

Abstract
The ability to count biomolecules such as cancer-biomarker miRNAs with the naked eye is seemingly impossible in molecular diagnostics. Here, we show an ultrasensitive naked-eye-counting strategy for quantifying miRNAs by employing T7 phage—a bacteria-specific virus nanoparticle—as a surrogate. The phage is genetically engineered to become fluorescent and capable of binding a miRNA-capturing gold nanoparticle (GNP) in a one-to-one manner. Target miRNAs crosslink the resultant phage–GNP couple and miRNA-capturing magnetic microparticles, forming a sandwich complex containing equimolar phage and miRNA. The phage is then released from the complex and developed into one macroscopic fluorescent plaque in a Petri dish by plating it in a host bacterial medium. Counting the plaques by the naked eye enables the quantification of miRNAs with detection limits of ~3 and ~5 aM for single-target and two-target miRNAs, respectively. This approach offers ultrasensitive and convenient quantification of disease biomarkers by the naked eye.
Molecular diagnostics aims to detect specific DNA, RNA or proteins to help diagnose and monitor disease, assess disease risk, and determine what treatments work best for individual patients. Therefore, its development enables the advancement of personalized and precision medicine1. However, the ability to quantify these molecules using an intuitive and simple method remains a challenge. The widely used methods in clinics, including enzyme-linked immune-sorbent assay (ELISA) and immunohistochemistry (IHC) for proteins, as well as polymerase chain reaction (PCR) for DNA or RNA, provide only a relative measurement method that must be calibrated by a standard curve. In recent years, nanotechnology has been applied in the field of molecular diagnostics, and generated many promising nanostructure-based diagnostic methods, such as gold nanoparticle (GNP) labelling2, GNP-based colorimetric discrimination3, Raman spectroscopic fingerprinting4 and Bio-barcode detection5–8. Labelling oligonucleotide targets with GNPs can discriminate the single-base mismatches due to the altered sharp melting profiles of targets2. GNP probes labelled with oligonucleotides and Raman-active dyes provide multiplexing and rationing capabilities for DNA and RNA detection owing to the advantage of the Raman-active dyes being a narrow-band spectroscopic fingerprint3,4. Bio-barcode strategies using diverse molecules, such as organic molecules5, peptides6 and DNA7,8, as a surrogate of the target of interest can reach a detection limit as low as 1 aM, characteristic of the sensitivity of PCR. However, so far, none of the detection methods can achieve the goal of counting target molecules with the naked eye. A methodology allowing direct counting of target molecules with ultrahigh sensitivity would be extremely beneficial for clinical diagnostics.
Counting strategy
Herein, we report a naked-eye-counting method (Fig. 1a) for quantifying miRNAs, with sensitivity comparable to PCR. This method relies on the use of two probes. One is a GNP probe dually modified with a recombinant GNP-binding fluorescent T7 phage and an oligonucleotide complementary to one segment of the target miRNAs. The other is a magnetic microparticle (MMP) probe functionalized with an oligonucleotide complementary to another segment of the same miRNAs. The target miRNAs are recognized and thus captured by the two probes to form a sandwich complex due to DNA/RNA hybridization. Then a gold-binding peptide (GBP) that can competitively bind to the GNPs is added to release the T7 phages from the GNPs in the complex, followed by magnetic separation to remove the GNP–MNP complex and purify the T7 phages from the complex. Subsequently, the fluorescent T7 phages are plated on a host bacterial medium in a Petri dish to develop fluorescent plaques (Fig. 1a). Because the number of T7 phages is equal to not only that of the target miRNAs before separation from the complex, but also that of the developed macroscopic eye-visible plaques, the quantity of target molecules is easily determined by directly counting the fluorescent plaques in a Petri dish. The innovative feature of this strategy is that T7 phages are assembled with GNPs in a ‘one-to-one’ manner to form a T7–GNP complex under appropriate conditions and serve as a surrogate that correlates the number of target miRNAs and eye-visible plaques.
Figure 1. General concepts of the counting strategy.
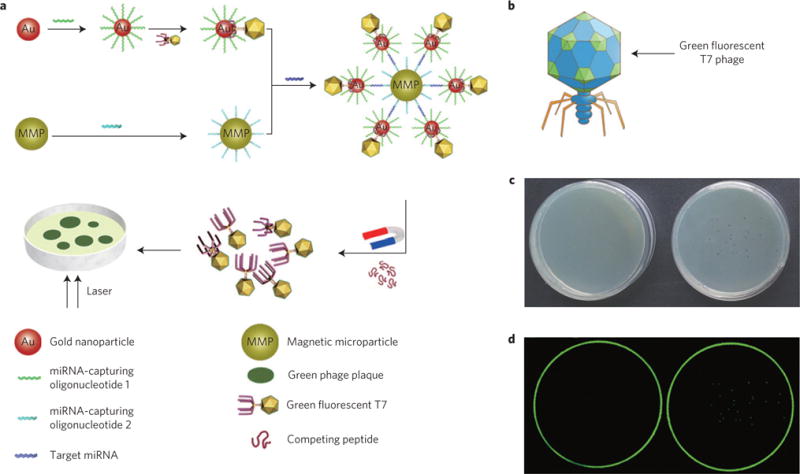
a, Schematic representation of the counting strategy (not drawn to scale). A T7–GNP probe is formed by first conjugating a thiolated miRNA-capturing oligonucleotide onto a GNP, which is then specifically bound with fluorescent T7 phage displaying GBP at the terminus of its tail fibres to form a T7–GNP couple in a one-to-one format. Another probe, the MMP probe, is prepared by conjugation of another biotinylated miRNA-capturing oligonucleotide onto a MMP. Then the MMP probe and the T7–GNP probe are mixed in the presence of the target miRNA. The recognition and capture of the target miRNA by the two different miRNA-capturing oligonucleotides on the GNP and MMP probes leads to the formation of a sandwich complex. The two different miRNA-capturing oligonucleotides are complementary and thus bind to two different segments (at two distal ends) of the same miRNA target molecule, favouring the formation of the sandwich complex. When a high concentration of GBP, which can compete for the binding sites on GNPs with T7 phages, is added to the complex, the T7 phages are released from the complex. Then the T7 phages and the rest of the sandwich are magnetically separated. By plating the purified T7 phages in the Petri dish, plaques are formed. Because the number of fluorescent plaques is equal to that of the T7 phages, which is originally equal to the number of miRNA target molecules in the complex, counting the number of fluorescent plaques that can be seen in the Petri dish by eye results in quantification of the miRNA. b, Schematic diagram of recombinant fluorescent T7 phage. The green colour on the capsid of recombinant fluorescent T7 phage represents the green fluorescence protein. c, Photographs of a Petri dish showing the presence of plaques when green fluorescence T7 phages are plated on the host bacteria media (right) and the absence of plaques when no phage is used to infect bacteria (left). d, Images of the same Petri dish shown in c under the fluorescence scanner. The Petri dish on the right shows green plaques whereas the one on the left does not. An excitation wavelength of 488 nm is used in detecting the green fluorescent plaques derived from the green fluorescent T7 phage–GNP probes.
Phage is a virus that specifically infects bacteria. We, along with others, have used non-lytic phages or plant viruses—that is, viruses that do not kill their host cells during infection (such as M13 phage or tobacco mosaic virus), to assemble metal or inorganic nanoparticles9–15 and detect target molecules such as cancer biomarkers16,17. Lytic T7 phage (Fig. 1b), the virus that kills and breaks open bacteria during infection, is another widely used protein/peptide display platform. It has a unique structure, different from a non-lytic phage such as M13. It consists of a spherical protein capsid (encapsulating DNA) with an inner diameter of 55 nm attached to six tail fibres (2 nm in diameter and 32 nm in length)18. Both the capsid and tail fibres can be genetically modified to display foreign peptides or proteins. Hence, T7 phage is an ideal surrogate species owing to its nanoscale size and ease of genetic modification19. In addition, it shows rapid reproductive ability; one phage can be developed into one clear plaque in its host Petri dish within 3 h at 37 °C.
Double display of fluorescent protein and GBP on T7 phage
We first modified two loci of the T7 phage genome, one is gene 10 (encoding a coat protein constituting the spherical capsid) for displaying fluorescent protein, and the other is gene 17 (encoding a coat protein forming the tail fibres) for displaying GBP (Supplementary Fig. 1). Specifically, by inserting a gene encoding a fluorescent protein, such as enhanced green fluorescent protein (EGFP) or red fluorescent protein (RFP), into the cloning regions of T7Select10-3b vector (Novagen), the fluorescent protein is incorporated into the spherical capsid of the T7 phage, causing the macroscopic plaques derived from it to emit either green or red light under a fluorescence scanner. Similarly, inserting a gene encoding GBP into gene 17 results in the display of GBP near the tips of the six tail fibres, allowing the tails of the T7 phage to acquire the gold-binding ability needed to construct the T7–GNP probe, where one GNP is attached to one T7 phage (Fig. 1a). The key design feature of the ‘one-to-one’ T7–GNP assembly is that one plaque, which is derived from one T7 phage released from the sandwich complex, represents one target molecule (that is, miRNA). Consequently, the exact number of captured target molecules in the complex corresponds to the number of plaques (Fig. 1a).
It is well established that various fluorescent proteins can be fused to the capsid (product of gene 10) of the T7 phage20,21. Consequently, T7 phages displaying different fluorescent proteins can be used to represent different molecular targets of interest, enabling our strategy to achieve multiplexed detection. In this study, two types of fluorescent phage particles were obtained by inserting the EGFP or RFP gene between the BamH I and Xho I sites of T7 Select10-3b vector (Supplementary Fig. 2a). As a result of the plating experiment with the constructed EGFP-displaying T7 phages, plaques developed from the recombinant T7 phages were visible to the naked eye (Fig. 1c, right) and appeared clearly green in the Petri dish under a fluorescence scanner at an excitation wavelength of 488 nm (Fig. 1d, right). However, no plaques were formed when no T7 phages were present (Fig. 1c,d, left picture). Furthermore, EGFP-displaying T7 phages were amplified by infecting host bacteria. An agarose (1.0%) gel-electrophoresis image resulting from the PCR reaction with the recombinant T7 phages as a template confirmed that the EGFP sequence was fused to gene 10 (Supplementary Fig. 3). Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS–PAGE) and western-blotting analyses further confirmed the successful construction of the green fluorescent T7 phage (Supplementary Fig. 4). The red fluorescent T7 phages were constructed in a similar manner.
To make the fluorescent T7 phage capable of binding a GNP to form a T7–GNP couple (Fig. 1a), we displayed a reported GBP (VSGSSPDS; ref. 22) at the terminus of the six tail fibres of the phage by inserting its gene into gene 17 (Supplementary Fig. 2b,c), as the display of the peptide does not affect the infection capability of T7 phage23. Specifically, GBP was fused to the C terminus of the product of gene 17 of the recombinant fluorescent T7 phage genome24 by overlapping PCR. Towards this end, the genome of the fluorescent T7 phages was first extracted from the lysate of host bacteria BLT5403 with the genome extraction kit (Genmed), and then cleaved into three segments by the restriction enzymes Alw I and Pm1 I. The two fragments (Supplementary Figs 2c and 5), fragment 1 and 2, were collected and connected with the modified version of gene 17 (Supplementary Figs 2b and 6IV) to construct the designed recombinant T7 genome. The modified version of gene 17 was obtained by overlapping PCR technology with fragment 3 (Supplementary Figs 2b and 6II) and fragment 4 (Supplementary Figs 2b and 6III). Successful overlapping was confirmed by PCR (Supplementary Fig. 6IV). After the two fragments were connected with the modified version of gene 17 using a ligase, the reaction product was packaged in vitro with T7 Packaging Kit (Novagen), and then allowed to infect BLT5403 culture. The lysate supernatant was collected to develop the positive plaques. The band size of the PCR product of one of the positive clones was consistent with the expected modification, which confirmed the successful construction of the desired T7 phage (Supplementary Fig. 7II).
Formation of probes for counting
To demonstrate our counting strategy, we chose miRNAs as a target analyte. miRNAs are a family of small non-coding regulatory RNAs and can serve as promising biomarkers of various types of cancer25–28. Quantitative PCR (qPCR) is widely used nowadays in miRNA assays because of its sensitivity. However, it is still limited by its high cost and false positivity29. Therefore, there is a pressing need for a new method that can reliably quantify miRNAs with lower cost and high fidelity. First, let-7a (5′-UGAGGUAGUAG GUUGUAUAGU-3′), a promising biomarker of non-small-cell lung cancer30, was chosen as a single miRNA target. Accordingly, the T7–GNP probe was prepared by initial functionalization of the GNPs with a thiolated miRNA-capturing oligonucleotide 1 (5′-AAAAAAAAAAAAAAAACTATACAACC-3′, 1), followed by binding with the GNP-binding fluorescent T7 phages through the GBP displayed at the tip of their tail fibres (Fig. 1a). The GNPs used were ~20 nm in size and showed good dispersibility (Supplementary Fig. 8a). The 1-modified GNPs on the TEM grid were aggregated into an evenly spaced planar structure because negatively charged oligonucleotides coated on the GNPs made them repel each other (Supplementary Fig. 8b). This result indicates that a high density of miRNA-capturing oligonucleotide has been coated on the GNPs. The 1-modified GNPs (Supplementary Fig. 9II) before hybridization had a red colour, similar to that of the as-synthesized GNPs (Supplementary Fig. 9I), which was in agreement with the surface plasmon resonance (SPR) band at 522 nm (Supplementary Fig. 10, red and blue curve). The hybridization capability of the 1-modified GNPs is the basis for the T7–GNP probes to capture target miRNA. To demonstrate such capability, 1-modified GNPs (2.5 nM) and GNPs (2.5 nM) functionalized by thiolated miRNA-capturing oligonucleotide 2 (5′-TACTACCTCAAAAAAAAAAAAAAAA-3′, 2) were incubated in the presence of excess let-7a (1 μM). Subsequently, the resultant red mixture turned blue (Supplementary Fig. 9IV), in contrast to the control in the absence of let-7a (Supplementary Fig. 9III). The blue shift is due to aggregation of the GNPs caused by hybridization (Supplementary Fig. 8c). This result confirms that these oligonucleotide-modified GNPs have an excellent ability to capture complementary miRNAs.
To assemble 1-modified GNPs and fluorescent T7 phages in a ‘one-to-one’ manner to form T7–GNP probes (Fig. 1a), the GNP-binding fluorescent T7 phages were mixed with excess 1-modified GNPs. The resultant T7–GNP complexes were then purified by CsCl gradient centrifugation, followed by dialysis against an assay buffer (0.2 M NaCl, 10 mM phosphate buffer, 0.1% Tween 20, pH 7.4). The gradient centrifugation was found useful in purifying T7–GNP complexes with a T7/GNP molar ratio of 1 and in removing the excess GNPs (Supplementary Fig. 11a). Transmission electron microscopy (TEM) images showed that T7–GNP probes were formed and in rare cases two T7 phages were attached to one obviously bigger GNP (Supplementary Fig. 11b). After optimizing the GNP synthesis protocol to avoid GNPs with larger diameters, we obtained uniform GNPs with a diameter of 20 nm (Supplementary Fig. 8a) for preparing T7–GNP probes. Successful purification of the T7–GNP probes (Supplementary Fig. 12III) after gradient centrifugation was confirmed by TEM. TEM observation clearly showed that GNPs were indeed bound with the T7 phages in a ‘one-to-one’ manner (Fig. 2b). However, when T7 phages did not display GBP at their tail fibres, T7–GNP complexes were not formed (Fig. 2a). In addition, oligonucleotide-modified GNPs (Supplementary Fig. 8b) and T7 phages (with anionic coat proteins31) are both anionic and thus tended to repel each other. Therefore, they could not be bound together unless T7 phages were engineered to display GBP. In addition, the ultraviolet–visible absorption of T7–GNP probes (Supplementary Fig. 10, green curve) at 280 nm was obviously different from that of GNPs (Supplementary Fig. 10, red curve) or 1-modified GNPs (Supplementary Fig. 10, blue curve) owing to the presence of T7 phage in the T7–GNP probes. Surprisingly, T7–GNP probes stored at 4 °C for nearly one year still maintained the one-to-one combination of T7 phages and GNPs (Fig. 2c), indicating the excellent stability of our probes.
Figure 2. Preparation and characterization of T7–GNP probes.
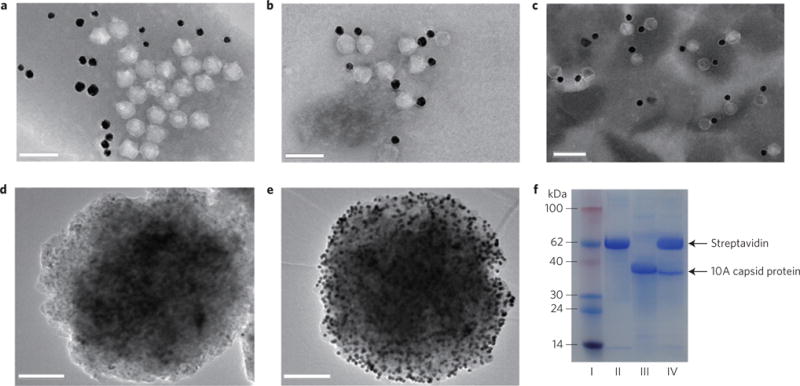
a, TEM image showing that when T7 phages not displaying GBP were mixed with oligonucleotide-modified GNPs under gentle shaking conditions for 5 h at 25 °C, the T7 phages did not have specific binding interactions with GNPs. b, TEM image of T7–GNP probes showing that when the engineered GNP-binding T7 phages were mixed with oligonucleotide-modified GNPs under the same conditions, T7 phages and GNPs were specifically bound together to form a couple in a one-to-one manner owing to the display of GBP at the tails of T7 phages. c, TEM image of T7–GNP probes stored for up to 12 months. d, TEM image of the MMP generated from a mixture solution containing 40 μl of 2-functionalized MMP probes at 2.5 mg ml−1, 40 μl of 1-functionalized GNPs at 2.5 nM and 20 μl of an assay buffer for 30 min, followed by washing six times with magnetic separation. e, TEM image of the MMP–GNP complex magnetically separated from a hybridization mixture containing 40 μl of 2-functionalized MMP probes at 2.5 mg ml−1, 40 μl of 1-functionalized GNPs at 2.5 nM and 20 μl of let-7a at 1 μM. The data show that the MMP probe has good ability to hybridize with target miRNA. f, SDS–PAGE analysis of different samples. I, protein marker. II, MMP probes. III, engineered T7 phages. IV, MMP probes separated by a magnetic separator in the presence of target miRNA molecules. These results confirmed the presence of T7 phage in the sandwich complexes. Scale bars, 100 nm (a–c); 200 nm (d, e).
To form MMP probes, commercial MMPs (Dynabeads MyOne Streptavidin T1, Invitrogen), superparamagnetic beads (~1 μm in diameter) coated with a monolayer of recombinant streptavidin and blocked with bovine serum albumin, were modified with a biotinylated 2 through avidin–biotin interaction. In a sandwich assay the resultant 2-modified MMP probes, let-7a and 1-modified GNPs were incubated and the MMP probes were then magnetically separated. We found that the magnetically separated MMP probes were decorated with hundreds of GNPs (Fig. 2e) as a result of the hybridization between let-7a and the miRNA-capturing oligonucleotides on the GNPs and MMPs. However, no binding between GNPs and MMP probes was seen in the absence of let-7a (Fig. 2d). These results confirmed that the MMP probes were successfully prepared.
Counting single and double miRNAs
The feasibility of the counting strategy was tested for the detection of single miRNAs. In the proof-of-concept experiments, 10 μl of let-7a (1 μM) and 40 μl of each probe (MMP probes at 2.5 mg ml−1 and T7–GNP probes at 2.5 nM) were mixed to form sandwich complexes. The formation of the sandwich complexes allowed the T7–GNP probes (complexed with MMP probes) to be driven towards the tube walls by a magnetic field, generating a colour change in the solution due to the removal of GNPs from the solution phase (Supplementary Fig. 13II). However, under the same magnetic field, the solution in the absence of let-7a remained red (Supplementary Fig. 13III) because T7–GNP probes were not associated with MMPs to form the sandwich complexes and remained in the solution phase. SDS–PAGE was employed to verify the presence of both MMPs and T7 phages in the sandwich complex (Fig. 2f). After the sandwich complexes were magnetically collected, T7 phage could be released from the complexes by adding excess synthesized GBP that could competitively bind to the GNPs, as indicated by using SDS–PAGE and agarose electrophoresis analysis (Supplementary Figs 14 and 15). We also verified that the sandwiches contained T7 phages and GNPs in a one-to-one ratio by adding an excess oligonucleotide perfectly complementary to let-7a to break the sandwich complex and by using TEM imaging (Supplementary Fig. 16a) and SDS–PAGE analysis (Supplementary Fig. 16b).
To count the single miRNA target, the sample of let-7a was diluted down to 30 aM (equivalent to ~180 molecules in 10 μl), and 97 individual fluorescent plaques were visible under the fluorescence scanner (Fig. 3c). When the let-7a concentration was reduced to 3 aM (~20 molecules in 10 μl), 11 green fluorescent plaques (Fig. 3b) were still visible. No plaques (Fig. 3a) were observed in the absence of let-7a, indicating the lack of false positivity. In the absence of target miRNAs, MMP probes were not prone to adsorb T7–GNP probes or T7 phages, owing to the repulsive interaction between the negatively charged oligonucleotides on the surface of MMPs and GNPs, and the negatively charged capsid of the T7 phage. Parallel experiments with various concentrations indicated that the counting strategy had good reproducibility in quantifying let-7a at each specific concentration (Fig. 3d and Supplementary Fig. 17a). To test the accuracy of the method, qPCR was used to quantify the samples with miRNA concentrations of 3 aM or 30 aM. The qPCR analysis showed that the concentrations of the samples were accurate (Supplementary Fig. 18), and the capturing efficiency of the counting strategy was approximately 50% (Fig. 3d and Supplementary Fig. 17b). The capturing efficiency could be used to calculate the actual number of target molecules, which approximately doubles the number of plaques counted in the Petri dish.
Figure 3. Determination of both single-target and multiple-target miRNAs by the counting strategy.
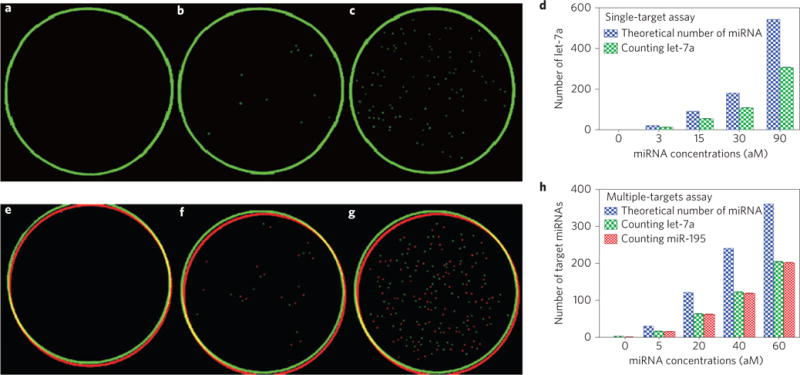
a–d, Single-target mRNA counting. a, In the absence of let-7a. b, In the presence of 3 aM of let-7a. c, In the presence of 30 aM of let-7a. The green circles in a–c originate from the border of the Petri dishes during fluorescent scanning. d, Statistical histogram of the number of target miRNA counted, which is equal to the number of green plaques counted, using the counting strategy in the presence of the single target let-7a at different concentrations. e–h, Multiple-target miRNA counting. e, In the absence of let-7a and miR-195. f, In the presence of 5 aM each of let-7a and miR-195. g, In the presence of 30 aM each of let-7a and miR-195. The red and green circles in e–g originate from the border of the Petri dishes during fluorescent scanning. h, Statistical histogram of the number of target miRNA counted, which is equal to the number of green/red plaques counted (corresponding to let-7a and miR-195, respectively) using the counting strategy in the presence of both let-7a and miR-195, each at different concentrations. The analysis shows that the theoretical target number of the miRNA is approximately twice that of the number of plaques counted—that is, Y ≈2X or X ≈50%Y —suggesting a capturing efficiency of ~50%. N =6, p <0.05.
The ability to distinguish multiple targets simultaneously in microlitres of samples is a desirable feature for future clinical applications. As diverse fluorescent phages could serve as labels for different targets, we constructed GNP-binding red fluorescent T7 phages as the second target’s surrogate. The miR-195 (5′-UAGCAGCACAGAAAUAUUGGC-3′), an important regulator in human hepatocellular carcinoma32, was chosen along with let-7a for multiple miRNAs assay. The procedure for preparing the second T7–GNP probe is similar to that for the first probe, except for the conjugation of a new oligonucleotide (5′-AAAAAAAAAAGCCAATATTTC-3′) with GNPs for capturing the new miRNA, and the complexation of the resultant GNPs with GNP-binding red fluorescent T7 phage to form a couple. Likewise, the second MMP probe was functionalized with a new miRNA-capturing oligonucleotide (5′-TGTGCTGCTAAAAAAAAAAA-3′) that can form a sandwich structure with miR-195 and the second T7–GNP probe. After preparing the second pair of probes for detecting target miR-195, we explored the performance of the counting strategy when both let-7a and miR-195 coexisted in a sample. A 10 μl mixture of the two target miRNAs, each at 30 aM, was first used as the sample. The result indicated that 101 green plaques and 94 red plaques were simultaneously detected in a Petri dish (Fig. 3g). Furthermore, the detection limit for the two targets was found to be 5 aM (~30 molecules in 10 μl). At this concentration, there were 15 green plaques and 11 red plaques in a Petri dish (Fig. 3f), whereas no fluorescent plaques were visible in the absence of let-7a and miR-195 (Fig 3e). Good reproducibility of the counting strategy and a target capturing efficiency of ~50% were confirmed for simultaneous quantification of two targets at various concentrations (Fig. 3h and Supplementary Fig. 19).
It is very important for a bioanalytical method to quantify miRNAs with single-base mutation, because a large number of miRNAs have similar sequences. Hence, we then proceeded to demonstrate that our counting method can distinguish let-7a from single-nucleotide polymorphisms. Our data (Supplementary Fig. 20) suggest that the counting strategy could distinguish let-7a from its single-base mutation variants at 29 °C. The data suggest that the probes modified with the sequence perfectly complementary to let-7a did not bind to the mutant with a single mismatch in the middle site of the sequence at 29 °C, even at a higher concentration of the mutant. Because some miRNAs happen to be the scrambled version of other miRNAs in the human body, the capability of our method in distinguishing them is also very important. Thus, we further assayed two samples, one containing the target miRNA (miR-21) at 20 aM and its scrambled version (miR-95) at 200 aM, and the other containing only 200 aM of miR-95 (Supplementary Fig. 21). The result showed that our counting method worked well in distinguishing the target miRNA and its scrambled sequence (Supplementary Fig. 21b,c).
To demonstrate that our method can be used in clinics, we further applied it to quantify miRNAs in clinical human samples including cells (using cancer cells as an example), body fluids (using serum as an example) and tissues (using tumour tissue from cancer patients as an example). First, we employed the counting method to quantify miRNAs with an altered level of expression in cancer cells due to chemical treatment. Briefly, we cultured HepG2 cancer cells (purchased from the Institute of Biochemistry and Cell Biology of the Chinese Academy of Science and then verified not to be contaminated by mycoplasma) until the cells occupied 80% of the culture plate, and then treated them with 300 ng TNF-α per plate. We then harvested the cells and extracted miRNAs from the cells using a commercial miRNA extraction kit (Mivana mirna isolation kit, Ambion). We selected two miRNAs (has-miR-210-5p and has-miR-1246) for detection from the extracted miRNAs. Finally, we assayed the two miRNAs in these samples using both the counting method and the qPCR method. The change in the expression level of the two miRNAs determined by our counting method is consistent with that determined by qPCR (Fig. 4a and Supplementary Fig. 22). The data confirm that the counting strategy is capable of detecting the change in the level of miRNAs expression in cancer cells. Second, we used the counting method to detect miRNAs from spiked human serum. Briefly, we removed one or two target miRNAs (let-7a and miR-195) from the human serum by first adding MMPs functionalized with the oligonucleotide sequence complementary to the target miRNAs and then using an external magnetic field to remove the miRNA-bound MMPs (Supplementary Table 1). Then we added target miRNAs in a known amount to serum devoid of the corresponding target to form serum samples with known concentrations of the target. We finally applied the counting method to detect a single target (let-7a) and two targets (let-7a and miR-195) from the spiked serum samples. The results (Fig. 4b,c and Supplementary Fig. 23) show that the counting method is reproducible and accurate in assaying the serum samples. Third, we detected miRNAs in tumour tissue and its nearby healthy tissue (para-carcinoma tissue) of lung-cancer patients using the counting method. It should be noted that such detection can give clues about tumour development and prognosis after tumour treatment. Briefly, we used a block of tumour-tissue sample and a piece of para-carcinoma tissue from the same patient with non-small-cell lung cancer and explored the possibility of assaying these tissues using our counting method. We chose two miRNAs (hsa-let-7b-5p and has-miR-21-5p) because their expression level was different in tumour and para-carcinoma tissue33,34. Then we employed our counting method to assay these samples and compared the data from the counting method and the control method (qPCR). The new results (Fig. 4d and Supplementary Fig. 24) undoubtedly show that the counting strategy is robust in counting miRNAs in tissue samples from the patients Moreover, the efficiency of capturing miRNAs remains approximately 50% (Fig. 4b–d). Collectively, these experiments (Fig. 4 and Supplementary Figs 22–24) prove that the counting strategy is capable of precisely detecting miRNAs from the clinical samples (cells, tissues, and body fluids) with high sensitivity.
Figure 4. Quantification of various real samples (cells, serum and tissues) by the counting strategy.
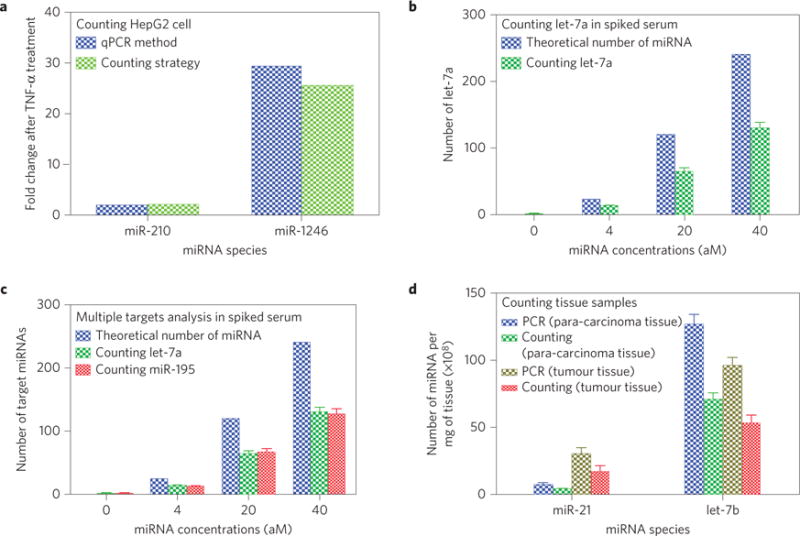
a, Fold change of the levels of each of the two miRNAs in HepG2 cancer cells after TNF-α treatment determined by our counting strategy and the qPCR method. A comparison between the two methods shows that the counting method can reliably determine the altered levels of miRNA expression due to chemical treatment of the cancer cells. b, Number of target miRNA (let-7a) in 10 μl of spiked human serum at various nominal concentrations determined by the counting method, as compared to the theoretical number. c, Number of target miRNAs (let-7a and miR-195) in 10 μl of spiked human serum at various nominal concentrations determined by the counting method, as compared to the theoretical number. To prepare the spiked serum samples in b and c, serum from a healthy person was made devoid of the target miRNAs (let-7a in b or both let-7a and miR-195 in c) by using a MMP probe functionalized with the sequence complementary to the target miRNAs to capture and magnetically remove the target miRNAs. Then a known amount of target miRNAs was added to form serum samples with specific concentrations of the target miRNAs. d, Numbers of two target miRNAs in 1 mg of the tissue (tumour or the surrounding healthy tissue) from a lung-cancer patient determined by the counting method and by qPCR. A comparison of the data from both methods confirms that the counting method is reliable in determining the level of miRNAs in the tissues of human cancer patients. The number of miRNAs was calculated by averaging the number obtained from three independent counting tests (p <0.05 with respect to the control).
Concluding remarks
Our counting strategy for quantifying miRNAs is in principle simpler and easier to perform than the qPCR technique. We estimate that both methods take a similar time to achieve detection, yet the counting strategy is less costly. A reliable qPCR method requires normalization of the data using a housekeeping gene as an endogenous control, which is expected to have constant quantity. Unfortunately, an ideal housekeeping gene has not yet been found35–37. Although our strategy for counting single or multiple miRNAs with good reproducibility is suitable only for counting miRNAs at less than fM concentrations owing to the limited capacity to accommodate plaques in a Petri dish, samples at a higher concentration can be quantified after diluting them first or by employing a spotting robot to spot nanolitres of samples. In short, using this naked-eye-counting strategy, one can quantify miRNAs and easily perform the assay in a few hours because macroscopic plaques can be developed in less than 3 h. Furthermore, if integrated into an automatic fluorescence-scanning analysis system, the counting would take even less time. Importantly, because the strategy combines the advantages of nanoparticles and biological species, it could be readily expanded to count other species, such as DNA, antigens and viruses. The counting strategy may open up new avenues for the ultrasensitive detection of disease biomarkers and thus should find potential applications in clinical diagnosis—in particular, in the early detection of diseases such as cancer.
Methods
Construction of fluorescent T7 phage by displaying EGFP or RFP on the capsid
First, the green fluorescent gene was obtained by PCR with primers (forward: 5′-ATATCGGGATCCGATGGTGAGCAAGGGCGAGGA-3′, reverse: 5′-ATCGCTCGAGTTACTTGTACAGCTCGTCC-3′, the bold nucleotides are the BamH I and Xho I recognition sites, respectively) and using the pEGFP vector (Clontech) as templates (Supplementary Fig. 2a). Second, the green fluorescent gene fragments and the T7 Select10-3b DNA (Novagen) were digested with BamH I and Xho I restriction enzymes (New England Biolabs) in respective reactions and subjected to agarose gel electrophoresis. Third, the two fragments from T7 Select10-3b DNA digestion and the EGFP fragment from PCR were recovered by Gel Extraction Kit (Axygen) and mixed in one reaction tube for ligation with T4 ligase (New England Biolabs). The resulting product was packaged in vitro with T7 Packaging Kit, followed by infecting host bacteria BLT5403 grown to an OD600nm of 0.6–1.0 to obtain a large number of the recombinant green fluorescent T7 phages in the lysate by centrifugation. A plating assay was performed to examine the formation of green fluorescent plaques with the recombinant GFP-displaying T7 phage lysate. Briefly, first 10 μl of the lysate was mixed with 200 μl of host BLT5403 (Novagen) grown to an OD600nm of 0.6–1.0 and 3 ml of molten top agar solid Luria-Bertani (LB) medium, then the mixture was poured onto a solid agar Petri dish with carbenicillin to develop fluorescent plaques. A fluorescence scanner was employed to check whether fluorescent plaques were present in the Petri dish and determine their number.
Construction of the red fluorescent T7 phage is similar to that described above, except for the use of the DsRed gene sequence from pDsred-N1 (Clontech) to replace the EGFP of the recombinant T7 genome in the same BamH I and Xho I restriction sites adjacent to the 3′-terminal of gene 10 with primers (forward: 5′-ATATCGGGATCCGATGGCCTCCTCCGAGAACGTCA-3′, reverse: 5′-ATCGCTCGAGTTACAGGAACAGGTGGT-3′). The successful construction of the RFP-displaying T7 phage was verified by the PCR product with both gel electrophoresis and DNA sequencing.
Construction of fluorescent T7 phage displaying gold-binding peptide
First, the DNA genome of the engineered fluorescent T7 phage was extracted from the lysate of host bacteria BLT5403 with the genome extraction kit (Genmed), and then cleaved into three fragments by the restriction enzymes Alw I and Pm1 I. The two fragments, termed fragment 1 and 2, were collected and connected with the modified version of gene 17, which was obtained by overlapping PCR with fragments 3 and 4 to construct the recombinant T7 genome with the gene encoding the GBP incorporated (Supplementary Fig. 2,b,c). Fragment 3 (~200 bp) was generated by PCR using a pair of primers (forward: 5′-TAGATCGGATCTCCGCTTCCGCAATATCTGG-3′; reverse: 5′-TTATGAGTCAGGTGATGAACCTGATACCTCGTTCTCCACCATG-3′; the underlined nucleotide is Alw I recognition site) and using T7 genome DNA as a template. Fragment 4 (~1,800 bp) was generated by PCR using a pair of primers (forward: 5′-GTATCAGGTTCATCACCTGACTCATAATTGGTAAATCACAAGGAAAGAC-3′; reverse: 5′-GGAATTCACGTGTCCTTGGGTACAGAGCAG-3′; the bold nucleotide is the oligonucleotide sequence responsible for coding the GBP ‘VSGSSPDS’. The underlined nucleotide is the Pml I recognition site). The modified version of gene 17 was implemented by overlapping PCR using a pair of primers (forward: 5′-TAGATCGGATCTCCGCTTCCGCAATATCTGG-3′; reverse: 5′-GGAATTCACGTGTCCTTGGGTACAGAGCAG-3′) and using the above fragment 3 and 4 as templates. The PCR product was connected with the two larger fragments (fragment 1 and 2) using T4 ligase to generate the recombinant T7 phage genome with genes encoding fluorescent protein and GBP incorporated (Supplementary Fig. 2c). Then the ligation product was packaged in vitro with T7 Packaging Kit and allowed to infect BLT5403 culture to produce the recombinant T7 phages. Finally, the T7 phages displaying both fluorescent protein and GBP were collected to develop the plaques and further verified by PCR and sequencing.
Amplification and purification of T7 phages packaged in vitro
After packaging in vitro, the T7 phages were added to 3 ml of BLT5403 culture grown to an OD600nm of 0.6–1.0 and the culture was shaken for 2–3 h until lysis. The lysate supernatant was collected by centrifugation at 10,000g for 10 min. 10 μl of the supernatant was decanted into 0.2 ml of BLT5403 grown at the exponential phase, followed by mixing with 3 ml of melted top agar at 45 °C. The contents of the tube were immediately mixed and poured into a standard 10 cm Petri dish containing 10 ml of solid LB medium and 500 μg of carbenicillin. The plate was allowed to stand for several minutes until the top agar hardened. Then it was inverted in an incubator and incubated for 3 h or overnight at 37 °C to develop plaques.
A few green or red fluorescent plaques in a Petri dish were picked using pipette tips and transferred into host BLT5403 culture grown to a OD600nm of 0.6–1.0 for amplification. After a few hours the recombinant T7 phage particles were purified from clarified lysate by precipitation with polyethylene glycol (PEG 8000) and dissolved in a small amount of buffer (1 M NaCl, 10 mM Tris-HCl, pH 8.0, 1 mM EDTA). The concentrated phage solution was layered atop four layers of CsCl solutions with different densities in a clear ultracentrifuge tube, followed by banding in a CsCl step gradient by centrifugation at 4 °C for 2 h at 288,000g (Beckman, Optima L-100XP, SW41 rotor). The purified phages, which aggregated clearly into a thin and turbid band of the centrifugation tube, were collected and subjected to subsequent verification or further purified by dialysis against an assay buffer (0.2 M NaCl, 10 mM phosphate buffer, 0.1% Tween 20, pH 7.4).
Quantifying miRNAs by our naked-eye-counting strategy
10 μl of miRNAs samples at a certain concentration were prepared. 40 μl of MMP probes (2.5 mg ml−1) in the aforementioned assay buffer and 50 μl of the assay buffer were both added into the sample solution, followed by incubation at 37 °C for 30 min under gentle shaking to generate complexes of MMP probes and target miRNAs. The complexes were then separated magnetically and washed six times with the assay buffer, followed by mixing with 40 μl of T7–GNP probes (2.5 nM) and 60 μl of the assay buffer at 37 °C for another 30 min. The resulting sandwich complexes were again extracted magnetically and washed with the assay buffer a further six times. T7 phages were released from the sandwich complexes by adding 0.1 ml of 2 μM synthesized GBP, followed by incubation for at least 15 min under gentle shaking at 37 °C. After centrifugation, the supernatant containing the liberated T7 phages was plated on a Petri dish with the host bacteria BLT5403, then incubated at 37 °C for 3 h or overnight. The resulting Petri dishes were observed under a fluorescent scanner (Typhoon Trio, GE). Green and red fluorescent plaques were scanned at excitation wavelengths of 488 and 633 nm, respectively. The number of the plaques was counted by the software IQTL 7.0.
Supplementary Material
Acknowledgments
This work was supported by the National Key Basic Research Program of China (2011CB933503), the Special Funds of the National Natural Science Foundation of China for Basic Research Projects of Scientific Instruments (61127002), the Basic Research Program of Jiangsu Province (BK2011036) and the Jiangsu Province Funds for Distinguished Young Scientists (BK20140049). Y.Z. and C.M. are grateful for financial support from the National Institutes of Health (EB015190 and CA200504), the National Science Foundation (CMMI-1234957), the Department of Defense Congressionally Directed Medical Research Programs, the Oklahoma Center for Adult Stem Cell Research (434003) and the Oklahoma Center for the Advancement of Science and Technology (HR14-160).
Footnotes
Author contributions
X.Z. and C.M conceived the experiments. X.Z., P.C., C.M. and Y.Z. performed the experiments. W.L. assisted with the experiments, X.Z., Y.Z. and C.M. wrote the manuscript and analysed the data. N.G. and C.M. designed and supervised the project. All authors discussed the results and commented on the manuscript.
Additional information
Supplementary information is available in the online version of the paper.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Hamburg MA, Collins FS. The path to personalized medicine. New Engl J Med. 2010;363:301–304. doi: 10.1056/NEJMp1006304. [DOI] [PubMed] [Google Scholar]
- 2.Taton TA, Mirkin CA, Letsinger RL. Scanometric DNA array detection with nanoparticle probes. Science. 2000;289:1757–1760. doi: 10.1126/science.289.5485.1757. [DOI] [PubMed] [Google Scholar]
- 3.Valentini P, et al. Gold-nanoparticle-based colorimetric discrimination of cancer-related point mutations with picomolar sensitivity. ACS Nano. 2013;7:5530–5538. doi: 10.1021/nn401757w. [DOI] [PubMed] [Google Scholar]
- 4.Cao YC, Jin R, Mirkin CA. Nanoparticles with Raman spectroscopic fingerprints for DNA and RNA detection. Science. 2002;297:1536–1540. doi: 10.1126/science.297.5586.1536. [DOI] [PubMed] [Google Scholar]
- 5.Qiu F, Jiang D, Ding Y, Zhu J, Huang LL. Monolayer-barcoded nanoparticles for on-chip DNA hybridization assay. Angew Chem Int Ed. 2008;47:5009–5012. doi: 10.1002/anie.200800435. [DOI] [PubMed] [Google Scholar]
- 6.Zhou X, Cao P, Tian Y, Zhu J. Expressed peptide assay for DNA detection. J Am Chem Soc. 2010;132:4161–4168. doi: 10.1021/ja9081827. [DOI] [PubMed] [Google Scholar]
- 7.Nam JM, Stoeva SI, Mirkin CA. Bio-bar-code-based DNA detection with PCR-like sensitivity. J Am Chem Soc. 2004;126:5932–5933. doi: 10.1021/ja049384+. [DOI] [PubMed] [Google Scholar]
- 8.Stoeva SI, Lee JS, Smith JE, Rosen ST, Mirkin CA. Multiplexed detection of protein cancer markers with biobarcoded nanoparticle probes. J Am Chem Soc. 2006;128:8378–8379. doi: 10.1021/ja0613106. [DOI] [PubMed] [Google Scholar]
- 9.Mao C, et al. Viral assembly of oriented quantum dot nanowires. Proc Natl Acad Sci USA. 2003;100:6946–6951. doi: 10.1073/pnas.0832310100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mao C, et al. Virus-based toolkit for the directed synthesis of magnetic and semiconducting nanowires. Science. 2004;303:213–217. doi: 10.1126/science.1092740. [DOI] [PubMed] [Google Scholar]
- 11.Liu A, Abbineni G, Mao C. Nanocomposite films assembled from genetically engineered filamentous viruses and gold nanoparticles: Nanoarchitecture- and humidity-tunable surface plasmon resonance spectra. Adv Mater. 2009;21:1001–1005. [Google Scholar]
- 12.Mao C, Wang F, Cao B. Controlling nanostructures of mesoporous silica fibers by supramolecular assembly of genetically modifiable bacteriophages. Angew Chem Int Ed. 2012;51:6411–6415. doi: 10.1002/anie.201107824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F, Nimmo S, Cao B, Mao CB. Oxide formation on biological nanostructures via a structure-directing agent: Towards an understanding of precise transcription. Chem Sci. 2012;3:2639–2645. doi: 10.1039/C2SC00583B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eber FJ, Eiben S, Jeske H, Wege C. Bottom-up-assembled nanostar colloids of gold cores and tubes derived from tobacco mosaic virus. Angew Chem Int Ed. 2013;52:7203–7207. doi: 10.1002/anie.201300834. [DOI] [PubMed] [Google Scholar]
- 15.Cao B, Zhu Y, Wang L, Mao CB. Controlled alignment of filamentous supramolecular assemblies of biomolecules into centimeter-scale highly ordered patterns by using nature-inspired magnetic guidance. Angew Chem Int Ed. 2013;52:11750–11754. doi: 10.1002/anie.201303854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mao C, Liu A, Cao B. Virus-based chemical and biological sensing. Angew Chem Int Ed. 2009;48:6790–6810. doi: 10.1002/anie.200900231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohan K, Donavan KC, Arter JA, Penner RM, Weiss GA. Sub-nanomolar detection of prostate-specific membrane antigen in synthetic urine by synergistic, dual-ligand phage. J Am Chem Soc. 2013;135:7761–7767. doi: 10.1021/ja4028082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuervo A, et al. Structural characterization of the bacteriophage T7 tail machinery. J Biol Chem. 2013;288:26290–26299. doi: 10.1074/jbc.M113.491209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan LY, Kosuri S, Endy D. Refactoring bacteriophage T7. Mol Syst Biol. 2005;1:2005.0018. doi: 10.1038/msb4100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slootweg EJ, et al. Fluorescent T7 display phages obtained by translational frameshift. Nucleic Acids Res. 2006;34:e137. doi: 10.1093/nar/gkl600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsuboyama M, Maeda I. Combinatorial parallel display of polypeptides using bacteriophage T7 for development of fluorescent nano-bioprobes. J Biosci Bioeng. 2013;116:28–33. doi: 10.1016/j.jbiosc.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 22.Huang Y, et al. Programmable assembly of nanoarchitectures using genetically engineered viruses. Nano Lett. 2005;5:1429–1434. doi: 10.1021/nl050795d. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Doval C, Raaij MJV. Structure of the receptor-binding carboxy-terminal domain of bacteriophage T7 tail fibers. Proc Natl Acad Sci USA. 2012;109:9390–9395. doi: 10.1073/pnas.1119719109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunn JJ, Studier FW. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J Mol Biol. 1983;166:477–535. doi: 10.1016/s0022-2836(83)80282-4. [DOI] [PubMed] [Google Scholar]
- 25.Johnson SM, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 26.Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nature Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 27.Lin PY, Yu SL, Yang PC. MicroRNA in lung cancer. Br J Cancer. 2010;103:1144–1148. doi: 10.1038/sj.bjc.6605901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bousquet M, Harris MH, Zhou B, Lodish HF. MicroRNA miR-125b causes leukemia. Proc Natl Acad Sci USA. 2010;107:21558–21563. doi: 10.1073/pnas.1016611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monya B. qPCR: Quicker and easier but don’t be sloppy. Nature Methods. 2011;8:207–212. [Google Scholar]
- 30.Kumar MS, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA. 2008;105:3903–3908. doi: 10.1073/pnas.0712321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serwer P, Pichler ME. Electrophoresis of bacteriophage T7 and T7 capsids in agarose gels. J Virol. 1978;28:917–928. doi: 10.1128/jvi.28.3.917-928.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu T, et al. MicroRNA-195 suppresses tumorigenicity and regulates G1/S transition of human hepatocellular carcinoma cells. Hepatology. 2009;50:113–121. doi: 10.1002/hep.22919. [DOI] [PubMed] [Google Scholar]
- 33.Jusufović E, et al. let-7b and miR-126 are down-regulated in tumor tissue and correlate with microvessel density and survival outcomes in non-small-cell lung cancer. PLoS ONE. 2012;7:e45577. doi: 10.1371/journal.pone.0045577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu ZL, Wang H, Liu J, Wang ZX. MicroRNA-21 (miR-21) expression promotes growth, metastasis, and chemo- or radioresistance in non-small cell lung cancer cells by targeting PTEN. Mol Cell Biochem. 2013;372:35–45. doi: 10.1007/s11010-012-1443-3. [DOI] [PubMed] [Google Scholar]
- 35.Thellin O, et al. Housekeeping genes as internal standards: Use and limits. J Biotechnol. 1999;75:291–295. doi: 10.1016/s0168-1656(99)00163-7. [DOI] [PubMed] [Google Scholar]
- 36.Vandesompele J, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:research0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dheda K, et al. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. BioTechniques. 2004;37:112–119. doi: 10.2144/04371RR03. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.