

Abstract
Resident bacteria in the densely populated human intestinal tract must efficiently compete for carbohydrate nutrition. The Bacteroidetes, a dominant bacterial phylum in the mammalian gut, encode a plethora of discrete polysaccharide utilization loci (PULs) that are selectively activated to facilitate glycan capture at the cell surface. The most well-studied PUL-encoded glycan-uptake system is the starch utilization system (Sus) of Bacteroides thetaiotaomicron. The Sus includes the requisite proteins for binding and degrading starch at the surface of the cell preceding oligosaccharide transport across the outer membrane for further depolymerization to glucose in the periplasm. All mammalian gut Bacteroidetes possess analogous Sus-like systems that target numerous diverse glycans. In this review, we discuss what is known about the eight Sus proteins of B. thetaiotaomicron that define the Sus-like paradigm of nutrient acquisition that is exclusive to the Gram-negative Bacteroidetes. We emphasize the well-characterized outer membrane proteins SusDEF and the α-amylase SusG, each of which have unique structural features that allow them to interact with starch on the cell surface. Despite the apparent redundancy in starch-binding sites among these proteins, each has a distinct role during starch catabolism. Additionally, we consider what is known about how these proteins dynamically interact and cooperate in the membrane and propose a model for the formation of the Sus outer membrane complex.
Keywords: Starch, Bacteroides thetaiotaomicron, Starch utilization system, Sus, Gut microbiota
Introduction
The consortium of bacteria that inhabit the mammalian gastrointestinal tract has a profound influence on host development and health [1–4]. A notable function of these microbes is the digestion and fermentation of both endogenous (i.e., host derived) and dietary carbohydrates into short chain fatty acids that offer a physiological benefit to the host [2, 5]. The bacteria that have adapted to persist and thrive in this densely populated ecosystem have evolved efficient strategies to harvest glycans, and it is the competition and synergy among species for their preferred glycans that drive the diet-dependent changes observed in the gut community structure [6–9].
Starch is produced by plants as an energy storage compound and is the dominant carbohydrate component of most Western style diets. It is produced by the plants as granules made up of two polymers of glucose, the linear α(1,4)-linked amylose and the branched amylopectin with an α(1,4)-linked backbone and α(1,6) branch points [10]. A recent analysis of the glycolytic potential encoded within the genomes of gut bacteria using the Carbohydrate-Active enZYme (CAZy) database (www.CAZy.org) reflected that genes encoding starch-processing enzymes from the glycoside hydrolase family 13 (GH13) are among the most represented in the microbiota [11]. This broad potential for starch utilization is distributed among the Bacteroidetes, Firmicutes, and Actinobacteria, the three most abundant phyla of gut bacteria. Humans are able to efficiently process most starch and it is only the resistant starch fraction of a more crystalline nature which survives transit to the large intestine where it is exposed to the gut microbiota [12]. The ability of these organisms to each thrive on specific forms of starch (e.g., complex resistant starch granules, soluble maltooligosaccharides, and amylopectin) is dependent upon both the specific activity of their GH13 enzymes and the types of glycan-uptake systems that work in concert with these enzymes. The enzymes used by the microbiota for starch degradation fall into three broad classes: amylases/neopullulanases that act upon the α(1,4)-linkages, pullulanases that act upon the α(1,6)-linkages, and α-glucosidases that act upon both types of linkages releasing glucose, typically from oligosaccharides [13]. To study these enzymes a variety of model substrates are used. This includes isolated amylose, amylopectin and maltooligosaccharides, pullulan, an α(1,6)-linked maltotriose polysaccharide, and cyclodextrins which are circularized oligosaccharides that mimic the helical shape of amylose and amylopectin (see summary in Table 1).
Table 1.
Characteristics of starch-related carbohydrates
| Carbohydrate | Description |
|---|---|
| Amylose | A starch polymer comprised of α(1,4)-linked glucose. The α(1,4) glycosidic linkage creates a helical conformation in solution and in the starch granule. Amylose helices can pack together creating insoluble crystalline regions within a starch granule |
| Amylopectin | The branched starch molecule differentiates itself from amylose by containing α(1,6)-linkage branch points along the α(1,4)-linked glucose backbone. These branches prevent the tight packing of neighboring helices resulting in amorphous regions within the starch granule and enhanced solubility |
| Maltooligosaccharides | Oligosaccharides of starch that are typically generated by amylolytic enzymes operating on the full-length polysaccharide. Purified oligosaccharides of known length allow for the more precise study of protein-carbohydrate-binding and activity |
| Pullulan | A linear starch-like polysaccharide containing repeating units of α(1,6)-linked maltotriose. The α(1,6)-linkages may mimic branch points in amylopectin and is sometimes used to determine an enzyme’s tolerance or activity towards those branch points |
| Cyclodextrins | Cyclic oligosaccharides of α(1,4)-linked glucose that mimic the curvature of a starch helix. The extent of this curvature, and similarly the molecule’s constrained geometry, decreases as the number of glucoses in the oligosaccharide increases. Most commonly used cyclodextrins include α-cyclodextrin and β-cyclodextrin that contain six and seven glucose residues, respectively, because of their similarity to the curvature of a starch helix |
| Resistant starch (RS) | Starch that is impervious to degradation by human dietary amylases due to inaccessibility, crystallinity, chemical modifications, or complex formation with lipids. RS becomes available to colonic microorganisms that are either equipped with the molecular machinery to degrade RS themselves or are available to crossfeed from RS-degrading organisms |
In addition to the diversity of enzymes complements employed, the strategies used to capture hydrolyzed starch in the gut are a function of the unique physiology of the respective microorganisms [14–17]. The Gram-positive Firmicutes and Actinobacteria take up monosaccharides and oligosaccharides via a variety of transport systems including ATP-binding cassette transporters, major facilitator superfamily, and phosphotransferase systems [18, 19]. Many of these transporters are encoded within putative operons that include one or more extracellular GH13 enzymes to hydrolyze starch at the cell surface [14, 17, 20]. In contrast, the genomes of most Bacteroidetes, the dominant Gram-negative phylum in the mammalian gut, have far fewer of these classically studied carbohydrate-uptake systems [21]. Rather the Bacteroidetes package their glycolytic potential within discrete gene clusters termed polysaccharide utilization loci (PUL) that encode glycoside hydrolases, glycan-binding proteins and a TonB-dependent transporter [15]. These PUL-encoded proteins work together in the outer membrane to capture and transport glycans, including starch.
Abigail Salyers’ seminal work on the carbohydrate-degrading phenotypes of human gut Bacteroides species laid the foundation for the modern study of this clade of bacteria. One of Salyers’ first studies revealed that 22 isolates of Bacteroides thetaiotaomicron shared the ability to grow on amylose and amylopectin [22]. Further investigation of starch utilization by B. thetaiotaomicron VPI-5482 revealed that starch-binding to the cell surface was a pre-requisite to growth on starch, and was mediated by several proteins in the outer membrane [23]. Through their efforts to determine the molecular basis of starch utilization, the Salyers lab identified the first PUL, an eight-gene cluster in B. thetaiotaomicron 2 that encodes all of the required proteins for starch adherence to the cell, as well as a surface amylase and TonB-dependent transporter to coordinate starch hydrolysis with maltooligosaccharide import into the cell (Fig. 1). This gene cluster was named the starch utilization system (Sus) for its apparent function, and is composed of susRABCDEFG [24, 25]. SusR is an inner membrane-spanning sensor/regulator protein that recognizes maltose, a disaccharide of glucose, in the periplasm and triggers the rapid upregulation of the sus genes [25]. The outer membrane lipoproteins SusDEF facilitate the binding of starch to the cell surface, and bound starch is then hydrolyzed by the α-amylase SusG [26–28]. The resulting maltooligosaccharides are shuttled into the periplasm via SusC, a TonB-dependent transporter [29], and further depolymerized by the neopullulanase SusA and α-glucosidase SusB [30, 31].
Fig. 1.

Overview of the starch utilization system (Sus) in B. thetaiotaomicron. The sus locus is transcribed from two divergent promoters. Transcription of susR occurs independently from the rest of the locus, which allows the inner membrane-spanning protein SusR to sense the disaccharide inducer, maltose, in the periplasm and subsequently drive the transcription of susABCDEFG. Starch is bound to the surface of the cell by the starch-binding outer membrane lipoproteins SusDEF. Subsequent hydrolysis by a similarly membrane-tethered α-amylase, SusG, generates oligosaccharides small enough to transit through the TonB-dependent transporter. Once in the periplasm, SusA and SusB, a neopullulanase and α-glucosidase, respectively, process oligosaccharides into glucose. The monosaccharide is then shuttled into the cytoplasm by an unknown transporter. The stoichiometry and assembly of the Sus is unknown
A decade or more after Salyers’ discovery, bacterial genome sequencing revealed that all gut Bacteroidetes possess PULs, and each PUL confers the ability to grow on a different glycan [15, 32]. All PULs encode homologues of the proteins SusCD, glycan-binding lipoproteins akin to SusEF, and a cadre of glycoside hydrolases for the utilization of a distinct glycan [15]. Based upon this commonality, all PUL-encoded proteins are believed to form a “Sus-like” system of proteins in the outer membrane that work together to target a specific glycan. PULs encoding Sus-like systems have been identified for the uptake of diverse substrates such as xyloglucan, arabinoxylan, α-mannan, inulin, and porphyran, among others [33–39]. Organisms such as B. thetaiotaomicron and Bacteroides ovatus dedicate ~18 % of their genomes to PULs, highlighting the importance of the PUL-encoded glycan uptake strategy to the adaptation of these organisms to the gut [39]. The repertoire of different PULs encoded by an organism dictates the metabolic lifestyle of the bacterium in the gut [39, 40].
The Sus of B. thetaiotaomicron remains the best-studied PUL-encoded glycan uptake system to date, and is often the prototypical system by which the function of homologous PUL-encoded proteins are compared or inferred. Here we summarize the structural and functional work to date on the Sus proteins of B. thetaiotaomicron VPI-5482, with a particular focus on the outer membrane proteins SusDEFG. These are not only the most well-characterized proteins in the system, but together they exemplify the molecular strategy that the Bacteroidetes utilize to sense and acquire carbohydrate nutrition. These studies have shaped a general model of the “Sus-like” paradigm that dominates glycan catabolism by the mammalian gut Bacteroidetes.
SusD: an α-helical carbohydrate-binding protein
Starch adherence to the cell surface is the first step in starch utilization by B. thetaiotaomicron [23]. Salyers and colleagues used a polar insertion at susE (ΩsusE) to create B. thetaiotaomicron that expressed only SusC and SusD, and noted that this mutant bound radioactively labeled starch at ~70 % the levels of wild-type cells. This strain grew normally on starch if complemented with SusG (ΩsusE::susG) [27]. However, neither SusC nor SusD alone could drive starch adsorption, as B. thetaiotaomicron cells expressing only one of these two proteins displayed barely detectable levels of starch-binding [41]. Furthermore, while neither isolated SusC nor SusD protein bound to amylose resin, mixing of the proteins prior to incubation with amylose resin resulted in the retention of both proteins [26]. The adaptation of a TonB-dependent transporter, a family of proteins historically associated with iron uptake, for starch utilization as well as the requirement of the co-receptor protein SusD marked two novel features of this system.
While SusD aids in starch-binding to the cell surface, it has no amino acid sequence similarity to known carbohydrate-binding modules (CBMs), and is notably larger (65 kDa) than any carbohydrate-binding protein. However, bacterial genome sequencing revealed the ubiquitous inclusion of homologs of susD as well as susC within all PULs of the gut Bacteroidetes, suggesting a conserved function for the encoded proteins in glycan uptake [38]. The crystal structure of SusD revealed an abundance of α-helices, with a single starch-binding site [42]. SusD is tethered to the outer membrane via a lipidated N-terminal cysteine preceded by a 16-residue flexible linker, effectively projecting the protein above the surface of the membrane like a balloon on a string. (As discussed in later sections, lipidation followed by a flexible linker is a conserved feature of SusEFG as well). The most definitive feature of SusD and its homologs is the conservation of four helix-turn-helix motifs known as tetratricopeptide repeats (TPR), that form a right-handed superhelix along one face of the protein [43, 44] (Fig. 2a, in darker colors). TPR motifs are ubiquitous in nature and most commonly support protein complex formation by serving as a site for protein–protein interactions [44]. The TPR portion of SusD-like proteins is almost structurally invariant and serves as a scaffold for the more variable remainder of the protein that includes the ligand-binding site [43].
Fig. 2.

Molecular structure of SusD with maltooligosaccharides. a Superposition of SusD (blue, PDB 3CK9) with bound maltoheptaose (blue sticks) and the SusD homolog BT1043 (gray, PDB 3EHN) that targets mucosal glycans with bound N-acetyllactosamine (black sticks). The conservation of the eight tetratricopeptide repeat helices is highlighted in darker colors for both proteins. The RMSD for these proteins is 2.8 Å over 324 aligned residues (12.6 % sequence identity). b Superposition of the structure of SusD with bound maltoheptaose (blue) and maltotriose (pink), highlighting the plasticity within the binding site. Residues that move upon binding of a longer α-glucan are in bold print. c SusD crystallized with α-cyclodextrin revealed protein dimerization. The affinity of starch-binding to the cell surface may be enhanced by an avidity effect, whereby multiple SusD proteins cooperate to bind the polymer
The starch-binding site of SusD is a shallow pocket containing an arc of aromatic amino acids that complement the shape of an amylose helix [42]. The crystal structure of SusD with maltoheptaose reveals these residues as W96, W320, and Y296, with hydrogen bonding of the O2 and O3 hydroxyls of adjacent glucose residues via the side chains of R81 and N101 [42] (Fig. 2b). These molecular determinants of starch recognition are shared with most starch-binding CBMs [45, 46], as well as the surface starch-binding sites of some GH13 enzymes [47, 48]. Thus, the glycan-binding site of SusD may be an example of convergent evolution whereby proteins of distinct evolutionary lineage and hence structure evolve similar functions [49]. A unique feature of maltooligosaccharide recognition by SusD is the flexibility of two loops near the binding cleft, one of which includes Y296 that is part of the aromatic arc. The crystal structure of SusD with maltotriose suggests that glycan binding is initiated at W98 and W320, followed by a shift in these two flexible loops, one of which moves away from the binding site to allow Y296 to shift into position. This plasticity in the binding site of SusD may allow the flexible recognition of the α-glucan helix in the context of naturally occurring α(1,6)-branching.
The affinity of SusD for maltoheptaose (K D ~ 1.0 mM) is much worse than most starch-binding CBMs that recognize maltoheptaose with low μM affinity [50–52]. Moreover, SusD displays negligible affinity for maltopentaose, and no detectable recognition of smaller sugars [42]. The binding affinity of SusD is akin to the surface starch-binding site of barley α-amylase [53], or that of the low-affinity starch-binding CBM45 family [54]. However, SusD binds β-cyclodextrin with a K D ~ 0.15 mM, highlighting that this protein is more adapted to recognize a constrained helical structure such as a starch polymer over a flexible oligosaccharide [42]. At the cell surface, it is unknown how the interaction of SusD with SusC influences the cell’s affinity for starch and maltooligosaccharides. The crystal structure of SusD with α-cyclodextrin displayed a glycan-induced dimerization, which hints at the potential of multiple SusDs to interact with a single ligand [42] (Fig. 2c). Such an avidity effect could enhance the ability of SusD to facilitate starch-binding to the cell surface. Another possibility is that SusC increases the affinity of SusD for starch, either by inducing a conformational change in SusD that enhances binding, or by extending the protein-carbohydrate interaction in a complex between the two proteins [27].
Despite its relatively weak affinity for its ligand, SusD has a critical role in starch utilization: cells with an in-frame deletion of susD (ΔsusD) cannot grow on starch or maltooligosaccharides larger than maltopentaose, and display an intermediate growth phenotype on maltopentaose and maltotetraose [42]. The Sus proteins are dispensable for growth on maltotriose and maltose, which presumably enter the outer membrane via a non-specific porin [24]. More recent work following the discovery of SusE and SusF as additional starch-binding proteins has revealed that the importance of SusD may extend beyond its ability to bind starch, as detailed in a later section.
SusG is a novel GH13 amylase required for starch utilization
Bacterial growth on polysaccharides including starch requires cell surface or extracellular glycoside hydrolases to break down the polymer into oligosaccharides that can be transported into the cell [55]. In B. thetaiotaomicron, SusG is the only cell surface amylase that is required for growth on starch [28]. Like SusD, SusG is tethered to the surface at an N-terminal cysteine followed by a 20 residue flexible linker before the first β-strand is formed [56]. While SusG displays the typical GH13 amylase family protein fold comprised of A, B, and C domains, a CBM58 is uniquely inserted within the B-domain sequence (Fig. 3a, CBM in pink) [56]. The CBM58 is extended from the rest of the catalytic domain via two short β-strands and does not interact with the rest of the protein, creating an overall bilobed appearance. The unique placement of this CBM within the catalytic fold contrasts with the typical N- or C-terminal placement of a starch-binding CBM within GH13 enzymes where the CBM can pack against the catalytic domain, sometimes shaping the active site or allowing dimerization [57–60].
Fig. 3.

SusG is an amylase with a unique CBM insertion. a Structure of the catalytically inactive mutant of SusG D498 N (PDB 3K8L) with bound maltoheptaose. CBM58 (residues 216–335) is highlighted in pink, and maltooligosaccharides bound at CBM58, the active site, and the surface starch-binding site are depicted as spheres. The orientation of the oligosaccharide from the nonreducing end (O4) to reducing end (O1) is indicated. b Close-up view of the active site in the catalytically inactive mutant of SusG D498 N (PDB 3K8L) with bound maltoheptaose. Hydrogen-bonding interactions (≤3.5 Å) are depicted as dashed lines, and Glc residues are labeled from the non-reducing to reducing end
The crystal structure of the catalytically inactive D498 N mutant of SusG with maltoheptaose revealed ligand binding to the CBM58, the active site, and an unexpected surface starch-binding site (SBS) adjacent to the active site [56]. Maltoheptaose binding at CBM58 is 45 Å away and on the opposite side of the protein from the active site, while maltoheptaose bound at the SBS is ~5 Å from a glucose residue of maltoheptaose sitting at the +2 subsite (Fig. 3b). That the SBS is distinct from the active site is demonstrated by the opposite orientation of the maltoheptaose molecules bound at the two sites: the reducing ends of each chain are directed towards each other. This orientation also makes it unlikely that a discrete starch helix can interact with both sites simultaneously.
Both CBMs and SBSs provide a proximity effect by localizing the starch polymer near the catalytic site to enhance catalysis [61, 62]. While both sites display the canonical dual aromatic residue platform that recognizes the shape of the α(1,4)-linked glucan, the manner in which maltooligosaccharide is bound at both sites is different. At CBM58, maltoheptaose is bound with the pitch of the helix parallel to the surface of the protein, whereas maltoheptaose at the surface starch-binding site is directed with the pitch of the helix into the plane of the protein. This difference in binding may differentiate the utility of these sites for starch utilization by B. thetaiotaomicron. We initially hypothesized that elimination of the SBS via site-directed mutagenesis would enhance catalysis, allowing a starch polymer better access to the catalytic site. However, the elimination of glycan binding at the SBS did not significantly affect activity on the colorimetric substrate p nitrophenol-maltohexaose, or soluble amylopectin and pullulan, but did reduce activity on insoluble corn starch, demonstrating that this site is important for the recognition of an insoluble substrate. When CBM58 was deleted from the enzyme, the activity of the enzyme against insoluble substrates decreased, but activity towards soluble amylopectin increased by threefold. While these data demonstrate that the CBM58 enhances the enzyme’s ability to localize to an insoluble starch molecule B. thetaiotaomicron in pure culture cannot grow on insoluble starch such as resistant starch [63]. Therefore, in the context of growth, CBM58 may have a different role in starch utilization, perhaps by helping to sequester oligosaccharides released by the active site, or in passing these sugars on to the SusCD complex.
To the best of our knowledge, SusG is the only GH13 with a CBM inserted within the catalytic domain. However, this interrupted domain structure was first noted in rumen Prevotella ruminicola GH10 xylanases [64] and more recently in the GH10 xylanases from B. ovatus [34] and B. intestinalis [65]. While the full-length protein structures of these GH10 enzymes have not been determined we hypothesize that like SusG, the CBMs are simply appended from the catalytic domain with minimal disruption of the GH10 protein fold. In many GH13 enzymes that have one or more CBMs, the CBM is located at the N- or C- terminus and in some cases facilitates dimerization, with the CBM shaping the catalytic cleft of the neighboring polypeptide [57–60]. In SusG, the remote location of CBM58 relative to the active site permits a wider catalytic cleft, a feature that may contribute to the enzyme’s broad activity towards amylopectin, pullulan, amylose, maltooligosaccharides, and to a much lesser extent, cyclodextrins [28, 56]. Pullulan degradation by SusG produces panose, and this product reflects the unique ability of SusG’s active site to accommodate α(1,6) glycosidic bonds while still solely acting on α(1,4) linkages [56]. This flexible recognition of various α-glucans may reflect the adaptation of B. thetaiotaomicron’s single extracellular GH13 to support growth on a variety of glycan structures that the cell might encounter in a human diet consisting of starch from various sources.
Excluding its CBM58, SusG is most similar in sequence and structure to the Halothermothrix orenii AmyA, a member of the GH13_36 subfamily that features enzymes that have amylase activity against α(1,6) containing glucans [66, 67]. The active site of SusG is typical of endo-amylases that hydrolyze endogenous α(1,4) glycosidic bonds using an acid–base double displacement mechanism [68]. In the crystal structure of a catalytically inactive SusG mutant (D498 N), maltoheptaose occupies subsites −4 through +3 via an extensive network of direct hydrogen bonding between the O2 and O3 hydroxyls of glucose and polar side chains lining the active site [56] (Fig. 3b). Aromatic stacking between H112 and Glc at the −2 subsite and between Y114 and Glc4 at the −1 subsite likely helps position the chain for efficient hydrolysis. D388 is positioned for nucleophilic attack on the C1 of Glc4; our structure of the active enzyme with acarbose captured this β-glucosyl-D388 covalent intermediate [56]. E431 interacts with the O4 of Glc3, acting as the catalytic acid to protonate the leaving oligosaccharide and activating water to split the β-glucosyl-D388 intermediate. D498 is likely important in stabilizing this intermediate [69].
The products liberated from complete starch degradation by SusG in vitro are glucose and maltose [56], yet B. thetaiotaomicron does not require SusC or SusD to grow on glucose and maltose. This disparity suggests that SusC and SusD have been maintained throughout this organism’s evolution because they are required to import oligosaccharides larger than di- or mono-saccharides. It is possible that in the context of the other Sus proteins at the cell surface, SusG liberates maltooligosaccharides larger than di- or mono-saccharides. While the typical size of the glycan that traverses the SusC porin is unknown, growth on maltoheptaose requires SusC and SusD, but not SusG [70], suggesting that maltooligosaccharides at least seven glucose units long can pass through the porin. How SusC works together with SusDEFG to import α-glucans remains a current area of investigation. It has been suggested that the SusC-like proteins from PULs that target chondroitin sulfate [71], xylan [34] and α-mannan [35] also transport larger fragments of their cognate glycan.
The α-glucans that arrive in the periplasm are processed by the neopullulanase SusA and the α-glucosidase SusB to yield glucose, which is imported via an undefined inner membrane transporter [23, 30, 72]. SusA and SusB are essential for starch utilization, and together account for most of the starch-degrading activity from whole-cell lysates compared to SusG alone [28, 31]. The crystal structure of SusB revealed that this GH97 enzyme hydrolyzes maltooligosaccharides via an inverting mechanism, yielding β-d-glucose as a product, which contrasts with the typical retaining mechanism within this glycosidase family of enzymes [73]. SusG has a relatively low affinity for starch (K m ~ 3.1 mM) compared to SusA (K m ~ 0.125 mM) [28]. The discrepancy between these two enzyme affinities may reflect SusG’s dependence on the starch-binding proteins SusDEF to bring starch within proximity of its active site, or perhaps SusG has evolved to act broadly on a variety of starch substrates at the expense of retaining high specific affinity for one substrate.
SusE and SusF bind starch via multiple carbohydrate-binding domains
Initial work by the Salyers lab established that SusCD mediate the majority of starch-binding to the cell surface, and comprise together with SusG the “minimal Sus” required for B. thetaiotaomicron growth on starch [26, 27]. Conflicting data suggested that the two remaining lipoproteins encoded within the sus operon, SusE and SusF, enhance starch-binding to the cell surface, although their amino acid sequences did not imply a function for these proteins in glycan capture [26]. Genome sequencing later revealed the presence of genes encoding such “putative lipoproteins” within most PULs of B. thetaiotaomicron and B. ovatus [39], as well as the majority of human gut-adapted Bacteroidetes hinting at a conserved function for these proteins within PUL-encoded Sus-like systems [15]. SusE and SusF belong to one of the most overrepresented protein families within the human gut microbiome, and the enrichment of these types of proteins in this niche underscore their functional importance to these bacteria [74].
The crystal structures of SusE and SusF reveal a multimodular structure comprised of a tandem array of immunoglobulin (Ig)-like folds that bind starch in a manner quite reminiscent of starch-targeting CBMs [75] (Fig. 4a, b). SusF contains an N-terminal Ig-like fold proceeded by three β-sandwich domains—Fa, Fb, and Fc—arranged in an S-like configuration (Fig. 4b) [46]. The placement of a proline residue at the midpoint between consecutive domains suggests a lack of conformational flexibility along the length of the protein. SusF is tethered to the membrane via a lipidated cysteine followed by 18 amino acids that create a flexible linker, but the rigidity of the folded protein may help project it off of the membrane to facilitate starch capture. In contrast, SusE has only two starch-binding domains—Eb and Ec, which are similar to the Fb and Fc domains of SusF—and an N-terminal Ig-like domain that was not observed in the electron density (Fig. 4a, c). A prediction of the SusE N-terminal domain structure suggests it is similar to that of SusF, with a longer, flexible linker between the N-terminal and Eb domains (see model in Fig. 5). The N-terminal domains of SusE and SusF do not contribute to starch-binding [75]. In both proteins, the final two C-terminal domains are packed in a back-to-back arrangement via a hydrophobic interface. The individual starch-binding domains of SusE and SusF share the most structural homology with the X25 domain of Bacillus acidopullyticus pullulanase [76] (Fig. 4d). While the crystal structure of this pullulanase with oligosaccharide did not reveal the X25 domain as a CBM, a superposition of the X25 domain with the SusE/F domains reveals conservation of the starch-binding residues, suggesting that this X25 may bind glycan (Fig. 4e).
Fig. 4.
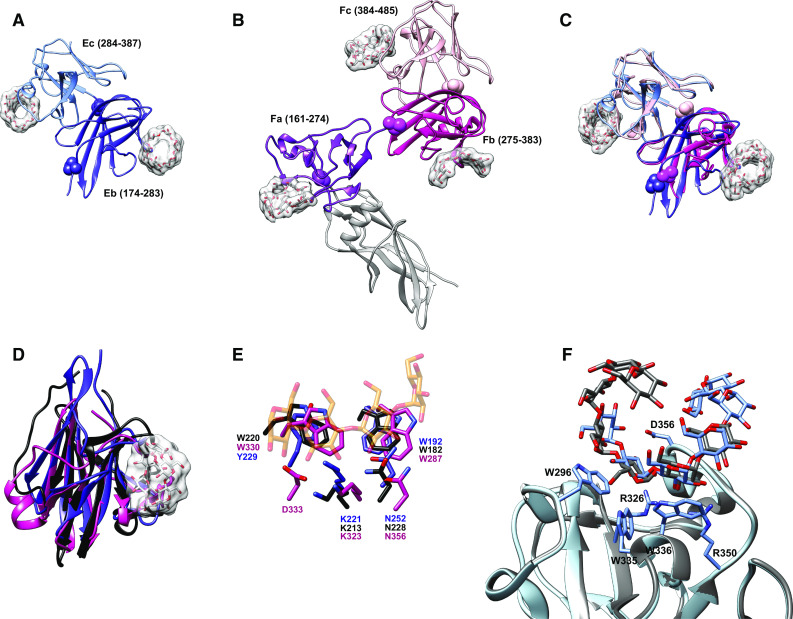
Structures of the SusE and SusF proteins. a Structure of SusE with bound α-cyclodextrin (PDB 4FEM), with the starch-binding domains Eb and Ec in different colors. Proline residues between the domains are highlighted as spheres. b Structure of SusF with bound maltoheptaose (PDB 4FE9), with the starch-binding domains Fa, Fb, and Fc in different colors. Proline residues between domains are highlighted as spheres. c Overlay of the Eb/Ec and Fb/Fc domains of SusE and SusF, colored as in panels a and b. d Superposition of the Eb domain (blue), Fb domain (pink) and the X25 domain (black, residues 161-270 of PDB 2WAN) from the Bacillus acidopullulyticus pullulanase [76]. e Close-up of the starch-binding sites in Eb and Fb from the overlay in panel d, demonstrating that these residues are conserved within the X25 module of the pullulanase (PDB 2WAN). Residues and labels are colored as in panel d, and the portion of the α-cyclodextrin bound to Eb is displayed as transparent orange and red sticks. f Overlay of the two positions that maltoheptaose occupied at the Ec binding site of SusE (PDB 4FCH), demonstrating how a longer single-helical stretch of amylose could be accommodated
Fig. 5.
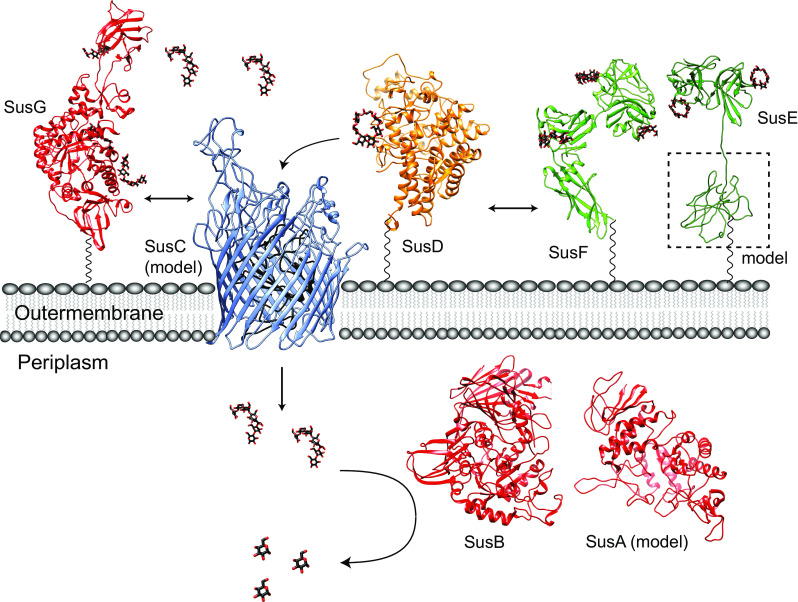
Sus protein structures and model of dynamic assembly. Ribbon diagrams of the crystal structures and homology models for the seven Sus proteins involved in starch utilization in B. thetaiotaomicron, colored as in Fig. 1. The flexible amino acid sequences that link SusDEFG to the membrane are depicted as a black wavy lines, as these residues were not resolved in the crystal structures. SusG (PDB 3K8L) dynamically interacts with SusD (PDB 3CK9) and SusC (ITASSER structure prediction [85, 86]). SusE (PDB 4FEM; ITASSER prediction of the N-terminal domain, with modeling of the linker sequence) and SusF (PDB 4FE9) may directly interact with each other and with the SusCD complex, as suggested by Salyers [27]. Maltooligosaccharides are transported through the SusC TonB-dependent transporter where they are further hydrolyzed to glucose by the action of SusB (PDB 2JKA) and SusA (model from Modpipe [87] using template PDB 3DHU). Maltooligosaccharides and glucose are depicted as black and red sticks
SusE and SusF are not described as CBMs as this would conflict with the definition of a CBM as a domain appended to a carbohydrate-active enzyme. However, although SusE and SusF are independent proteins physically separate from the α-amylase SusG, they may provide a similar functionality to a traditional CBM in the context of the Sus. The design of the Sus outer membrane protein complex, whereby glycan capture and carbohydrate degradation is spread over multiple polypeptides, is vaguely reminiscent of the cellulosomal architecture [27]. Cellulosomes are multiprotein structures comprised of enzymes and some distinct CBMs that assemble into a complex for efficient cellulose deconstruction [77, 78] and a similar system for starch hydrolysis has recently been identified in the Firmicute Ruminococcus bromii [16]. The cellulosome is held together by a system of complementary protein–protein interaction domains called cohesins and dockerins, motifs that are not found in the Sus. However, Salyers and others have demonstrated that proteins within Sus and Sus-like systems also interact [27, 79, 80].
The five starch-binding domains shared between SusE and SusF display a similar starch-binding architecture featuring two aromatic amino acids that provide a hydrophobic interface for α-glucan binding, plus additional residues that hydrogen-bond with the hydroxyl groups of the glucose residues to stabilize the interaction (Table 2). Subtle differences in the arrangement of glycan-binding residues likely explains the somewhat different affinities of each domain for maltoheptaose versus α-cyclodextrin (Table 2) [75]. All of the SusEF domains show weaker binding for glucosyl-α(1,6)-maltotriosyl-α(1,6)-maltotriose (GMM), a linear oligosaccharide of pullulan, compared to maltoheptaose suggesting that SusE and SusF have not been adapted for α(1,6) recognition. The most divergent domain between SusE and SusF is Ec, which displays the highest affinity for maltoligosaccharides. In this domain a loop within this binding site defined by residues 353–357 caps one end of the α-glucan binding site. In this crystal structure, maltoheptaose was shared across a crystallographic symmetry axis between chain A from one asymmetric unit and chain B from another. Superposition of these chains simulated a 10-glucose long maltooligosaccharide that is wound up and over this capping loop (Fig. 4f). We suggest that this binding site has been adapted to preferentially recognize single-helical regions of starch, a feature that may help the Sus complex to target partially denatured regions of starch that could be more easily hydrolyzed by SusG.
Table 2.
Starch-binding residues within each domain of SusF and SusE
| Glc4 | Glc3 | Glc2 | Glc1 | αCD (μM)a | M7 (μM)a | GMM (μM)a | |
|---|---|---|---|---|---|---|---|
| Fa | W222 | K221 | W177, D231,N206 | – | 775 | 361 | 990 |
| Fb | – | W330, D333, K323 | W287, N356 | – | 460 | 310 | 2710 |
| Fc | W442, R456, D461 | W441, R428, Q399 | W396 | – | 465 | 97 | 507 |
| Eb | Y229, K221 | W192, N252 | – | – | 386 | 1024 | 3584 |
| Ec | W296 | R326 | W336, R350 | N353 | 97 | 17 | 641 |
Glc designates the glucose monomer within a maltooligosaccharide, numbered 1-4 from the non-reducing end of sugar. Bold residues denote the aromatic platform, and all other residues are involved in hydrogen-bonding with the glucose hydroxyl groups
aAverage K d for α-cyclodextrin (αCD), maltoheptaose (M7), and glucosyl-α(1,6)-maltotriosyl-α(1,6)-maltotriose for each domain as reported in Cameron et al. [75]
Interestingly, despite the tandem arrangement of starch-binding domains in both SusE and SusF, only the domains of SusE synergize and display enhanced binding to insoluble corn starch via an avidity effect; SusE mutants with a single functional domain display greatly decreased binding [75]. In comparison, the native full-length SusF protein binds insoluble cornstarch nearly as well as a site-directed mutant possessing only a functional Fc domain (i.e., both the Fa and Fb sites were ablated) [75]. Thus the individual domains of SusF do not appear to enhance overall protein binding to starch, but rather the Fc domain is largely responsible for the SusF starch-binding affinity. Although there are distinct structural and biochemical differences between SusE and SusF, it is not yet clear what functional differences these proteins have in the context of the Sus or B. thetaiotaomicron’s ability to utilize starch in the gut.
Differentiating the roles of the SusDEFG starch-binding sites in starch utilization
Among SusDEFG there are eight starch-binding sites present on the surface of B. thetaiotaomicron, yet the roles of these sites within the Sus are distinct. The most critical starch-binding protein is SusD, as an in-frame deletion of susD (ΔsusD) results in the loss of growth on starch [42]. In this mutant, transcription of susEFG occurs at wild-type levels, supporting the hypothesis that the growth defect is due to the loss of SusD, and presumably, the loss of starch-binding to the cell surface conferred by SusD.
At the time that we created the ΔsusD strain, we did not know that SusEFG also possessed starch-binding domains. To determine if the loss of growth on starch in the ΔsusD strain was due to the loss of starch-binding by SusD, we compared the growth of the ΔsusD to a ΔsusD strain complemented with the allele for SusD* (ΔsusD::susD*), a site-directed mutant of the SusD binding site that eliminates starch-binding in vitro [70]. The ΔsusD::susD* cells grow on starch (5 mg/ml) when sus transcription is activated by the addition of a small amount of maltose (0.5 mg/ml) to the media. Furthermore, ΔsusD::susD* cells can grow on maltoheptaose with wild-type growth kinetics and without the need for maltose induction in contrast to ΔsusD cells. We concluded from these experiments that the presence of SusD was essential for growth on starch, although a SusD that also binds starch facilitates more efficient growth without the need for transcriptional activation by maltose. Indeed, quantification of sus transcript from both wild-type and SusD* expressing cells exposed to various concentrations of maltooligosaccharides revealed that the SusD* cells required 100- to 1000-fold higher concentrations of glycan than wild-type cells to achieve wild-type transcriptional activation. The role of SusD in sugar sensing is likely indirect; we speculate that starch/maltooligosaccharide binding by SusD enhances the rate of import through SusC, leading to an accumulation of sugar in the periplasm that is sensed by SusR resulting in sus transcriptional activation. These data from the SusD* growth experiments support a role for SusD in starch utilization that hinges upon its interaction with SusC. The physical presence of SusD may help stabilize SusC, or otherwise permit the assembly of a larger complex containing the rest of the Sus proteins.
Unlike SusD, starch-binding by SusEF does not contribute to α-glucan sensing by inducing sus expression, although the sus transcriptional response is somewhat diminished when a deletion of both SusEF from the cell surface is combined with mutations in either the SusG SBS (ΔsusG SURF) or CBM58 (ΔsusG-CBM58) [70]. Rather, SusEF influence the growth of B. thetaiotaomicron in a substrate-dependent manner. Growth on high molecular weight, highly branched maize amylopectin is impeded in cells with a combined deletion in SusEF and one of the SusG starch-binding sites [70]; this growth defect was not observed on potato starch that has a lower molecular weight. This observation lead to the hypothesis that the multiple starch-binding sites of SusEFG aid in acquiring high molecular weight starch species through the thick capsule layer of B. thetaiotaomicron. Like most human gut-adapted Bacteroides species, B. thetaiotaomicron produces a polysaccharide capsule several hundred nanometers thick [81], which likely protects the cell from the host immune response, but could impose a diffusion barrier to nutrients that must reach the cell surface. While the Sus proteins do not protrude above the capsule layer, they seem to aid in the capture of starch through this extracellular matrix, as a ΔsusEFG SURF or ΔsusEFG-CBM58 mutant in an acapsular strain of B. thetaiotaomicron does not display a growth defect on maize amylopectin [70].
The specialized roles of SusDEFG in starch utilization are apparent in vivo as well. To test how the individual Sus proteins adapt the cell to scavenge starch in the host intestinal tract, germ-free mice were co-colonized with wild-type, ΔsusC, and either ΔsusD::susD*, or ΔsusEFG SURF B. thetaiotaomicron [ 70 ]. Mice were fed a diet high in resistant starch and additionally colonized with or without Ruminococcus bromii, a resistant starch-degrading species that may cross-feed maltooligosaccharides to B. thetaiotaomicron [7, 63]. The ΔsusC and ΔsusD::susD* mutants were outcompeted by the wild-type strain in the presence or absence of R. bromii. Interesting, the ΔsusEFG SURF mutant fared as well as wild-type B. thetaiotaomicron when R. bromii was absent, but was quickly outcompeted when R. bromii was also present. Here, R. bromii may increase the abundance of larger maltooligosaccharides that require further processing prior to transport. These data suggest that B. thetaiotaomicron’s multiple starch-binding sites have evolved to optimize nutrient acquisition within the competitive polymicrobial environment of the colon.
The Sus complex dynamically assembles in the presence of starch
The observed cooperation between starch-binding sites during starch degradation and import implies that the Sus proteins are working closely together to optimize starch utilization. Salyers and colleagues demonstrated that SusCD interact, and that SusE may also interact with this complex [27]. Additionally, both SusEF are less sensitive to proteolytic degradation when expressed together on the cell surface, suggestive of complex formation [27]. In the Bacteroidetes Capnocytophaga canimorsus, affinity purification of the SusC-like transporter GpdC, required for host N-glycan utilization, resulted in the co-purification of the SusD-like protein GpdD, the surface glycosylase GpdG, and a periplasmic sialidase [79]. This suggests that interactions among Sus-like proteins may be conserved across different glycan utilization systems within the Bacteroidetes. However, the nature of these protein–protein interactions has not been defined.
The dynamic movement of SusG on the cell surface has been captured by single-molecule fluorescence imaging in live B. thetaiotaomicron [80]. In these experiments, a mutant of SusG was created by replacing the CBM58 with a HaloTag (HT) protein, which was fluorescently labeled by the dye tetramethyl rhodamine [82, 83]. We tracked the diffusion of this tagged SusG (SusG-HT) in live B. thetaiotaomicron cells in the presence of glucose and starch. Under all conditions, we observed both freely diffusing and slow-moving SusG-HT. We hypothesize that these slow-moving species occur due to the interaction of SusG with other Sus proteins. In addition, the net movement of SusG-HT decreased in the presence of amylopectin compared to glucose, likely due to the interaction of SusG-HT with the polymer. However, SusG-HT mobility in the presence of starch increased in both ΔsusD and ΔsusEF cells, presumably because SusG was not able to associate with these proteins [80]. We believe these data suggest that SusG dynamically associates with other Sus proteins, resulting in a slow-moving population of molecules, and that during growth on starch the polysaccharide may effectively “crosslink” the Sus proteins, promoting or stabilizing their interactions.
Summary and working model
The starch utilization system of B. thetaiotaomicron is composed of eight genes, five of which encode proteins that localize to the outer membrane of the cell where starch is first encountered. These proteins collectively bind and degrade large starch polysaccharides so that smaller oligosaccharides can be shuttled into the cell for further hydrolysis and energy harvest. Delineation of the biochemical and structural features of the individual Sus proteins has facilitated the development of a working model for how the Sus proteins, and likely homologous proteins from other Sus-like systems within the Bacteroidetes, interact to metabolize carbohydrate nutrition (Fig. 5). In this model, the SusCD proteins are key for initial starch sensing, and together promote the efficient uptake of maltooligosaccharides. SusC and D likely interact frequently as the essential unit for glycan uptake, while the interactions of these proteins with SusEFG may be more dynamic. SusEF as well as the starch-binding sites within SusG support starch binding at the cell surface through the polysaccharide capsule. The redundancy of cell surface starch-binding sites likely enhances the capture of dietary starch, and maltooligosaccharides generated by other species in the gut. Finally, the dynamic assembly of the Sus proteins may enhance starch capture by allowing each protein additional degrees of freedom for optimal starch-binding.
The structure of the sus operon of B. thetaiotaomicron is not completely conserved as there are several variations of predicted starch-targeting PULs among other well-studied Bacteroides species (Fig. 6). In particular, the number of SusE/F homologs, and conservation of the SusG protein varies extensively. For example, Bacteroides fragilis encodes one SusE, and a SusG homolog, both of which are longer than their homologs in B. thetaiotaomicron, and have limited identity over the length of the polypeptide. In addition, many predicted Sus PULs do not include obvious susA or susB genes within the same operon. How these variations in operon structure, protein sequence (and hence structure) affect starch utilization in these organisms is unknown. However, this comparison highlights that the proteins encoded by the susC and susD genes are the most well conserved, underscoring their central function in glycan uptake.
Fig. 6.
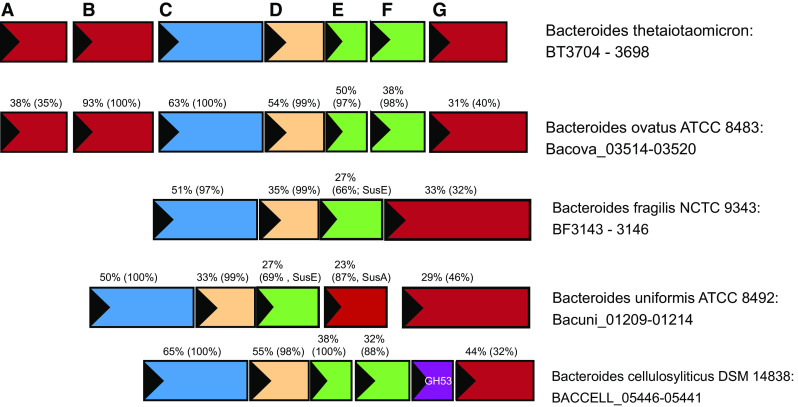
Sus operon structure across Bacteroides species. Selected sus operons from the type strains of several human gut Bacteroides species are displayed, identified via conservation of the operon structure surrounding a susG homolog. Numbers displayed above each gene indicate the percent identity of the encoded protein and in parentheses the percent coverage of the match to the homologous protein from B. thetaiotaomicron. For example, 63 % (100 %) above the susC homolog from B. ovatus indicates that the SusC homolog from B. ovatus is 63 % identical to the B. thetaiotaomicron SusC and that this match covers 100 % of the protein sequence
The Sus is a model system for glycan uptake by mammalian gut Bacteroidetes, and the repertoire of Sus-like systems encoded within the genomes of these organisms dictates their glycan utilization profile [40, 84]. As the field moves toward a molecular-level understanding of the organization and function of other Sus-like systems, we will see how this basic paradigm as outlined for the Sus of B. thetaiotaomicron has been adapted for the capture of diverse glycans from the gut environment.
Acknowledgments
This work was supported by funds from a pilot/feasibility Grant from the University of Michigan Gastrointestinal Peptides Research Center (DK 034933) awarded to N.M.K., as well as the Host Microbiome Initiative at the University of Michigan Medical School (N.M.K.). M.H.F. was partially supported by a predoctoral fellowship from the Cellular Biotechnology Training Program (T32GM008353).
References
- 1.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9(5):313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol. 2008;6(2):121–131. doi: 10.1038/nrmicro1817. [DOI] [PubMed] [Google Scholar]
- 3.Wardwell LH, Huttenhower C, Garrett WS. Current concepts of the intestinal microbiota and the pathogenesis of infection. Curr Infect Dis Rep. 2011;13(1):28–34. doi: 10.1007/s11908-010-0147-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci USA. 2002;99(24):15451–15455. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Natarajan N, Pluznick JL. From microbe to man: the role of microbial short chain fatty acid metabolites in host cell biology. Am J Physiol Cell Physiol. 2014;307(11):C979–C985. doi: 10.1152/ajpcell.00228.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10(5):323–335. doi: 10.1038/nrmicro2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ze X, Le Mougen F, Duncan SH, Louis P, Flint HJ. Some are more equal than others: the role of “keystone” species in the degradation of recalcitrant substrates. Gut Microbes. 2013;4(3):236–240. doi: 10.4161/gmic.23998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carmody RN, Gerber GK, Luevano JM, Jr, Gatti DM, Somes L, Svenson KL, Turnbaugh PJ. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe. 2015;17(1):72–84. doi: 10.1016/j.chom.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeeman SC, Kossmann J, Smith AM. Starch: its metabolism, evolution, and biotechnological modification in plants. Annu Rev Plant Biol. 2010;61:209–234. doi: 10.1146/annurev-arplant-042809-112301. [DOI] [PubMed] [Google Scholar]
- 11.El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol. 2013;11(7):497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- 12.Birt DF, Boylston T, Hendrich S, Jane JL, Hollis J, Li L, McClelland J, Moore S, Phillips GJ, Rowling M, Schalinske K, Scott MP, Whitley EM. Resistant starch: promise for improving human health. Adv Nutr. 2013;4(6):587–601. doi: 10.3945/an.113.004325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hii SL, Tan JS, Ling TC, Ariff AB. Pullulanase: role in starch hydrolysis and potential industrial applications. Enzyme Res. 2012;2012:921362. doi: 10.1155/2012/921362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cockburn DW, Orlovsky NI, Foley MH, Kwiatkowski KJ, Bahr CM, Maynard M, Demeler B, Koropatkin NM. Molecular details of a starch utilization pathway in the human gut symbiont Eubacterium rectale. Mol Microbiol. 2015;95(2):209–230. doi: 10.1111/mmi.12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martens EC, Koropatkin NM, Smith TJ, Gordon JI. Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J Biol Chem. 2009;284(37):24673–24677. doi: 10.1074/jbc.R109.022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ze X, Ben David Y, Laverde-Gomez JA, Dassa B, Sheridan PO, Duncan SH, Louis P, Henrissat B, Juge N, Koropatkin NM, Bayer EA, Flint HJ. Unique organization of extracellular amylases into amylosomes in the resistant starch-utilizing human colonic firmicutes bacterium Ruminococcus bromii . MBio. 2015 doi: 10.1128/mBio.01058-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duranti S, Turroni F, Lugli GA, Milani C, Viappiani A, Mangifesta M, Gioiosa L, Palanza P, van Sinderen D, Ventura M. Genomic characterization and transcriptional studies of the starch-utilizing strain Bifidobacterium adolescentis 22L. Appl Environ Microbiol. 2014;80(19):6080–6090. doi: 10.1128/AEM.01993-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, Shah N, Wang C, Magrini V, Wilson RK, Cantarel BL, Coutinho PM, Henrissat B, Crock LW, Russell A, Verberkmoes NC, Hettich RL, Gordon JI. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci USA. 2009;106(14):5859–5864. doi: 10.1073/pnas.0901529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu Y, Ding Y, Ren C, Sun Z, Rodionov DA, Zhang W, Yang S, Yang C, Jiang W. Reconstruction of xylose utilization pathway and regulons in Firmicutes. BMC Genom. 2010;11:255. doi: 10.1186/1471-2164-11-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scott KP, Martin JC, Chassard C, Clerget M, Potrykus J, Campbell G, Mayer CD, Young P, Rucklidge G, Ramsay AG, Flint HJ. Substrate-driven gene expression in Roseburia inulinivorans: importance of inducible enzymes in the utilization of inulin and starch. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4672–4679. doi: 10.1073/pnas.1000091107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu J, Mahowald MA, Ley RE, Lozupone CA, Hamady M, Martens EC, Henrissat B, Coutinho PM, Minx P, Latreille P, Cordum H, Van Brunt A, Kim K, Fulton RS, Fulton LA, Clifton SW, Wilson RK, Knight RD, Gordon JI. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 2007;5(7):e156. doi: 10.1371/journal.pbio.0050156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salyers AA, Vercellotti JR, West SE, Wilkins TD. Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl Environ Microbiol. 1977;33(2):319–322. doi: 10.1128/aem.33.2.319-322.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson KL, Salyers AA. Biochemical evidence that starch breakdown by Bacteroides thetaiotaomicron involves outer membrane starch-binding sites and periplasmic starch-degrading enzymes. J Bacteriol. 1989;171(6):3192–3198. doi: 10.1128/jb.171.6.3192-3198.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tancula E, Feldhaus MJ, Bedzyk LA, Salyers AA. Location and characterization of genes involved in binding of starch to the surface of Bacteroides thetaiotaomicron . J Bacteriol. 1992;174(17):5609–5616. doi: 10.1128/jb.174.17.5609-5616.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D’Elia JN, Salyers AA. Effect of regulatory protein levels on utilization of starch by Bacteroides thetaiotaomicron . J Bacteriol. 1996;178(24):7180–7186. doi: 10.1128/jb.178.24.7180-7186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shipman JA, Berleman JE, Salyers AA. Characterization of four outer membrane proteins involved in binding starch to the cell surface of Bacteroides thetaiotaomicron . J Bacteriol. 2000;182(19):5365–5372. doi: 10.1128/JB.182.19.5365-5372.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho KH, Salyers AA. Biochemical analysis of interactions between outer membrane proteins that contribute to starch utilization by Bacteroides thetaiotaomicron . J Bacteriol. 2001;183(24):7224–7230. doi: 10.1128/JB.183.24.7224-7230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shipman JA, Cho KH, Siegel HA, Salyers AA. Physiological characterization of SusG, an outer membrane protein essential for starch utilization by Bacteroides thetaiotaomicron . J Bacteriol. 1999;181(23):7206–7211. doi: 10.1128/jb.181.23.7206-7211.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reeves AR, D’Elia JN, Frias J, Salyers AA. A Bacteroides thetaiotaomicron outer membrane protein that is essential for utilization of maltooligosaccharides and starch. J Bacteriol. 1996;178(3):823–830. doi: 10.1128/jb.178.3.823-830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith KA, Salyers AA. Characterization of a neopullulanase and an alpha-glucosidase from Bacteroides thetaiotaomicron 95-1. J Bacteriol. 1991;173(9):2962–2968. doi: 10.1128/jb.173.9.2962-2968.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D’Elia JN, Salyers AA. Contribution of a neopullulanase, a pullulanase, and an alpha-glucosidase to growth of Bacteroides thetaiotaomicron on starch. J Bacteriol. 1996;178(24):7173–7179. doi: 10.1128/jb.178.24.7173-7179.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD, Gordon JI. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307(5717):1955–1959. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 33.Larsbrink J, Rogers TE, Hemsworth GR, McKee LS, Tauzin AS, Spadiut O, Klinter S, Pudlo NA, Urs K, Koropatkin NM, Creagh AL, Haynes CA, Kelly AG, Cederholm SN, Davies GJ, Martens EC, Brumer H. A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature. 2014;506(7489):498–502. doi: 10.1038/nature12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogowski A, Briggs JA, Mortimer JC, Tryfona T, Terrapon N, Lowe EC, Basle A, Morland C, Day AM, Zheng H, Rogers TE, Thompson P, Hawkins AR, Yadav MP, Henrissat B, Martens EC, Dupree P, Gilbert HJ, Bolam DN. Glycan complexity dictates microbial resource allocation in the large intestine. Nat Commun. 2015;6:7481. doi: 10.1038/ncomms8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cuskin F, Lowe EC, Temple MJ, Zhu Y, Cameron EA, Pudlo NA, Porter NT, Urs K, Thompson AJ, Cartmell A, Rogowski A, Hamilton BS, Chen R, Tolbert TJ, Piens K, Bracke D, Vervecken W, Hakki Z, Speciale G, Munoz-Munoz JL, Day A, Pena MJ, McLean R, Suits MD, Boraston AB, Atherly T, Ziemer CJ, Williams SJ, Davies GJ, Abbott DW, Martens EC, Gilbert HJ. Human gut Bacteroidetes can utilize yeast mannan through a selfish mechanism. Nature. 2015;517(7533):165–169. doi: 10.1038/nature13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, Sonnenburg JL. Specificity of polysaccharide use in intestinal Bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141(7):1241–1252. doi: 10.1016/j.cell.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hehemann JH, Kelly AG, Pudlo NA, Martens EC, Boraston AB. Bacteria of the human gut microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes. Proc Natl Acad Sci USA. 2012;109(48):19786–19791. doi: 10.1073/pnas.1211002109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bjursell MK, Martens EC, Gordon JI. Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. J Biol Chem. 2006;281(47):36269–36279. doi: 10.1074/jbc.M606509200. [DOI] [PubMed] [Google Scholar]
- 39.Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP, Abbott DW, Henrissat B, Gilbert HJ, Bolam DN, Gordon JI. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 2011;9(12):e1001221. doi: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McNulty NP, Wu M, Erickson AR, Pan C, Erickson BK, Martens EC, Pudlo NA, Muegge BD, Henrissat B, Hettich RL, Gordon JI. Effects of diet on resource utilization by a model human gut microbiota containing Bacteroides cellulosilyticus WH2, a symbiont with an extensive glycobiome. PLoS Biol. 2013;11(8):e1001637. doi: 10.1371/journal.pbio.1001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reeves AR, Wang GR, Salyers AA. Characterization of four outer membrane proteins that play a role in utilization of starch by Bacteroides thetaiotaomicron . J Bacteriol. 1997;179(3):643–649. doi: 10.1128/jb.179.3.643-649.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koropatkin NM, Martens EC, Gordon JI, Smith TJ. Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure. 2008;16(7):1105–1115. doi: 10.1016/j.str.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bolam DN, Koropatkin NM. Glycan recognition by the Bacteroidetes Sus-like systems. Curr Opin Struct Biol. 2012;22(5):563–569. doi: 10.1016/j.sbi.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 44.D’Andrea LD, Regan L. TPR proteins: the versatile helix. Trends Biochem Sci. 2003;28(12):655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 45.Machovic M, Janecek S. The evolution of putative starch-binding domains. FEBS Lett. 2006;580(27):6349–6356. doi: 10.1016/j.febslet.2006.10.041. [DOI] [PubMed] [Google Scholar]
- 46.Machovic M, Janecek S. Starch-binding domains in the post-genome era. Cell Molec Life Sci. 2006;63(23):2710–2724. doi: 10.1007/s00018-006-6246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cockburn D, Wilkens C, Ruzanski C, Andersen S, Nielsen J, Smith AM, Field RA, Willemoes M, Hachem MA, Svensson B. Analysis of surface binding sites (SBSs) in carbohydrate active enzymes with focus on glycoside hydrolase families 13 and 77–a mini-review. Biologia. 2014;69(6):705–712. doi: 10.2478/s11756-014-0373-9. [DOI] [Google Scholar]
- 48.Abbott DW, van Bueren AL. Using structure to inform carbohydrate binding module function. Curr Opin Struct Biol. 2014;28:32–40. doi: 10.1016/j.sbi.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 49.Taylor ME, Drickamer K. Convergent and divergent mechanisms of sugar recognition across kingdoms. Curr Opin Struct Biol. 2014;28:14–22. doi: 10.1016/j.sbi.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boraston AB, Healey M, Klassen J, Ficko-Blean E, Lammerts van Bueren A, Law V. A structural and functional analysis of alpha-glucan recognition by family 25 and 26 carbohydrate-binding modules reveals a conserved mode of starch recognition. J Biol Chem. 2006;281(1):587–598. doi: 10.1074/jbc.M509958200. [DOI] [PubMed] [Google Scholar]
- 51.Chou WI, Pai TW, Liu SH, Hsiung BK, Chang MD. The family 21 carbohydrate-binding module of glucoamylase from Rhizopus oryzae consists of two sites playing distinct roles in ligand binding. Biochem. 2006;J396(3):469–477. doi: 10.1042/BJ20051982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williamson MP, Le Gal-Coeffet MF, Sorimachi K, Furniss CS, Archer DB, Williamson G. Function of conserved tryptophans in the Aspergillus niger glucoamylase 1 starch binding domain. Biochemistry. 1997;36(24):7535–7539. doi: 10.1021/bi9702896. [DOI] [PubMed] [Google Scholar]
- 53.Robert X, Haser R, Mori H, Svensson B, Aghajari N. Oligosaccharide binding to barley alpha-amylase 1. J Biol Chem. 2005;280(38):32968–32978. doi: 10.1074/jbc.M505515200. [DOI] [PubMed] [Google Scholar]
- 54.Glaring MA, Baumann MJ, Abou Hachem M, Nakai H, Nakai N, Santelia D, Sigurskjold BW, Zeeman SC, Blennow A, Svensson B. Starch-binding domains in the CBM45 family-low-affinity domains from glucan, water dikinase and alpha-amylase involved in plastidial starch metabolism. FEBS J. 2011;278(7):1175–1185. doi: 10.1111/j.1742-4658.2011.08043.x. [DOI] [PubMed] [Google Scholar]
- 55.Flint HJ, Scott KP, Duncan SH, Louis P, Forano E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes. 2012;3(4):289–306. doi: 10.4161/gmic.19897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koropatkin NM, Smith TJ. SusG: a unique cell-membrane-associated alpha-amylase from a prominent human gut symbiont targets complex starch molecules. Structure. 2010;18(2):200–215. doi: 10.1016/j.str.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 57.Fritzsche HB, Schwede T, Schulz GE. Covalent and three-dimensional structure of the cyclodextrinase from Flavobacterium sp. no. 92. Eur J Biochem. 2003;270(10):2332–2341. doi: 10.1046/j.1432-1033.2003.03603.x. [DOI] [PubMed] [Google Scholar]
- 58.Hondoh H, Kuriki T, Matsuura Y. Three-dimensional structure and substrate binding of Bacillus stearothermophilus neopullulanase. J Mol Biol. 2003;326(1):177–188. doi: 10.1016/S0022-2836(02)01402-X. [DOI] [PubMed] [Google Scholar]
- 59.Kamitori S, Kondo S, Okuyama K, Yokota T, Shimura Y, Tonozuka T, Sakano Y. Crystal structure of Thermoactinomyces vulgaris R-47 alpha-amylase II (TVAII) hydrolyzing cyclodextrins and pullulan at 2.6 A resolution. J Molec Biol. 1999;287(5):907–921. doi: 10.1006/jmbi.1999.2647. [DOI] [PubMed] [Google Scholar]
- 60.Lee HS, Kim MS, Cho HS, Kim JI, Kim TJ, Choi JH, Park C, Lee HS, Oh BH, Park KH. Cyclomaltodextrinase, neopullulanase, and maltogenic amylase are nearly indistinguishable from each other. J Biol Chem. 2002;277(24):21891–21897. doi: 10.1074/jbc.M201623200. [DOI] [PubMed] [Google Scholar]
- 61.Cockburn D, Nielsen MM, Christiansen C, Andersen JM, Rannes JB, Blennow A, Svensson B. Surface binding sites in amylase have distinct roles in recognition of starch structure motifs and degradation. Int J Biol Macromol. 2015;75:338–345. doi: 10.1016/j.ijbiomac.2015.01.054. [DOI] [PubMed] [Google Scholar]
- 62.Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J. 2004;382(Pt 3):769–781. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ze X, Duncan SH, Louis P, Flint HJ. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012;6(8):1535–1543. doi: 10.1038/ismej.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Flint HJ, Whitehead TR, Martin JC, Gasparic A. Interrupted catalytic domain structures in xylanases from two distantly related strains of Prevotella ruminicola . Biochim Biophys Acta. 1997;1337(2):161–165. doi: 10.1016/S0167-4838(96)00213-0. [DOI] [PubMed] [Google Scholar]
- 65.Zhang M, Chekan JR, Dodd D, Hong PY, Radlinski L, Revindran V, Nair SK, Mackie RI, Cann I. Xylan utilization in human gut commensal bacteria is orchestrated by unique modular organization of polysaccharide-degrading enzymes. Proc Natl Acad Sci USA. 2014;111(35):E3708–E3717. doi: 10.1073/pnas.1406156111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sivakumar N, Li N, Tang JW, Patel BK, Swaminathan K. Crystal structure of AmyA lacks acidic surface and provide insights into protein stability at poly-extreme condition. FEBS Lett. 2006;580(11):2646–2652. doi: 10.1016/j.febslet.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 67.Janecek S, Svensson B, MacGregor EA. Alpha-Amylase: an enzyme specificity found in various families of glycoside hydrolases. Cell Molec Life Sc. 2014;71(7):1149–1170. doi: 10.1007/s00018-013-1388-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qian M, Nahoum V, Bonicel J, Bischoff H, Henrissat B, Payan F. Enzyme-catalyzed condensation reaction in a mammalian alpha-amylase. High-resolution structural analysis of an enzyme-inhibitor complex. Biochemistry. 2001;40(25):7700–7709. doi: 10.1021/bi0102050. [DOI] [PubMed] [Google Scholar]
- 69.McCarter JD, Withers SG. Mechanisms of enzymatic glycoside hydrolysis. Curr Opin Struct Biol. 1994;4(6):885–892. doi: 10.1016/0959-440X(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 70.Cameron EA, Kwiatkowski KJ, Lee BH, Hamaker BR, Koropatkin NM, Martens EC. Multifunctional nutrient-binding proteins adapt human symbiotic bacteria for glycan competition in the gut by separately promoting enhanced sensing and catalysis. MBio. 2014;5(5):e01441–01414. doi: 10.1128/mBio.01441-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raghavan V, Lowe EC, Townsend GE, 2nd, Bolam DN, Groisman EA. Tuning transcription of nutrient utilization genes to catabolic rate promotes growth in a gut bacterium. Mol Microbiol. 2014;93(5):1010–1025. doi: 10.1111/mmi.12714. [DOI] [PubMed] [Google Scholar]
- 72.Smith KA, Salyers AA. Cell-associated pullulanase from Bacteroides thetaiotaomicron: cloning, characterization, and insertional mutagenesis to determine role in pullulan utilization. J Bacteriol. 1989;171(4):2116–2123. doi: 10.1128/jb.171.4.2116-2123.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gloster TM, Turkenburg JP, Potts JR, Henrissat B, Davies GJ. Divergence of catalytic mechanism within a glycosidase family provides insight into evolution of carbohydrate metabolism by human gut flora. Chem Biol. 2008;15(10):1058–1067. doi: 10.1016/j.chembiol.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ellrott K, Jaroszewski L, Li W, Wooley JC, Godzik A. Expansion of the protein repertoire in newly explored environments: human gut microbiome specific protein families. PLoS Comput Biol. 2010;6(6):e1000798. doi: 10.1371/journal.pcbi.1000798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cameron EA, Maynard MA, Smith CJ, Smith TJ, Koropatkin NM, Martens EC. Multidomain carbohydrate-binding proteins involved in Bacteroides thetaiotaomicron starch metabolism. J Biol Chem. 2012;287(41):34614–34625. doi: 10.1074/jbc.M112.397380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Turkenburg JP, Brzozowski AM, Svendsen A, Borchert TV, Davies GJ, Wilson KS. Structure of a pullulanase from Bacillus acidopullulyticus . Proteins. 2009;76(2):516–519. doi: 10.1002/prot.22416. [DOI] [PubMed] [Google Scholar]
- 77.Bayer EA, Lamed R, White BA, Flint HJ. From cellulosomes to cellulosomics. Chem Rec. 2008;8(6):364–377. doi: 10.1002/tcr.20160. [DOI] [PubMed] [Google Scholar]
- 78.White BA, Lamed R, Bayer EA, Flint HJ. Biomass utilization by gut microbiomes. Annu Rev Microbiol. 2014;68:279–296. doi: 10.1146/annurev-micro-092412-155618. [DOI] [PubMed] [Google Scholar]
- 79.Renzi F, Manfredi P, Mally M, Moes S, Jeno P, Cornelis GR. The N-glycan glycoprotein deglycosylation complex (Gpd) from Capnocytophaga canimorsus deglycosylates human IgG. PLoS Pathog. 2011;7(6):e1002118. doi: 10.1371/journal.ppat.1002118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karunatilaka KS, Cameron EA, Martens EC, Koropatkin NM, Biteen JS. Superresolution imaging captures carbohydrate utilization dynamics in human gut symbionts. MBio. 2014;5(6):e02172. doi: 10.1128/mBio.02172-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martens EC, Roth R, Heuser JE, Gordon JI. Coordinate regulation of glycan degradation and polysaccharide capsule biosynthesis by a prominent human gut symbiont. J Biol Chem. 2009;284(27):18445–18457. doi: 10.1074/jbc.M109.008094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3(6):373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 83.Los GV, Wood K. The HaloTag: a novel technology for cell imaging and protein analysis. Methods Mol Biol. 2007;356:195–208. doi: 10.1385/1-59745-217-3:195. [DOI] [PubMed] [Google Scholar]
- 84.Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008;4(5):447–457. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pieper U, Webb B, Dong GQ, Schneidman-Duhovny D, Fan H, Kim SJ, Khuri N, Spill Y, Weinkam P, Hammel M, Tainer J, Nilges M, Sali A. ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2014;42:336–346. doi: 10.1093/nar/gkt1144. [DOI] [PMC free article] [PubMed] [Google Scholar]