

Abstract
Neuromyelitis optica (NMO) is a rare disease, common in white females and rarely reported in Hispanic males. It is usually associated with recurrent demyelinating spectrum that is autoimmune in nature. The diagnosis is usually confirmed by antibody biomarkers; however, they can be negative and lead to more dilemma in diagnosis. Furthermore, the course of disease and prognosis are different in seronegative as compared to seropositive NMO. Treatment is similar in both subgroups with new approaches under investigation for seronegative NMO patients. We present an interesting case of a 37-year-old Hispanic male who presented with sudden onset of lower extremity weakness, numbness, blurry vision, and urinary retention. Magnetic resonance imaging (MRI) of the thoracic spine showed multiphasic demyelinating process involving the thoracic spinal cord. His brain MRI also revealed changes suggesting optic neuritis. The patient met the criteria for diagnosis of NMO by having optic neuritis and myelitis by imaging studies despite having negative aquaporin-4 antibodies (AQP4-Ab). His condition improved after plasma exchange. NMO can be difficult to distinguish from acute multiple sclerosis in the early stages of the disease. Having AQP4-Ab testing is important for diagnosis with imaging studies; however, negative antibody results cannot exclude the diagnosis, but rather group it in seronegative subtype. Ongoing studies and research suggest that seronegative NMO might have a different pathophysiology, manifestation, and prognosis.
Key Words: Neuromyelitis optica, Devic syndrome, Recurrent optic neuritis, Aquaporin-4 antibodies
Introduction
Neuromyelitis optica (NMO) is a rare, female predominance disease associated with recurrent autoimmune and demyelinating spectrum with cardinal manifestations. The diagnosis of NMO requires the following criteria: presence of optic neuritis, myelitis, involvement of spinal cord lesions in 3 or more segments by magnetic resonance imaging (MRI), initial MRI of the brain not meeting the criteria of multiple sclerosis, and seropositive aquaporin-4 antibodies (AQP4-Abs) [1]. Highly specific biomarker antibodies targeting the water channel protein AQP4 provided an insight into the immunopathology of NMO, which also served to predict relapse ratio [1,2]. There are many patients who meet the criteria for NMO with seronegative AQP4-Abs. An interesting finding is that this group showed no female predominance, a higher proportion of Caucasian ethnicity, monophasic disease, and younger age at the time of presentation [1]. In addition, seronegative NMO patients also present with simultaneous optic neuritis and myelitis at the beginning of the disease, less severe visual impairment, and common MRI finding consistent with deep gray matter involvement [2,3,4]. There are indications that seropositive and seronegative patients might differ with regard to clinical presentation or prognosis. According to the Marignier series, all seronegative patients had relapsed; however, they also had milder visual impairment [3]. Seropositive patients in another study were found to have severe outcome, specifically disability [5]. The clinical spectrum of NMO as defined by Wingerchuk et al. [1] includes cases of simultaneous optic neuritis and myelitis, in which the two events do not develop simultaneously.
Case Presentation
We present a 37-year-old Hispanic male with a past medical history of 2 episodes of bilateral optic neuritis and new onset of lower extremity weakness. He had previous admission with working diagnosis of multiple sclerosis and was treated with steroids.
Beside the lower extremity weakness, other signs and symptoms included numbness, blurry vision, and urinary retention. Vital signs were unremarkable except for mild elevated blood pressure of 153/98. Neurological exam revealed right lower extremity muscle power of 3/5, and 2/5 in the left lower extremity. Decrease sensation (including vibratory, thermal) was below the 7th thoracic vertebrae. Hyporeflexia was noted in both upper extremities, and Babinski's sign was positive bilaterally, with clonus at the right ankle.
MRI of thoracic spine demonstrated multiphasic demyelinating process involving the thoracic spinal cord (fig. 1, fig. 2). MRI of the brain also showed nonspecific changes with evidence in T1 imagining as noticed in fig. 3.
Fig. 1.
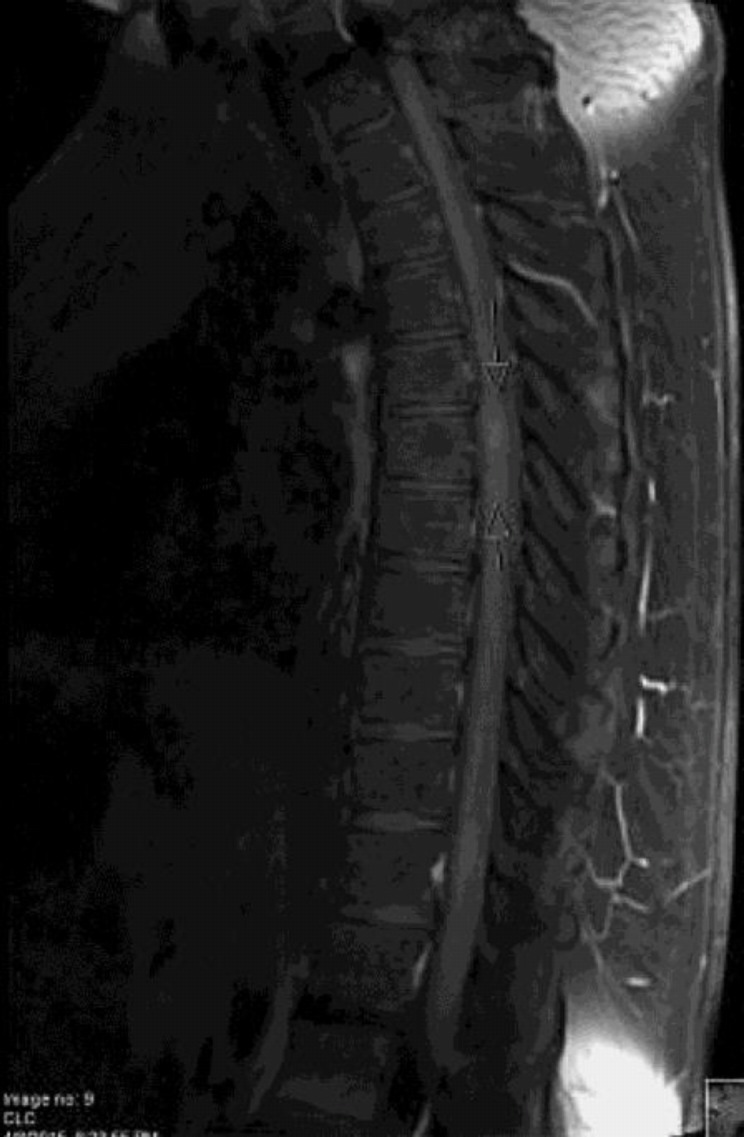
The T5-T7 lesion demonstrates imaging evidence of active inflammation.
Fig. 2.

T spine STIR imaging showing changes between the 2 arrows.
Fig. 3.

Brain T1 with contrast.
Multiple sclerosis was still in the differential diagnosis; however, MRI of the brain showed hyperintense signal and faint hyperenhancement within the intracanalicular and intracranial segments of the optic nerves bilaterally (fig. 4). Lumbar puncture was negative for oligoclonal bands ruling out multiple sclerosis.
Fig. 4.

Bilateral optic neuritis, multiple scattered foci involving the subcortical supratentorial white matter, sparing the corpus collosum, collosal septal junction, brainstem and cerebellum.
Our patient met the diagnostic criteria for optic NMO by having two of the absolute criteria (optic neuritis and myelitis) and two of the supportive criteria (brain MRI not meeting criteria for multiple sclerosis diagnosis, and longitudinally extensive transverse myelitis on T2-weighted imaging on MRI). Further workup was negative for AQP4-Ab. The patient was started on methylprednisolone 250 mg every 6 h for 5 days. Due to the severity of NMO with no improvement after steroids, the decision was made to start plasma exchange. After two treatment sections, the patient's condition improved and he regained his lower extremity strength. He completed five cycles of plasma exchange and was discharged with steroid therapy with follow-up in neurology clinic.
Discussion
NMO, also known as Devic's disease, is a rare condition that can be associated with systemic autoimmune disorders such as Sjögren's syndrome, systemic lupus erythematosus, thyroid autoimmune diseases, and myasthenia gravis [6]. It was previously considered a variant of multiple sclerosis but the discovery of AQP4-Ab grouped it as a separate diagnosis with different entity [1,2,3]. Imaging studies usually reveal longitudinally extensive transverse myelitis with T1 hypointensity, periependymal brainstem changes, and perivenous white matter lesions. It was also estimated that there is a correlation between segmental length of spinal cord lesions and expected disability [7]. The traditional treatment for acute attacks in seropositive NMO is intravenous corticosteroids with or without plasma exchanges achieving almost 60% recovery rate. NMO is highly associated with relapse rate (high AQP4-Ab titers), requiring the need to use a maintenance immunosuppressive therapy. Azathioprine with or without low-dose oral steroids is considered reasonably effective in reducing the frequency of attacks. Treatment for seronegative NMO is similar to the seropositive patients; however, recent studies showed rituximab to be an effective therapy for resistant or seronegative NMO patients and some consider it to be the first line of therapy [8,9].
Conclusion
NMO should be thought of in the differential diagnosis in any patient presenting with transverse myelitis and or optic neuritis regardless of the gender and race. Negative AQP4-Ab cannot exclude the diagnosis, but rather should trigger further evaluation of pathophysiology, manifestation, and prognosis. Treatment is similar between seropositive and seronegative NMO; however, new studies showed promising results using rituximab as an alternative first-line therapy. Future studies and research are still needed to evaluate the prognostic outcome of both groups.
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
None of the authors have any conflicts of interest to disclose.
References
- 1.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–815. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- 2.Pittock SJ, Lucchinetti CF. Neuromyelitis optica and the evolving spectrum of autoimmune aquaporin-4 channelopathies: a decade later. Ann N Y Acad Sci. 2016;1366:20–39. doi: 10.1111/nyas.12794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marignier R, Bernard-Valnet R, Giraudon P, Collongues N, Papeix C, Zéphir H, et al; NOMADMUS Study Group. Aquaporin-4 antibody-negative neuromyelitis optica: distinct assay sensitivity-dependent entity. Neurology. 2013;80:2194–2200. doi: 10.1212/WNL.0b013e318296e917. [DOI] [PubMed] [Google Scholar]
- 4.Fujihara K, Leite MI. Seronegative NMO: a sensitive AQP4 antibody test clarifies clinical features and next challenges. Neurology. 2013;80:2176–2177. doi: 10.1212/WNL.0b013e318296ea22. [DOI] [PubMed] [Google Scholar]
- 5.Akman-Demir G, Tüzün E, Waters P, Içöz S, Kürtüncü M, Jarius S, et al. Prognostic implications of aquaporin-4 antibody status in neuromyelitis optica patients. J Neurol. 2011;258:464–470. doi: 10.1007/s00415-010-5780-4. [DOI] [PubMed] [Google Scholar]
- 6.Wingerchuk DM, Weinshenker BG. The emerging relationship between neuromyelitis optica and systemic rheumatologic autoimmune disease. Mult Scler. 2012;18:5–10. doi: 10.1177/1352458511431077. [DOI] [PubMed] [Google Scholar]
- 7.Fan Y, Shan F, Lin SP, Long Y, Liang B, Gao C, et al. Dynamic change in magnetic resonance imaging of patients with neuromyelitis optica. Int J Neurosci. 2015;126:448–454. doi: 10.3109/00207454.2015.1055356. [DOI] [PubMed] [Google Scholar]
- 8.Jeong IH, Park B, Kim SH, Hyun JW, Joo J, Kim HJ. Comparative analysis of treatment outcomes in patients with neuromyelitis optica spectrum disorder using multifaceted endpoints. Mult Scler. 2016;22:329–339. doi: 10.1177/1352458515587752. [DOI] [PubMed] [Google Scholar]
- 9.Zéphir H, Bernard-Valnet R, Lebrun C, Outteryck O, Audoin B, Bourre B, et al. Rituximab as first-line therapy in neuromyelitis optica: efficiency and tolerability. J Neurol. 2015;262:2329–2335. doi: 10.1007/s00415-015-7852-y. [DOI] [PubMed] [Google Scholar]