1 (exon 3), BRAF (exon 15), and TP53 (exon 8), as described elsewhere 16. The primers used to amplify AKT1 (exon 2), RET (exon 11), and APC (the mutation cluster region in exon 15 where most of the mutations segregate) are listed in Table 3.
Table 3.
Primers used for Sanger sequencing
| Gene | Primers |
|---|---|
| Adenomatous polyposis coli (APC) EXON 15 PART 1 | Fw 5′‐TGAAGAGAAACGTCATGTGGA‐3′ |
| Rev 5′‐CTTTGCAAGTGGCAGCCTTT‐3′ | |
| APC EXON 15 PART 2 | Fw 5′‐AAGTGGTCAGCCTCAAAAGG‐3′ |
| Rev 5′‐GTGACACTGCTGGAACTTCG‐3′ | |
| APC EXON 15 PART 3 | Fw 5′‐GATCCTGTGAGCGAAGTTCC‐3′ |
| Rev 5′‐AACATGAGTGGGGTCTCCTG‐3′ | |
| APC EXON 15 PART 4 | Fw 5′‐ACACCCAAAAGTCCACCTGA‐3′ |
| Rev 5′‐ACTTCTCGCTTGGTTTGAGC‐3′ | |
| APC EXON 15 PART 5 | Fw 5′‐AGCTCAAACCAAGCGAGAAG‐3′ |
| Rev 5′‐TTTCCTGAACTGGAGGCATT‐3′ | |
| APC EXON 15 PART 6 | Fw 5′‐GCCTCCAGTTCAGGAAAATG‐3′ |
| Rev 5′‐ACAGGCAGCTGACTTGGTTT‐3′ | |
| APC EXON 15 PART 7 | Fw 5′‐AGCCCAGACTGCTTCAAAAT‐3′ |
| Rev 5′‐TGCCCCTCCTCTAACTCCTT‐3′ | |
| AKT1 EXON 2 | Fw 5′‐CGAAGGTCTGACGGGTAGAG‐3′ |
| Rev 5′‐CGCCACAGAGAAGTTGTTGA‐3′ | |
| RET EXON 11 | Fw 5′‐AGGGATAGGGCCTGGGCTTC‐3′ |
| Rev 5′‐GACCTGGTTCTCCATGGAGTC‐3′ |
The PCR products were submitted to direct sequencing using a 3500 DX Genetic Analyzer (Applied Biosystems, Foster City, CA) and then assessed with the ChromasPro software.
Next‐generation sequencing (NGS)
The 50‐gene Ion AmpliSeq Cancer Hotspot Panel v2 (Life Technologies) with the Ion‐Torrent™ Personal Genome Machine platform (Life Technologies, Foster city, CA, USA) was used in all experiments. This panel is designed to amplify 207 amplicons covering about 2800 COSMIC mutations from 50 oncogenes and tumor suppressor genes commonly mutated in human cancers (ABL1, AKT1, anaplastic lymphoma kinase (ALK), APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAS, GNAQ, HNF1A, HRAS, IDH1, IDH2, JAK2, JAK3, KDR/VEGFR2, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, VHL).
The Ion AmpliSeq Library Kit 2.0 (Life Technologies) was used to amplify 40 ng of DNA according to the manufacturer's instructions (MAN0006735 rev 5.0). The amplicons were partially digested with FuPa Reagent (Life Technologies), ligated to P1 and barcode adapters using DNA ligase. Barcoded libraries were purified using AMPure Beads XP (Beckman Coulter, Brea, CA, USA) and PCR‐amplified for a total of five cycles. After a second round of purification with AMPure Beads, the amplified libraries were measured for size and tested for quality using the Agilent Bio Analyzer DNA High Sensitivity kit (Agilent Technologies, Santa Clara, CA, USA), and quantified using the Qubit dsDNA HS kit (Invitrogen, Life Technologies, Carlsbad, CA, USA). Emulsion PCR and sample enrichment were completed using the IonOne Touch 2 instrument, the emulsion PCR master‐mix, the Ion sphere particles and Dynabeads MyOne Streptavidin C1 beads (Life Technologies), according to the manufacturer's instructions (Life Technologies). Sequencing was done on Ion Torrent PGM using Ion 316 Chips and the Ion‐PGM 200 sequencing kit (Life Technologies), according to the manufacturer's instructions.
PGM sequencing data were initially processed using the Ion Torrent platform‐specific Torrent Suite software to generate sequence readouts, align them on the reference genome Hg19, trim the adapter sequences, filter, and discard any poor signal‐profile readouts. Variant calling from the sequencing data was done with the Variant Caller plug‐in. The filtered variants were examined visually using the Integrative Genomic Viewer tool to check their quality level and confirm the variant's presence on both the “+” and the “−” strand. The resulting variants were then recorded using the Ensemble Variant Effect Predictor pipeline, COSMIC database, dbSNP database, and MyCancerGenome database (http://www.mycancergenome.org/). Information on the distribution of mutated genes in lung cancer was obtained from online catalogs of somatic mutations, such as Cosmic (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/), ClinVar of the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/clinvar/), the NCBI's Entrez Gene database (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?CMD=search&DB=gene), and the GeneCards of the Weizmann Institute of Science, Israel (http://www.genecards.org/).
Statistical analysis
The prognostic analyses focused on the effects of the CTNNB1, AKT1, and BRAF mutations in pediatric AF patients. The study endpoint was recurrence‐free survival (RFS), and the time to the event was calculated from the date of surgery to the date of relapse or death, or it was censored as at the date of the latest uneventful follow‐up. RFS curves were estimated using the Kaplan–Meier method and were compared statistically using the log‐rank test.
Results
Pediatric AF mutation analysis
β‐catenin and APC Sanger sequencing
Twenty‐eight patients (M/F: 18/10) with a median age of 12 years (range 2 months–18 years) were assessed. Two patients with tumors arising in the abdominal wall had Gardner syndrome. The whole blood sample of only one of these two syndromic patients was subjected few years ago to the mutational analysis of the whole APC gene that revealed a germline mutation, as expected. This analysis was not performed for the second patient during its follow‐up.
The 28 FFPE samples of pediatric AF were analyzed for CTNNB1 mutations by Sanger sequencing, which revealed this mutation in 18 (64%) cases (Table 1). The mutations found were as follows: ACC>GCC T41A (28%), TCT>TTT S45F (21%), and TCT>CCT S45P (18%) cases. Curiously, one case had both T41A and S45F mutations.
Since CTNNB1 and APC mutations are generally mutually exclusive in AF, as in other tumors, nine of the 10 remaining cases carrying a CTNNB1 wild type (wt) successfully underwent sequencing of APC. Since we analyzed FFPE samples, we sequenced exclusively the small part of the APC gene corresponding to the Mutated Cluster Region (MCR) and we found the missense mutation GAA>AAA E1544K in one tumor (10% of cases) (Table 1). The MCR of two patients with Gardner syndrome showed no mutation. Given that no peripheral blood lymphocytes were available for this retrospective study, it was impossible the sequencing of the whole APC to assess the occurrence of germline mutations in the two cases with Gardner syndrome, or to check for the absence of the E1544K APC mutation in normal tissue and thus exclude Gardner syndrome or FAP. The extended follow‐up nonetheless makes us confident of the clinical diagnosis of Gardner syndrome or sporadic AF.
NGS
To better characterize the genotype of pediatric AF, NGS was performed in 16 sporadic cases (six with T41A, two with S45F, four with S45P, one with T41A+S45F, and three with CTNNB1 wt) using a panel of 50 oncogenes and tumor suppressor genes.
In addition to confirming the findings for CTNNB1 and APC obtained by Sanger sequencing, NGS revealed further mutations in five cases (31%), including: AKT1 E17K (25%); BRAF V600E (12%) (Fig. 1A); TP53 R273H (6%); and RET V648I (6%) (Table 4).
Figure 1.
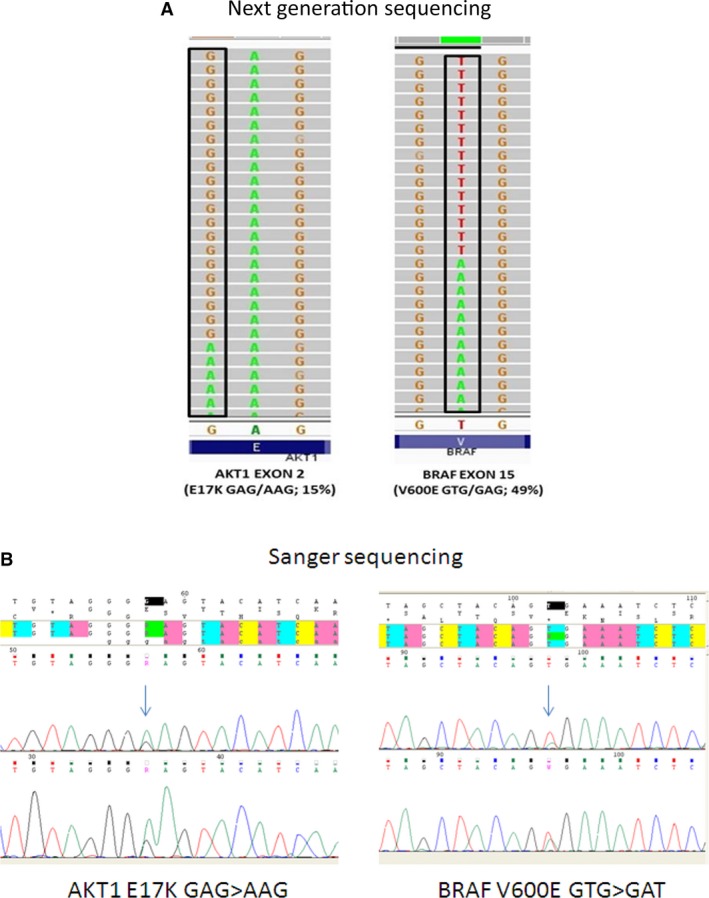
Mutational analysis in pediatric aggressive fibromatosis (AF). Next‐generation sequencing (NGS) (A) and Sanger sequencing (B) revealed AKT1 E17K and BRAF V600E mutations.
Table 4.
Next‐generation sequencing in pediatric aggressive fibromatosis
| Mutation | Polymorphism | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N. | CTNNB1 | AKT1 | BRAF | TP53 | RET | VEGFR2 | PI3KCA | TP53 | FGFR3 | KIT |
| 1 | T41A | E17K | V600E | R273H | I391M | |||||
| 2 | T41A | Q472H | ||||||||
| 3 | T41A | Q472H | ||||||||
| 4 | T41A | Q472H | I391M | |||||||
| 5 | T41A | P72R | ||||||||
| 6 | T41A | V600E | ||||||||
| 8 | T41A + S45F | Q472H | P72R | |||||||
| 9 | S45F | V648I | M541L | |||||||
| 11 | S45F | Q472H | P72R | |||||||
| 14 | S45P | E17K | Q472H | |||||||
| 15 | S45P | Q472H | ||||||||
| 17 | S45P | E17K | I391M | F386L | ||||||
| 18 | S45P | F386L | ||||||||
| 20 | wt | |||||||||
| 23 | wt | E17K | Q472H | I391M | ||||||
| 27 | wt | Q472H | I391M | M541L | ||||||
| 25% | 12% | 6% | 6% | 56% | 31% | 19% | 12% | 12% | ||
wt, wild type; Q472H, variant able to increase VEGFR2 phosphorylation after VEGFA stimulation in vitro [ 25 ]; M541L, KIT variant with controversial impact on fibromatosis response to imatinib 17, 18, 19; P72R, p53 variant with high apoptotic potential and cytoplasmic localization able to influence the cytosolic functions of the p53 protein 29.
We also found some known polymorphisms such as the Q472H VEGFR2 (56%), I391M PIK3CA (31%), P72R TP53 (19%), F386L FGFR3 (12%), and M541L c‐KIT (12%) variants (Table 4). In particular, the M541L variant of c‐KIT has already been described in adult AF, where an association with AF tumorigenesis is unlikely, and its impact on response to imatinib remains controversial 17, 18, 19.
AKT1, BRAF, TP53, and RET Sanger sequencing
All the mutations found by NGS were confirmed by Sanger resequencing. Given the sizable proportion of AKT1, BRAF, and TP53 mutations observed using NGS, these three genes were also sequenced directly in another 10 cases of pediatric AF. We were unable to analyze two cases because of the excessively small amount of tumor material available. AKT1 and BRAF mutations were found in 40% and 30% cases, respectively (Fig. 1B). TP53 analysis was successfully completed in only five cases, one of which (20%) carried the R273H mutation.
When the NGS and Sanger sequencing data were combined, after CTNNB1, AKT1 emerged as the most frequently mutated gene (31%) (Table 5). The missense mutation E17K was seen in 2/7 T41A (28%), 2/4 S45P (50%), and 4/9 wt cases (44%), but in none of the S45F mutant AF. The BRAF V600E activating mutation occurred in five cases (19%), that is in 3/7 T41A (43%) and 2/9 wt AF (22%), whereas the TP53 R273H mutation was observed in two (9%) cases. The APC mutation occurred in only one case (4%).
Table 5.
Combined Sanger and next‐generation sequencing results in pediatric aggressive fibromatosis
| Mutation | |||||
|---|---|---|---|---|---|
| N. | CTNNB1 | APC MCR | AKT1 | BRAF | TP53 |
| 1 | T41A | wt | E17Ka | V600Ea | R273Hd, nf |
| 2 | T41A | wt | wt | wt | wt |
| 3 | T41A | wt | wt | wt | wt |
| 4 | T41A | wt | wt | wt | wt |
| 5 | T41A | wt | wt | wt | wt |
| 6 | T41A | wt | wt | V600Ea | wt |
| 7 | T41A | ND | E17Ka | V600Ea | na |
| 8 | T41A + S45F | wt | wt | wt | wt |
| 9 | S45F | wt | wt | wt | wt |
| 10 | S45F | ND | wt | wt | na |
| 11 | S45F | wt | wt | wt | wt |
| 12 | S45F | wt | wt | wt | wt |
| 13 | S45F | ND | wt | wt | wt |
| 14 | S45P | wt | E17Ka | wt | wt |
| 15 | S45P | wt | wt | wt | wt |
| 16 | S45P | ND | na | na | na |
| 17 | S45P | wt | E17Ka | wt | wt |
| 18 | S45P | wt | wt | wt | wt |
| 19 | wt | wt | wt | V600Ea | R273Hd, nf |
| 20 | wt | wt | wt | wt | wt |
| 21 | wt | wt | wt | wt | na |
| 22 | wt | E1544K | E17Ka | wt | na |
| 23 | wt | wt | E17Ka | wt | wt |
| 24 | wt | wt | E17Ka | V600Ea | wt |
| 25 | wt | wt | na | na | na |
| 26 | wt | na | wt | wt | wt |
| 27 | wt | wt | wt | wt | wt |
| 28 | wt | wt | E17Ka | wt | na |
| 64% | 4% | 31% | 19% | 9% | |
Considering all the genes analyzed, there were 14 cases with one mutation, six with two, one with three, and one with four mutations, while five cases revealed none.
Adult AF mutational analysis
NGS
We analyzed 10 adult sporadic AFs selected on the grounds of their previously established CTNNB1 mutational status (three T41A, two S45F, one S45P, and four wt). No mutations other than CTNNB1 came to light (Table 2). Interestingly, NGS revealed the T41A mutation in a case classified as wt on Sanger sequencing (case number 10). The only polymorphism observed was Q472H VEGFR2 (40%).
AKT1, BRAF, and TP53 Sanger sequencing
Twenty‐three additional adult cases of sporadic AF (nine T41A; five S45F; three S45P, six wt) were submitted to Sanger sequencing of the three additional genes found mutated in pediatric AF (Table 2), and they all proved to be wt for AKT1, BRAF, and TP53.
Pooling the NGS and Sanger sequencing data, 36 sporadic adult AFs were analyzed successfully and no mutations other than CTNNB1 were found.
Recurrence‐free survival in pediatric AF
The prognostic analyses focused on the effects of β‐catenin, AKT1, and BRAF mutations in pediatric patients. With a median follow‐up of 93 months (range between 21–246 months), the estimated 3‐year and 5‐year RFS rates were 45.5%, for the series as a whole.
For the patients with CTNNB1 mutations, the estimated 3‐year and 5‐year RFS rates were: 20% at both time points for patients with the 45F mutation; 43% for patients with the 41A mutation; and 20% and 0% for patients with the 45P mutation. Patients who were wt for CTNNB1 mutations had a 3‐year and 5‐year RFS rate of 60% at both time points. (Fig. 2A). When wt and all CTNNB1 mutated patients were compared, the 3‐ and 5‐year RFS rates were 60% and 28%, respectively (P = 0.078; Fig. 2B).
Figure 2.
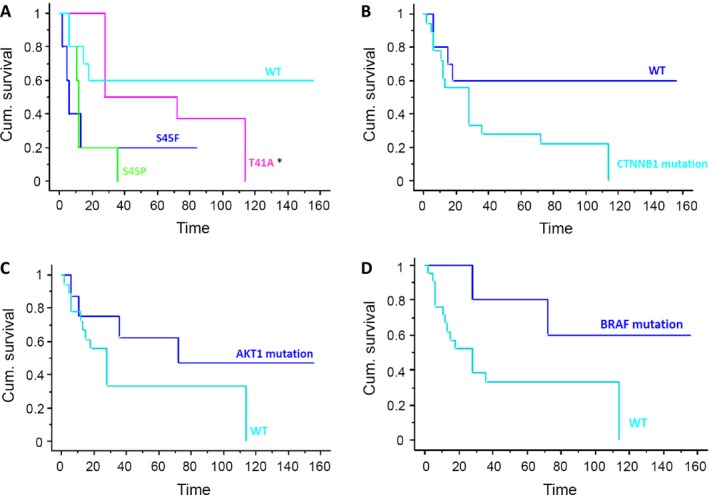
Recurrence‐free survival (RFS) curves in pediatric aggressive fibromatosis (AF). RFS is illustrated in patients by specific CTNNB1 mutations (A) CTNNB1 mutation versus wild type (B) AKT1 mutation (C) BRAF mutation (D).
Eight patients had a AKT1 mutation: the estimated 3‐year and 5‐year RFS rates for these patients were both 62.5%, as opposed to 30% for the AKT1 wt patients (P = 0.02469) (Fig. 2C).
Five patients had a BRAF mutation: the estimated 3‐year and 5‐year RFS rates for these patients were 80% and 60%, as opposed to 33% for the BRAF wt patients (P = 0.1058) (Fig. 2D).
Nine patients had Q472H VEGFR2 polymorphism: the estimated 3‐year and 5‐year RFS rates for H472 patients were both 22%, as opposed to 43% for Q472 patients (P = 0.38) (Fig. 3).
Figure 3.
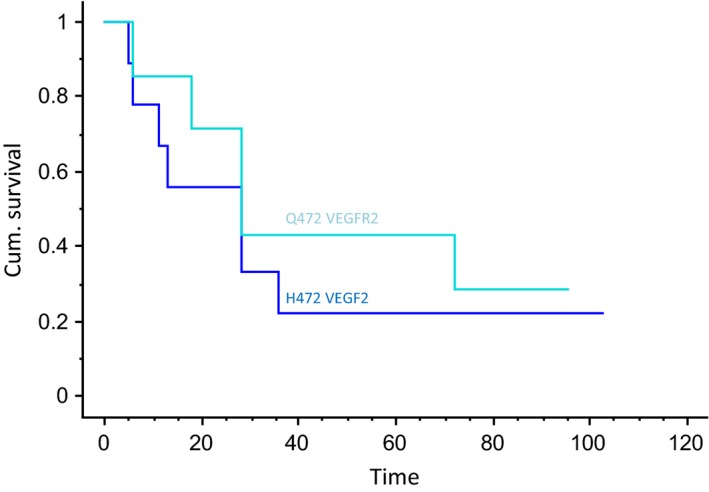
Recurrence‐free survival (RFS) curves in pediatric aggressive fibromatosis (AF) according to Q472H VEGFR2 polymorphism.
We reported that five patients did not have any aforementioned mutations (cases #20, 21, 25, 26, and 27): the estimated 3‐year and 5‐year RFS for this group was 40%, as opposed to 36% for mutated patients (P = 0.9).
Discussion
In our study, we obtained sequencing data on 50 cancer‐related genes in cases of pediatric AF and compared them with findings in the adult counterpart of AF, identifying significant differences mainly involving AKT1 and BRAF mutations in pediatric AF.
This retrospective series of 28 pediatric AFs was characterized for Wnt/beta‐catenin alterations. As in the adult counterpart 13, CTNNB1 genotyping revealed a particularly high rate (64%) of mutations in the two codons T41 and S45. In detail, the distribution of the types of mutation that we identified suggests a similar frequency between T41A (28%) and S45F (21%), unlike two previous studies reporting that T41A was the most common type of mutation seen in pediatric AF 11, 12, as in adult AF 13. We nonetheless confirmed that S45P (14%) was the least common mutation, in line with the literature 11. Based on our previous experience with 179 cases of completely resected, sporadic primary AF, judging from which tumors with the S45F mutation have a higher tendency for local recurrence 13, we explored the possible prognostic role of specific mutations in pediatric AF. We found no statistically significant difference in terms of RFS for wt cases, though there was evidence of a trend (Fig. 2B). Unfortunately, our results failed to demonstrate that S45F or any of the CTNBB1 mutations could be useful as a molecular indicator of a higher likelihood of recurrence (probably because the number of patients considered here was not enough to reveal differences relating to the different types of mutation). Similar data were obtained by Wang 11, whereas one study found the S45F mutation more frequent in recurrent pediatric AF than in primary tumors 12. This matter deserves further investigation to establish clearly whether these different types of mutation confer a different phenotype, and whether CTNNB1 mutational status might therefore be used as prognostic tool.
To broaden our limited molecular understanding of AF and seek potential new biomarkers for targeted therapy, we better characterized the pediatric AF genotype by performing NGS and Sanger sequencing, and comparing the results with findings in adult AF. To the best of our knowledge, this is the first report of the E17K AKT1 mutation (31%) occurring frequently in pediatric AF. This mutation was found in CTNNB1 wt and T41A and S45P mutant pediatric cases, but not in patients with the S45F mutation.
AKT1 kinase is one of the most frequently activated survival pathways in cancer, and the well‐studied E17K mutation in the pleckstrin homology (PH) domain of AKT1 stimulates downstream signaling and transforms cells, causing AKT1 activation by means of a rapid conformational drift that in turn increases the localization of the protein on the plasma membrane 20, 21. The presence of AKT1 mutation might have clinical implications based on the preclinical evidence that this mutant has been found resistant to AKT1/2 inhibitor, suggesting that pediatric AF patients probably would not benefit from this target treatment 20.
A small proportion of our CTNNB1 mutant or wt pediatric AF cases (19%) harbored the common BRAF V600E substitution that destabilizes this kinase's inactive conformation, leading to its constitutive activation. This finding deserves further investigation in order to explore BRAF as a potential therapeutic target in pediatric AF due to the current availability of several BRAF inhibitors.
We found a higher RFS rate for the AKT1 or BRAF mutated cases of no statistical significance.
The R273H TP53 mutation was observed in 2 (9%) cases. It is worth noting that—both in sporadic and FAP‐related adult AF—previous studies investigated p53 protein expression only by immunohistochemistry, showing that nuclear p53 immunostaining was associated with a higher risk of tumor recurrence 22, 23.
Interestingly, no AKT1, BRAF, or TP53 mutations were found in the series of 33 adult AFs that we selected on the grounds of their CTNNB1 status; this would suggest that such alterations are a distinctive feature of pediatric AF that should be validated on independent series.
One pediatric case (6%) also revealed the V648I mutation in the human RET oncogene, a rare substitution reported in medullary thyroid carcinoma, a setting in which it was judged to be “non‐transforming” in the light of elegant in silico and in vitro analyses, and this raised some doubts as to whether V648I might represent the driving force behind the tumor 24. Although in AF, we cannot say for sure whether the ability of V648I to induce a tumoral transformation is related to particular predisposing genetic conditions, this mutation seems to have a marginal role in pediatric AF.
In a sizable proportion of both pediatric (56%) and adult (40%) cases of AF, NGS also revealed the Q472H VEGFR2 polymorphism that was the only genetic variant capable of increasing protein phosphorylation after VEGFA stimulation in vitro 25. The effect of Q472H on VEGFR2 function may be due to a more efficient binding to the ligand. The role of VEGFR2 in carcinogenesis and tumor progression is related to its well‐known proangiogenic effects, and the PDGFR‐B expression that we observed by IHC in all pediatric AF cases may go in the same direction, along with COX expression (data not shown). These results are in line with our previous findings of PDGFRB expression/phosphorylation in frozen samples of adult AF, and of PDGFRA too (in some of them at least), probably mediated by their cognate ligands 26. Unfortunately, we were unable to test the activation of these two receptors in our pediatric AF series owing to a shortage of frozen material. We speculate that the PDGFR expression, along with the Q472H VEGFR2 variant and the mutation‐mediated BRAF activation, might have a proangiogenic role in pediatric AF, favoring a response to multitarget anti‐angiogenic agents found active in the adult counterpart 27, 28.
In conclusion, our results suggest that the mutational spectrum of pediatric AF is more complex than in adult AF, being rich in AKT1 and BRAF, as well as CTNNB1 gene mutations. These intriguing findings could have clinical implications and warrant further investigation on a new and larger cohort of patients. We hopefully expect that our study could represent the rationale for a future international collaboration between pediatric oncologists involved in a so rare and peculiar disease.
Conflict of Interest
None declared.
Acknowledgement
The authors thank the Italian Ministry of Health (n.265/RF‐2009 ‐1511297) that funded the research.
Cancer Medicine 2016; 5(6): 1204–1213
References
- 1. Goldblum, J. , and Fletcher J. A.. 2002. Desmoid‐type fibromatosis Pp. 83–84 in Fletcher C. D. M., Unni K. K., Mertens F., eds. World health organization classification of tumors: pathology and genetics of tumors of soft tissue and bone. International Agency for Research on Cancer Press, Lyon, France. [Google Scholar]
- 2. Reitamo, J. J. , Scheinin T. M., and Hayry P.. 1996. The desmoid syndrome: new aspects in the cause, pathogenesis and treatment of desmoid tumors. Am. J. Surg. 151:230–237. [DOI] [PubMed] [Google Scholar]
- 3. Giarola, M. , Wells D., Mondini P., et al. 1998. Mutations of adenomatous polyposis coli (APC) gene are uncommon in sporadic desmoid tumours. Br. J. Cancer 78:582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dormans, J. P. , Spiegel D., Meyer J., et al. 2001. Fibromatosis in childhood: the desmoid/fibromatosis complex. Med. Pediatr. Oncol. 37:126–131. [DOI] [PubMed] [Google Scholar]
- 5. Skapek, S. X. , Hawk B. J., Hoffer F. A., et al. 1998. Combination chemotherapy using vinblastine and methotrexate for the treatment of progressive desmoid tumor in children. J. Clin. Oncol. 16:3021–3027. [DOI] [PubMed] [Google Scholar]
- 6. Lackner, H. , Urban C., Benesch M., et al. 2004. Multimodal treatment of children with unresectable or recurrent desmoid tumors: an 11‐year longitudinal observational study. J. Pediatr. Hematol. Oncol. 26:518–522. [DOI] [PubMed] [Google Scholar]
- 7. Lackner, H. , Urban C., Kerbl R., Schwinger W., and Beham A.. 1997. Noncytotoxic drug therapy in children with unresectable desmoid tumors. Cancer 80:334–340. [DOI] [PubMed] [Google Scholar]
- 8. Meazza, C. , Bisogno G., Gronchi A., et al. 2010. Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer 116:233–240. [DOI] [PubMed] [Google Scholar]
- 9. Bonvalot, S. , Eldweny H., Haddad V., et al. 2008. Extra‐abdominal primary fibromatosis: aggressive management could be avoided in a subgroup of patients. Eur. J. Surg. Oncol. 34:462–468. [DOI] [PubMed] [Google Scholar]
- 10. Fiore, M. , Rimareix F., Mariani L., et al. 2009. Desmoid‐type fibromatosis: a front‐line conservative approach to select patients for surgical treatment. Ann. Surg. Oncol. 6:2587–2593. [DOI] [PubMed] [Google Scholar]
- 11. Wang, W. L. , Nero C., Pappo A., Lev D., Lazar A. J., and Lopez‐Terrada D.. 2012. CTNNB1 genotyping and APC screening in pediatric desmoid tumors: a proposed algorithm. Pediatr. Dev. Pathol. 15:361–367. [DOI] [PubMed] [Google Scholar]
- 12. Bo, N. , Wang D., Wu B., Chen L., Ruixue M. A.. 2012. Analysis of β‐catenin expression and exon 3 mutations in pediatric sporadic aggressive fibromatosis. Pediatr. Dev. Pathol. 15:173–178. [DOI] [PubMed] [Google Scholar]
- 13. Colombo, C. , Miceli R., Lazar A. J., et al. 2013. CTNNB1 45F mutation is a molecular prognosticator of increased postoperative primary desmoid tumor recurrence: an independent, multicenter validation study. Cancer 119:3696–3702. [DOI] [PubMed] [Google Scholar]
- 14. Salas, S. , Chibon F., Noguchi T., Terrier P., and Ranchere‐Vince D.. 2010. Molecular characterization by array comparative genomic hybridization and DNA sequencing of 194 desmoid tumors. Genes Chromosomes Cancer 49:560–568. [DOI] [PubMed] [Google Scholar]
- 15. Erben, P. , Nowak D., Sauer C., et al. 2012. Molecular analysis of desmoid tumors with a high‐density single‐nucleotide polymorphism array identifies new molecular candidate lesions. Onkologie 35:684–689. [DOI] [PubMed] [Google Scholar]
- 16. Pelosi, G. , Gasparini P., Cavazza A., et al. 2012. Multiparametric molecular characterization of pulmonary sarcomatoid carcinoma reveals a nonrandom amplification of anaplastic lymphoma kinase (ALK) gene. Lung Cancer 77:507–514. [DOI] [PubMed] [Google Scholar]
- 17. Grabellus, F. , Worm K., Sheu S. Y., Siffert W., Schmid K. W., and Bachmann H. S.. 2011. The prevalence of the c‐kit exon 10 variant, M541L, in aggressive fibromatosis does not differ from the general population. J. Clin. Pathol. 64:1021–1024. [DOI] [PubMed] [Google Scholar]
- 18. Tamborini, E. , Negri T., Miselli F., Lagonigro M. S., Pricl S., and Pilotti S.. 2006. Re: response of a KIT‐positive extra‐abdominal fibromatosis to imatinib mesylate and KIT genetic analysis. J. Natl. Cancer Inst. 98:1583–1584. [DOI] [PubMed] [Google Scholar]
- 19. Gonçalves, A. , Monges G., Yang Y., et al. 2006. Response of a KIT‐positive extra‐abdominal fibromatosis to imatinib mesylate and KIT genetic analysis. J. Natl. Cancer Inst. 98:562–563. [DOI] [PubMed] [Google Scholar]
- 20. Carpten, J. D. , Faber A. L., Horn C., et al. 2007. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448:439–444. [DOI] [PubMed] [Google Scholar]
- 21. Kumar, A. , and Purohit R.. 2013. Cancer associated E17K mutation causes rapid conformational drift in AKT1 pleckstrin homology (PH) domain. PLoS One 8:e64364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Colombo, C. , Foo W. C., Whiting D., et al. 2012. FAP‐related desmoid tumors: a series of 44 patients evaluated in a cancer referral center. Histol. Histopathol. 27:641–649. [DOI] [PubMed] [Google Scholar]
- 23. Gebert, C. , Hardes J., Kersting C., et al. 2007. Expression of beta catenin and p53 are prognostic factors in deep aggressive fibromatosis. Histopathology 50:491–497. [DOI] [PubMed] [Google Scholar]
- 24. Cosci, B. , Vivaldi A., Romei C., et al. 2001. In silico and in vitro analysis of rare germline allelic variants of RET oncogene associated with medullary thyroid cancer. Endocr. Relat. Cancer 18:603–612. [DOI] [PubMed] [Google Scholar]
- 25. Glubb, D. M. , Cerri E., Giese A., et al. 2011. Novel functional germline variants in the VEGF receptor 2 gene and their effect on gene expression and microvessel density in lung cancer. Clin. Cancer Res. 17:5257–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Signoroni, S. , Frattini M., Negri T., et al. 2007. Cyclooxygenase‐2 and platelet‐derived growth factor receptors as potential targets in treating aggressive fibromatosis. Clin. Cancer Res. 13:5034–5054. [DOI] [PubMed] [Google Scholar]
- 27. Martin‐Liberal, J. , Benson C., McCarty H., Thway K., Messiou C., and Judson I.. 2013. Pazopanib is an active treatment in desmoid tumour/aggressive fibromatosis. Clin. Sarcoma. Res 3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jo, J. C. , Hong Y. S., Kim K. P., et al. 2014. A prospective multicenter phase II study of sunitinib in patients with advanced aggressive fibromatosis. Invest. New Drugs 32:369–376. [DOI] [PubMed] [Google Scholar]
- 29. Panni, S. , Salvioli S., Santonico E., et al. 2015. The adapter protein CD2AP binds to p53 protein in the cytoplasm and can discrimate its polymorphic variants P72R. J. Biochem. 157:101–111. [DOI] [PubMed] [Google Scholar]
- 30. Poeta, M. L. , Manola J., Goldwasser M. A., et al. 2007. TP53 mutations and serviva in squamous‐cell carcinoma of the head and neck. N. Engl. J. Med. 357:2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kato, S. , Han S. Y., Liu W., et al. 2003. Understanding the function‐structure and function‐mutation relationships of p53 tumor suppressor protein by high‐resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 100:8424–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]