

Abstract
Thioamide residues can be effective, minimally-perturbing fluorescence quenching probes for studying protein folding and proteolysis. In order to increase the level of quenching, we have here explored the use of adjacent dithioamides. We have found that they are more effective fluorescence quenchers, as expected, but we have also observed unexpected changes in the thioamide absorption spectra that may arise from n-to-π* interactions of the thiocarbonyls. We have made use of the increased quenching to improve the fluorescence turn-on of thioamide protease sensors.
Graphical abstract
Fluorescence quenching can be very effectively used to study dynamic biological processes, both in vitro and in living systems.1, 2 For this purpose, a fluorophore and a quencher are needed to label the target protein or other biomolecule. In such applications, it is important to maintain the intrinsic structural and functional characteristics of the protein of interest.3 Thus, much effort is continuously put into the development of new fluorophores and quenchers. Our laboratory has developed the thioamide functional group as a minimally perturbing fluorescent quencher to monitor protein conformational changes as well as proteolysis.4–6
The thioamide is a single atom substitution of the native peptide bond, with relatively small changes to its key functional features. For example, the thioamide is a stronger hydrogen bond donor, but a slightly weaker acceptor than the native amide bond, sharing a similar planar structure with a rotational barrier of 15–20 kcal/mol.7–11 Importantly, the thioamide has been shown to be broadly compatible with α-helix, β-sheet, and polyproline type II (PPII) secondary structures, although position-dependent increases or decreases in stability have been reported.12–16 A strong π-π* transition band around 270 nm makes the thioamide a good photoswitch and a good Förster resonance energy transfer (FRET) acceptor for short wavelength fluorophores such as p-cyanophenylalanine (F*, Fig. 1).5, 17, 18 Additionally, thioamides are able to quench fluorophores such as tryptophan, 7-methoxycoumarin-4-ylalanine (μ, Fig. 1), acridon-2-ylalanine (δ, Fig. 1) and fluorescein via photo-induced electron transfer (PET).19, 20 However, the quenching efficiency for longer wavelength fluorophores is relatively limited. For instance, the maximum observed quenching by 65 mM thioacetamide is 84% for μ and 63% for δ.19 Further improvement of the quenching ability of the thioamide is of great interest, since this would bring greater sensitivity to protein folding and proteolysis studies.
Fig. 1.

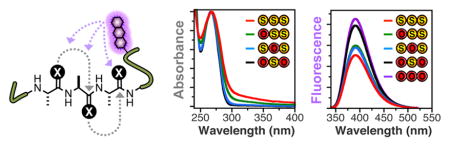
UV/Vis spectra of polyproline peptides containing multiple thioamides demonstrate non-additive changes in the thioamide absorption region (250–300 nm). All peptides contained multiple thioamide residues, varying numbers of prolines, and a 7-methoxycoumarin-4-ylalanine (μ) or p-cyanophenylalanine (F*) fluorophore. Left: Spectra of μ-containing peptides 1-7. The spectrum of a peptide containing two adjacent thioamides (5) is broadened compared to the spectrum of a peptide containing i, i+2 thioamides (6). Normalizing the absorbance at 270 nm makes clear the increased relative absorbance of 5 at longer wavelengths (Inset). Spectral broadening of the polythioamides can be even more clearly seen for peptides 8-12, containing F*. Particularly long wavelength features arising from thioamide-thioamide interactions are observed in the spectra of trithioamide peptides 7 and 12.
According to a collision-based description of dynamic electron-transfer, placing two thioamides near each other would increase the probability of quenching by effectively increasing the radius of the quenching moiety.21 This increase in quenching efficiency might be further modulated by an electronic interaction between the two thioamide moieties that could change the optical or electrochemical properties of the dithioamide relative to the corresponding single thioamides. To investigate these possibilities, we examined quenching by multiple thioamides in two model systems.
First, we designed a series of peptides in which an N-terminal μ was separated by two or four proline residues from two consecutive thioamide amino acid analogs in order to provide rigid spacers to ensure that interactions between the fluorophore and the quencher occurred primarily through space. We also synthesized control peptides containing single thioamides at each of the considered residues and an all oxoamide peptide. It was found that yields of the dithiopeptides were inconsistent when using standard 20% piperidine in DMF for deprotection, and that multiple products of identical mass (presumably attributable to epimerization) were observed. A recent report on the solid-phase synthesis of dithioamide α–amino acid peptides advocated the use of dry dichloromethane in coupling thioacylbenzotriazole precursors.22 In our hands, the use of dry dichloromethane did not suppress the production of these sideproducts. However, when deprotection conditions consisting of 2% 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 1% hydroxylbenzotriazole (HOBt) in DMF were used a single product was observed (ESI, Scheme S2, and Fig. S10). The peptide sequences are shown in Fig. 1, where the superscript “S” is used to denote the positions of thioamide residues.
Examining the UV/Vis spectra of the thioamide containing peptides showed that the absorbance of the i, i+1 dithioamide was different than the sum of the absorbances of the two corresponding monothioamides. For example, by analyzing the spectra of peptides 1-7, one can see that placing thioamides at adjacent i, i+1 positions induces broadening and red-shifting of the thioamide absorption. The spectrum of μP3PAAG (1) shows a typical absorption spectrum of a μ-containing peptide (Fig. 1, Left). The spectra of μP3PSAAG (2), μP3PASAG (3), and μP3PAASG (4) are all typical of μ- and thioamide-containing peptides, with slight changes in the spectral shape based on position. However, in comparing the spectra of μP3PASASG (5), an adjacent i, i+1 dithioamide, and μP3PSAASG (6), a “skipped” i, i+2 dithioamide, one can see a clear difference in their spectra. Normalizing the absorbance at 270 nm for peptides 5 and 6 allows one to more clearly see the increased relative absorbance of 5 in the 300–350 nm range (Fig. 1, Left Inset). Examination of the spectrum of trithiopeptide μP3PSASASG (7) shows that this red-shifting and spectral broadening can even spread into the 400 nm range.
Since the unexpected polythioamide electronic interaction overlapped with the μ absorbance maximum at 325 nm, we prepared peptides 8-12, containing an N-terminal F* residue. F* has no significant absorbance above 250 nm, so the red-shifted shoulder that arises for adjacent i, i+1 thioamides in dithiopeptide F*P3PASASG (11) and trithiopeptide F*P3PSASASG (12) is very clear (Fig. 1, Right Inset). The effect is independent of chromophore identity. Indeed, spectral broadening was also observed in δ-containing peptides 17-20 (ESI, Fig. S13).
While the focus of our work is in using the dithioamides to improve quenching assays, we were intrigued by this spectral red-shifting. Raines and coworkers have assembled a large body of evidence showing that the previously unappreciated n-to-π* interaction is a common and significant stabilizing force in protein secondary structure, particularly in PPII helices.23–25 Importantly, they have shown in model systems that n-to-π* interactions involving thioamides can be stronger than the corresponding interactions amongst oxoamides.25 We believe that our data provide the first direct spectroscopic evidence for the n-to-π* interaction. The n-to-π* interaction would be difficult to discern for an oxoamide since the oxoamide carbonyl absorption occurs at ~220 nm.26 We note that an n-to-π* transition within a single thioamide carbonyl occurs with an absorption at ~ 340 nm, which is different from the red-shift of the π-π* absorption seen here. Preliminary ab initio calculations using model dithiopeptides constrained to form an n-to-π* interaction show a sharing of electron density between the two thiocarbonyls and a decrease in the energy for the π-π* transition consistent with our experimental spectra (see ESI, Fig. S17–S18). We have carried out some preliminary denaturation studies to determine whether the PPII folds of our polythiopeptides are stabilized by n-to-π* interactions among the thioamides, but the peptides are short enough that secondary structures are difficult to discern by circular dichroism (data not shown). We will pursue the study of this thioamide-thioamide interaction in more depth in systems such as proline oligomers or collagen fibrils where stability effects will be easier to observe.
Comparing the emission intensities of peptides 1-7 as well as shorter peptides 13-16 demonstrated that μ fluorescence was quenched in a position dependent manner, with greater quenching from the proximal thioamides, consistent with previous measurements of thioamide-based PET quenching of Trp in model peptides (Fig. 2).20 As expected, the di- and trithioamide peptides showed higher levels of quenching. The steady state quenching efficiencies (EQSS) are summarized in Table S3 (see ESI). It is interesting to note that the putative n-to-π* interaction does not have a dramatic effect on quenching. This can be seen by a comparison of EQSS for the adjacent dithioamide 5 and the skipped dithioamide 6. We have also observed increased quenching by dithioamides in peptides containing F*, δ, and fluorescein (see ESI, Table S3 and Figs. S12, S13, S14).
Fig. 2.

Increased fluorescence quenching by dithioamides observed in steady state spectra and fluorescence lifetime measurements. Left: Fluorescence emission spectra (λex = 333 nm) of peptides 1-7 at 5 μM in 10 mM sodium phosphate, 150 mM NaCl, pH7.0 buffer. Right: Fluorescence emission spectra (λex = 333 nm) of peptides 13-16 at 5 μM in 10 mM sodium phosphate, 150 mM NaCl, pH7.0 buffer. Inset: Time-correlated single photon counting measurements of (λex = 340 nm, λem = 393 nm) of peptides 13-16 at 5 μM in the same buffer, shown fit to single exponential decays (see ESI for details). Instrument response function is shown in light purple.
The quenching of F* by thioamides is FRET-based, due to spectral overlap between F* emission and thioamide π-π* absorption. The broadening of this absorption band due to thioamide-thioamide interactions alters the spectral overlap integral, and thus the distance dependence of F* quenching. The Förster radius (R0), the distance of half-maximal energy transfer, increases from 14–16 Å for a typical F*-containing monothiopeptide (e.g., 9) to 18.0 Å for dithioamide 11, to 20.5 Å for trithioamide 12 (see ESI for R0 calculations).5 In contrast to F*, quenching of μ, δ, or fluorescein is PET-based for monothioamides, since the emissions of these fluorophores lack any spectral overlap with thioamide absorption. The polythioamide broadening effect observed in the absorption spectra of μ-containing peptides 5-7, and 16 dictates that a FRET-based mechanism will now contribute to the quenching. For example, if we assume that the three thioamides act as one chromophore unit, R0 for peptide 7 is 18.3 Å (see ESI for other R0 values). However, for all of the polythioamide peptides, it is likely that the n-to-π* conformers are only a subset of the conformational ensemble, so that there will also be contributions from conformers where they act as isolated monothioamides. It should also be noted that interactions between the thioamide units are likely to change their oxidation potentials, which would impact the free energy of electron transfer governing PET processes (FRET and PET discussed in more detail in the ESI). Further characterization will be necessary to determine whether we can deconvolute the various FRET and PET components to extract distance information in fluorophore/polythioamide interactions.
It is possible that the change in absorption spectrum also results from the formation of a ground-state complex between the polythioamide and the fluorophore, resulting in static quenching.21 Previous studies of the quenching of a variety of fluorophores in monothiopeptides had shown thioamide PET quenching to be predominantly dynamic, in which the fluorophore is excited, and then electron transfer for quenching.4, 19 We used fluorescence lifetime measurements to characterize the quenching efficiencies (EQτ) for peptides 13-24. Consistent with the steady state measurements, the lifetimes of dithiopeptides were shorter than those of the corresponding monothiopeptides (Fig. 2, Right, and ESI, Table S3). However, the lifetimes were not completely correlated with the steady state fluorescence intensities, which may result from a small static quenching component. We have investigated quenching at multiple concentrations in the μM range using peptides 17-24 (ESI, Fig. S19). We see no evidence for intermolecular static quenching effects or aggregation.
In a proof-of-principle demonstration, we took advantage of the higher levels of quenching by dithioamides to increase the sensitivity of fluorescent thiopeptide-based protease sensors.27 We designed a peptide substrate for trypsin (28) featuring μ at the C-terminus and two thioamide bonds in the N-terminus. The corresponding monothioamide and oxoamide control peptides (25-27) were also generated. As in the proline containing peptides, steady state and fluorescence lifetime data showed quenching by monothioamides in a distance dependent manner and stronger quenching in the dithioamide peptide (Fig. 3 and ESI, Table S2 and Fig. S16). Upon hydrolysis by trypsin, the fluorescence of the oxoamide peptide 25 showed no change, while both the mono- and dithioamide peptides showed strong increases in μ fluorescence (Fig. 3). The timecourse and identity of the peptide fragments were verified by HPLC and mass spectrometry analysis (ESI, Fig. S17). It should be noted that although the thioamide substitution in 26 (between P4 and P3 in protease nomenclature) does not perturb proteolytic kinetics, the thioamide between in 27 (between P3 and P2) slows hydrolysis slightly (ESI, Fig. S17). We do not observe any additional slowing of reaction kinetics in dithiopeptide 28.
Fig. 3.
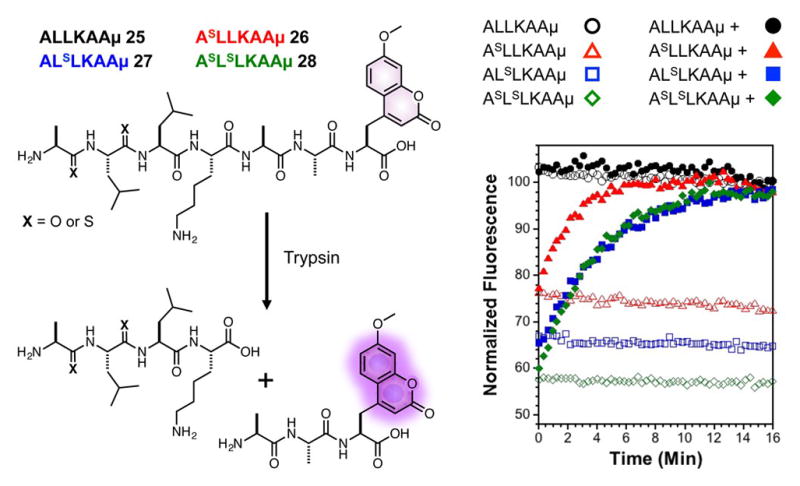
Dithioamide peptides provide improved fluorogenicity in a quenching assay. Time-dependent fluorescence measurements of peptides 25-28 (solid symbols) show an increase in fluorescence in the presence of trypsin, which can be used to monitor protease kinetics in real time. No changes in fluorescence are observed in the absence of protease (open symbols).
Rapid-mixing experiments using a stopped-flow apparatus allowed us to use the increased quenching of the dithioamide in making kinetic measurements of the cleavage of 28 by trypsin (see ESI, Fig. S18). Initial cleavage rates were measured for concentrations of 28 ranging from < 5 μM to > 50 μM. These data were fit to a standard Michaelis-Menten kinetic model to obtain kcat and Km,app values of 0.547 s−1 and 66.7 μM, respectively (see ESI). These values are consistent with previous studies of trypsin using similar substrates.28
In summary, we have demonstrated that one can easily increase the level of fluorogenicity in thioamide-based sensors by simply introducing an additional thioamide. While the examples here are restricted to short peptides, dithioamides can in principle be inserted into full-length proteins by native chemical ligation to generate fluorophore/dithioamide labeled proteins for folding studies, as our laboratory has done for single thioamides.29–31 Additionally, a recent report by Hecht and coworkers describing the incorporation of a dipeptidyl thioamide unit into proteins by mutant ribosomes raises the prospect of incorporating dithioamide units in a similar fashion.32 One may be concerned about the stability of dithioamides (and we have observed “messier” product distributions during peptide synthesis), but we note that several natural products contain adjacent polythioamides in both α-amino acid and β-amino acid peptides.33–35 The impact of multiple thiocarbonyl substitutions can extend beyond natural products and protein analogs to other classes of foldamers such as synthetic β-peptides, α- and β-peptoids, ureas, and azapeptides.36–41 We will continue to evaluate the prospects of dithioamides as fluorescence quenching probes as well as their usage in studying n-to-π* interactions in proteins.
Supplementary Material
Acknowledgments
This work was supported by funding from the University of Pennsylvania, as well as grants from the National Institutes of Health and the National Science Foundation (NIH NS081033 to EJP and NSF CHE-1150351 to EJP). Instruments supported by the NSF and NIH include: MALDI MS (NSF MRI-0820996), stopped flow fluorometer (NSF CHE-1337449), and NMR (NIH RR-022442). JJF is grateful for fellowship support from the NSF (DGE-1321851).
Footnotes
Electronic Supplementary Information (ESI) available: Synthesis of thioamide monomers and peptides, collection of spectroscopic data, peptide stability studies. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Eftink MR, Ghiron CA. Analytical Biochemistry. 1981;114:199. doi: 10.1016/0003-2697(81)90474-7. [DOI] [PubMed] [Google Scholar]
- 2.Giepmans BNG, Adams SR, Ellisman MH, Tsien RY. Science. 2006;312:217. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 3.Speight LC, Samanta M, Petersson EJ. Australian Journal of Chemistry. 2014;67:686. [Google Scholar]
- 4.Goldberg JM, Batjargal S, Chen BS, Petersson EJ. J Am Chem Soc. 2013;135:18651. doi: 10.1021/ja409709x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldberg JM, Batjargal S, Petersson EJ. J Am Chem Soc. 2010;132:14718. doi: 10.1021/ja1044924. [DOI] [PubMed] [Google Scholar]
- 6.Petersson EJ, Goldberg JM, Wissner RF. Phys Chem Chem Phys. 2014;16:6827. doi: 10.1039/c3cp55525a. [DOI] [PubMed] [Google Scholar]
- 7.Wiberg KB, Rush DJ. J Am Chem Soc. 2001;123:2038. doi: 10.1021/ja003586y. [DOI] [PubMed] [Google Scholar]
- 8.Dudek EP, Dudek GO. J Org Chem. 1967;32:823. [Google Scholar]
- 9.La Cour TFM, Hansen HAS, Clausen KIM, Lawesson SO. International Journal of Peptide and Protein Research. 1983;22:509. doi: 10.1111/j.1399-3011.1983.tb02122.x. [DOI] [PubMed] [Google Scholar]
- 10.Lee HJ, Choi YS, Lee KB, Park J, Yoon CJ. Journal of Physical Chemistry A. 2002;106:7010. [Google Scholar]
- 11.Choudhary A, Raines RT. Chembiochem: a European journal of chemical biology. 2011;12:1801. doi: 10.1002/cbic.201100272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Culik RM, Jo H, DeGrado WF, Gai F. J Am Chem Soc. 2012;134:8026. doi: 10.1021/ja301681v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miwa JH, Pallivathucal L, Gowda S, Lee KE. Org Lett. 2002;4:4655. doi: 10.1021/ol027056d. [DOI] [PubMed] [Google Scholar]
- 14.Miwa JH, Patel AK, Vivatrat N, Popek SM, Meyer AM. Org Lett. 2001;3:3373. doi: 10.1021/ol0166092. [DOI] [PubMed] [Google Scholar]
- 15.Reiner A, Wildemann D, Fischer G, Kiefhaber T. J Am Chem Soc. 2008;130:8079. doi: 10.1021/ja8015044. [DOI] [PubMed] [Google Scholar]
- 16.Newberry RW, VanVeller B, Raines RT. Chemical Communications. 2015;51:9624. doi: 10.1039/c5cc02685g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wildemann D, Schiene-Fischer C, Aumüller T, Bachmann A, Kiefhaber T, Lücke C, Fischer G. J Am Chem Soc. 2007;129:4910. doi: 10.1021/ja069048o. [DOI] [PubMed] [Google Scholar]
- 18.Bregy H, Heimgartner H, Helbing J. Journal of Physical Chemistry B. 2009;113:1756. doi: 10.1021/jp8089402. [DOI] [PubMed] [Google Scholar]
- 19.Goldberg JM, Speight LC, Fegley MW, Petersson EJ. J Am Chem Soc. 2012;134:6088. doi: 10.1021/ja3005094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldberg JM, Wissner RF, Klein AM, Petersson EJ. Chemical Communications. 2012;48:1550. doi: 10.1039/c1cc14708k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lakowicz JR. Principles of fluorescence spectroscopy. Springer; 2006. [Google Scholar]
- 22.Mukherjee S, Verma H, Chatterjee J. Org Lett. 2015;17:3150. doi: 10.1021/acs.orglett.5b01484. [DOI] [PubMed] [Google Scholar]
- 23.Bartlett GJ, Choudhary A, Raines RT, Woolfson DN. Nature Chemical Biology. 2010;6:615. doi: 10.1038/nchembio.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choudhary A, Gandla D, Krow GR, Raines RT. J Am Chem Soc. 2009;131:7244. doi: 10.1021/ja901188y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newberry RW, VanVeller B, Guzei IA, Raines RT. J Am Chem Soc. 2013;135:7843. doi: 10.1021/ja4033583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Judge RH, Moule DC, Goddard JD. Canadian Journal of Chemistry. 1987;65:2100. [Google Scholar]
- 27.Goldberg JM, Chen XS, Meinhardt N, Greenbaum DC, Petersson EJ. J Am Chem Soc. 2014;136:2086. doi: 10.1021/ja412297x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang ECW, Hung SH, Cahoon M, Hedstrom L. Protein Engineering. 1997;10:405. doi: 10.1093/protein/10.4.405. [DOI] [PubMed] [Google Scholar]
- 29.Batjargal S, Wang YJ, Goldberg JM, Wissner RF, Petersson EJ. J Am Chem Soc. 2012;134:9172. doi: 10.1021/ja2113245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wissner RF, Batjargal S, Fadzen CM, Petersson EJ. J Am Chem Soc. 2013;135:6529. doi: 10.1021/ja4005943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wissner RF, Wagner AM, Warner JB, Petersson EJ. Synlett. 2013;24:2454. doi: 10.1055/s-0033-1339853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maini R, Dedkova LM, Paul R, Madathil MM, Chowdhury SR, Chen SX, Hecht SM. J Am Chem Soc. 2015;137:11206. doi: 10.1021/jacs.5b03135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banala S, Sussmuth RD. Chembiochem: a European journal of chemical biology. 2010;11:1335. doi: 10.1002/cbic.201000266. [DOI] [PubMed] [Google Scholar]
- 34.Lincke T, Behnken S, Ishida K, Roth M, Hertweck C. Angew Chem Int Ed. 2010;49:2011. doi: 10.1002/anie.200906114. [DOI] [PubMed] [Google Scholar]
- 35.Hayakawa Y, Sasaki K, Nagai K, Shin-ya K, Furihata K. Journal of Antibiotics. 2006;59:6. doi: 10.1038/ja.2006.2. [DOI] [PubMed] [Google Scholar]
- 36.Engel-Andreasen J, Wich K, Laursen JS, Harris P, Olsen CA. J Org Chem. 2015;80:5415. doi: 10.1021/acs.joc.5b00048. [DOI] [PubMed] [Google Scholar]
- 37.Laursen JS, Engel-Andreasen J, Fristrup P, Harris P, Olsen CA. J Am Chem Soc. 2013;135:2835. doi: 10.1021/ja312532x. [DOI] [PubMed] [Google Scholar]
- 38.Gorske BC, Nelson RC, Bowden ZS, Kufe TA, Childs AM. J Org Chem. 2013;78:11172. doi: 10.1021/jo4014113. [DOI] [PubMed] [Google Scholar]
- 39.Nelli YR, Antunes S, Salauen A, Thinon E, Massip S, Kauffmann B, Douat C, Guichard G. Chemistry-a European Journal. 2015;21:2870. doi: 10.1002/chem.201405792. [DOI] [PubMed] [Google Scholar]
- 40.Ottersbach PA, Schnakenburg G, Guetschow M. Tetrahedron Letters. 2015;56:4889. [Google Scholar]
- 41.Kimmerlin T, Seebach D, Hilvert D. Helv Chim Acta. 2002;85:1812. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.