

Abstract
Rapamycin has been used as a clinical immunosuppressant for many years; however, the molecular basis for its selective effects on lymphocytes remains unclear. We investigated the role of two canonical effectors of the mammalian target of rapamycin (mTOR), ribosomal S6 kinases (S6Ks) and eukaryotic initiation factor 4E (eIF4E)–binding proteins (4E-BPs). S6Ks are thought to regulate cell growth (increase in cell size) and 4E-BPs are thought to control proliferation (increase in cell number), with mTORC1 signaling serving to integrate these processes. However, we found that the 4E-BP–eIF4E signaling axis controlled both the growth and proliferation of lymphocytes, processes for which the S6Ks were dispensable. Furthermore, rapamycin disrupted eIF4E function selectively in lymphocytes, which was due to the increased abundance of 4E-BP2 relative to that of 4E-BP1 in these cells and the greater sensitivity of 4E-BP2 to rapamycin. Together, our findings suggest that the 4E-BP–eIF4E axis is uniquely rapamycin-sensitive in lymphocytes, and that this axis promotes clonal expansion of these cells by coordinating growth and proliferation.
Introduction
In numerous animal organs, the control of cell growth (increase in size) and proliferation (increase in number) is separated, a mechanism that is thought to ensure correct organ and organismal size (1–3). Signaling by mammalian (or mechanistic) target of rapamycin (mTOR) complex 1 (mTORC1) is central to these processes, because mTORC1 inhibitors reduce both the growth and proliferation of most cells in response to multiple extracellular signals. (4). Two canonical mTORC1 substrates are the S6 kinases (S6K1 and S6K2) and the eukaryotic initiation factor 4E (eIF4E)–binding proteins (4E-BP1, 4E-BP2, and 4E-BP3) (5–7). mTORC1 activates S6Ks to promote biosynthetic pathways that are important for cell growth (7, 8). The mTORC1-mediated phosphorylation of 4E-BPs disrupts their inhibitory interaction with eIF4E, thus enabling efficient cap-dependent translation of mRNAs encoding cell cycle regulators (8, 9). Through these mechanisms, S6Ks promote cell growth, whereas the 4E-BP–eIF4E axis controls proliferation in a largely independent fashion in fibroblasts and other cell types (2, 3). However, the roles of S6Ks and 4E-BPs in immunosuppression by rapamycin have not been defined.
Lymphocyte blastogenesis is a unique process in which cells increase substantially in size during an extended growth phase, in preparation for the multiple rapid cell divisions required for clonal expansion. It has been proposed that cells, such as lymphocytes, that undergo clonal expansion may couple cell growth and proliferation through a common control mechanism (10). Deletion of the integral mTORC1 subunit raptor in T or B cells profoundly blocks growth and proliferation (11, 12), establishing that mTORC1 is essential for blastogenesis. Furthermore, rapamycin-treated T cells enter cell cycle with a long delay, which correlates with slower size increase (13); however, it is not known whether distinct mTORC1 effector arms control lymphocyte growth and proliferation as in other cell types.
Two classes of mTOR inhibitors have been used to investigate the cellular functions of mTORC1. The natural product rapamycin is an allosteric mTORC1 inhibitor that reduces the phosphorylation of mTORC1 substrates to varying degrees. For example, rapamycin suppresses the phosphorylation of S6K1 (at Thr389) more completely than that of 4E-BP1 (Thr37/46) (14, 15). In contrast, synthetic adenosine triphosphate (ATP)-competitive mTOR kinase inhibitors (TOR-KIs) fully block the phosphorylation of mTOR substrates (16, 17). The partial inhibition of 4E-BP1 phosphorylation by rapamycin results in a weaker anti-proliferative effect than that of TOR-KIs in fibroblasts and cancer cells despite both inhibitors having equivalent effects on cell size (16–18). In contrast, rapamycin profoundly inhibits the proliferation of primary T and B cells to a similar extent as do the TOR-KIs (11, 18). Identifying the key rapamycin-sensitive effectors of mTORC1 should shed light on the longstanding question of why this immunosuppressive drug has selective potency in lymphocytes (19). Furthermore, determining the mechanisms that regulate lymphocyte activation downstream of mTORC1 might reveal novel targets for immunosuppression. Here, we genetically dissected the function of S6Ks and the 4E-BP–eIF4E axis in primary T and B cells. We found that lymphocytes coordinate growth and proliferation through the 4E-BP–eIF4E effector arm of mTORC1 in a rapamycin-sensitive manner.
Results
S6K activity is dispensable for lymphocyte growth and proliferation
Lymphocyte-specific deletion of mTOR or raptor greatly impairs growth and proliferation in response to antigen receptor engagement (11, 12, 20, 21); however, the roles of mTORC1 substrates in lymphocyte blastogenesis have not been defined. The ability of rapamycin to suppress both growth and proliferation to a similar extent as that of the TOR-KIs in lymphocytes (Fig. 1) (11, 18) implies that the relevant substrates are rapamycin-sensitive. We tested the hypothesis that lymphocyte growth and proliferation are coupled through a single mTORC1 effector.
Fig. 1. Rapamycin and TOR-KIs inhibit lymphocyte growth and proliferation to the same extent.
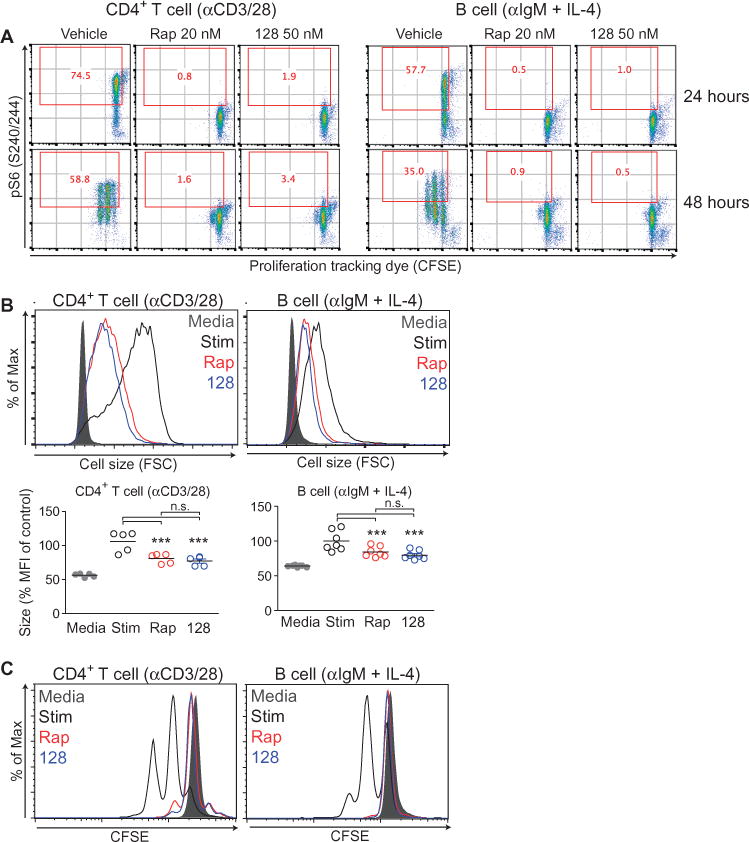
(A) Purified CD4+ T cells (left) and B cells (right) from wild-type (WT) C57/B6 mouse splenocytes were labeled with CFSE, pretreated with vehicle, 20 nM rapamycin (Rap), or 50 nM MLN0128 (128), and then were stimulated with anti-CD3 and anti-CD28 antibodies (for T cells) or with anti-IgM antibody and IL-4 (for B cells) for the indicated times. Cells were then fixed, permeabilized, and stained with anti-pS6 antibody to assess mTORC1 activity. Red numbers in each plot indicate the percentage of cells that stained positive for pS6. Data are representative of four independent experiments. (B) Top: The growth of the indicated cells at 24 hours after stimulation was measured by flow cytometric analysis of forward scatter (FSC). Bottom: The decrease in size compared to that of control cells was measured by calculating the percentage median fluorescent intensity (MFI) of FSC normalized to the stimulated condition for each experiment. Data are means ± SEM of three to seven experiments. (C) The proliferation of the indicated cells at 48 hours after stimulation was measured by flow cytometric analysis of CFSE dilution. Data are representative of at least three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001, by repeated-measures analysis of variance, measured versus the medium control.
We focused initially on S6Ks, which have a conserved role in controlling cell size (3, 22, 23) and are important for proliferation in certain cell types (24). Phosphorylation of S6K or its substrate ribosomal protein S6 is commonly used as a rapamycin-sensitive readout of mTORC1 activity, and we found that both processes were completely blocked by rapamycin in lymphocytes and other cell types (Fig. 1). In addition, differences in cell size among activated T cells correlate closely with the mTORC1-dependent phosphorylation of S6 in individual cells (25). Although inhibition of S6K has been proposed as a key mechanism by which rapamycin inhibits the entry of lymphocytes into the S phase of the cell cycle (26), there has been no evidence to support the idea that S6K inactivation in primary lymphocytes affects growth or proliferation. Lymphocytes express both S6K1 and S6K2, and single knockout mice of either S6K do not have obvious immune defects (22, 27). Mice lacking both S6K1 and S6K2 have a perinatal lethal phenotype, but with incomplete penetrance (27). Thus, we were able to obtain splenocytes from a limited number of surviving adult mice.
We measured the signaling, growth, and proliferation of S6K1/2 double knockout (S6K DKO) lymphocytes after antigen receptor engagement. As expected, phosphorylated ribosomal protein S6 (pS6) at Ser240/244, the measurement of which is a sensitive readout of S6K activity, was completely absent in activated S6K DKO T cells and B cells (Fig. 2A). However, both T cells and B cells from S6K DKO mice exhibited no substantial defect in their ability to grow or proliferate (Fig. 2, B and C). Furthermore, both the growth and proliferation of S6K DKO lymphocytes were still sensitive to rapamycin, indicating that lymphocyte growth and proliferation depend on other mTORC1 outputs. To address the possibility that S6K DKO lymphocytes had compensatory changes, we used a chemical genetic approach (fig. S1) in which S6K2 deficient (S6K2 KO) lymphocytes, which developed normally (fig. S1A), were treated with the highly selective S6K1 inhibitor LY2584702 (28) to completely suppress S6K activity (fig. S1B). Equivalent results were obtained with this approach (fig. S1C) and from experiments in which an S6K1 hypomorphic mouse model was crossed with S6K2 KO mice (fig. S2). We also assessed proliferation in an antigen-specific system in experiments with total splenocytes stimulated in vitro with the superantigen staphylococcal enterotoxin B (SEB), which activates all T cells bearing T cell receptors (TCRs) with a Vβ8 chain (fig. S1D). Again, S6K DKO T cells proliferated normally and were rapamycin-sensitive.
Fig. 2. S6Ks are dispensable for lymphocyte growth and proliferation.
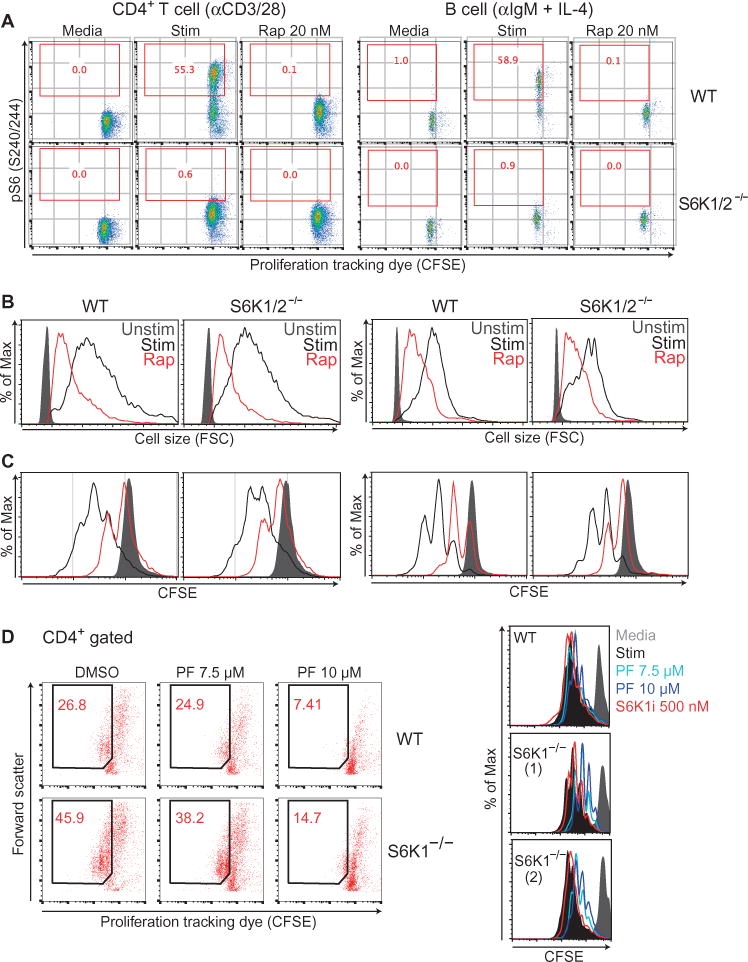
(A) CD4+ T cells (left) and B cells (right) from wild-type (WT) or S6K1/2−/− (DKO) mice were left unstimulated (Media) or were stimulated for 24 hours with anti-CD3 and anti-CD28 antibodies (for T cells) or with anti-IgM antibody and IL-4 (for B cells) in the presence or absence of 20 nM rapamycin (Rap). The cells were then analyzed by flow cytometry to detect pS6 (Ser240/244). Red numbers inside the plots indicate the percentage of pS6-positive cells. Data are representative of two experiments. (B) Cell growth at 24 hours was measured by flow cytometric analysis of FSC. Data are representative of two experiments (C) Cell proliferation at 72 hours after activation was measured by flow cytometric analysis of CFSE dilution. (D) Total splenocytes (left) or purified CD4+ T cells (right) from WT or S6K1−/− mice were labeled with CFSE and stimulated for 72 hours with anti-CD3 and anti-CD28 antibodies with or without the indicated concentration of PF-4708671 (PF). Cells were stained with anti-CD4 and proliferation was measured by flow cytometric analysis of CFSE dilution gating on CD4+ cells. All results are representative of two independent experiments. In addition to the comparisons of wild-type (WT) and S6K1/2−/− (DKO) mice in (A) to (C), equivalent results were observed in independent experiments using additional chemical and genetic approaches (figs. S1 and S2).
A study reported that phosphorylation of the S6K sites on ribosomal S6 protein is dispensable for T cell growth and proliferation in response to antigen (29). This study also reported that the S6K1 inhibitor PF-4708671 suppresses T cell activation. However, we found that at the concentration of PF-4708671 used in the study of Salmond et al. (10 μM), this compound inhibited the proliferation of S6K1-deficient T cells, indicating a potential off-target effect (Fig. 2D). In contrast, at the concentrations used for our chemical genetic approach to target all S6K activity (fig. S1), LY2584702 exhibited no effect in either wild-type or S6K1-knockout T cells (Fig. 2D). These results demonstrate that in the context of antigen receptor–driven growth and proliferation, S6K activity is dispensable.
The relevant mTORC1 effector is a kinasesubstrate
Two models could explain how rapamycin suppresses lymphocyte function independently of S6K inactivation: (i) by disrupting a noncatalytic function of mTOR complexes (30); or (ii) by inhibiting another mTORC1 substrate. The first possibility was suggested by the ability of rapamycin, but not TOR-KIs, to destabilize mTOR complexes (31). To test this model, we generated mouse strains in which the kinase activity of mTOR was abolished specifically in T cells or B cells by Cd4-Cre and Cd19-Cre driver mice, respectively (Fig. 3A; fig. S3, A to C). Despite the presence of mTOR protein, lymphocytes expressing kinase-inactive mTOR (mTOR-KI) displayed an absence of mTORC1 kinase activity as assessed by detection of the phosphorylation of S6 and 4E-BP1 (Fig. 3, A and B; fig. S3C). As reported previously for T cell– or B cell–specific mTOR knockout mice (mTOR-KO; ΔT or ΔB), kinase-inactive mTOR (mTOR-KI) mice displayed decreased frequencies, but not an absence, of mature CD4+ T cells and B cells (fig. S3D). However, activated mTOR-KI lymphocytes displayed profound reductions in growth and proliferation that were equivalent to those of lymphocytes from mTOR-KO mice (Fig. 3, B and C). Selective inactivation of mTORC1 through T cell– or B cell–specific depletion of raptor (Raptor-KO; ΔT or ΔB) phenocopied the reduced size and proliferation of lymphocytes from the mTOR-KI mice (Fig. 3C). Together, these results show that the critical mTOR effectors required for lymphocyte growth and proliferation are kinase substrates of mTORC1.
Fig. 3. The relevant downstream mTORC1 effector in lymphocytes is a kinase substrate.
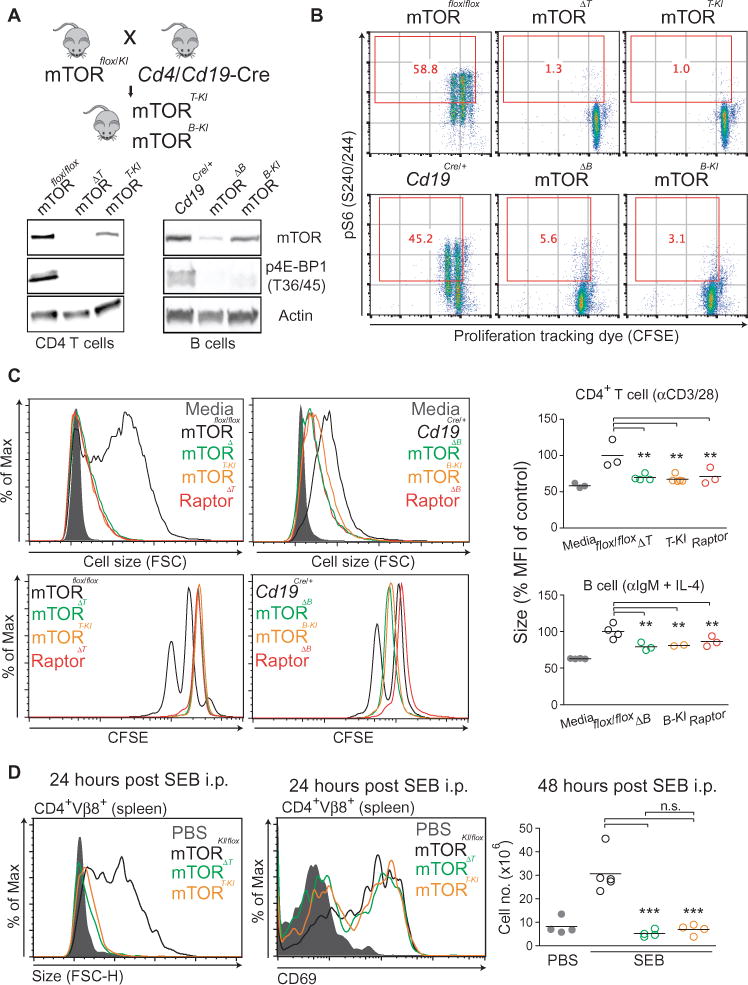
(A) Top: Strategy to generate T cell– or B cell–specific mTOR-KI mice. mTORKI/+ mice have a D2338A mutation in the mTOR kinase domain. These mice were crossed with mTORflox/flox mice to generate mTORflox/KI. Mice were further crossed to either Cd4Cre or Cd19Cre mice to delete the floxed mTOR allele only in T cells (mTOR-TKI) or B cells (mTOR-BKI), respectively. Bottom: CD4+ T cells were purified from mTOR-ΔT, mTOR-TKI, Raptor-ΔT, and control mTORflox/flox mice. B cells were purified from mTOR-ΔB, mTOR-BKI, Raptor-ΔB, and control Cd19Cre/+ mice. CD4+ T cells and B cells were stimulated for 24 hours as described earlier, and mTORC1 kinase activity was assessed by Western blotting analysis of p4EBP1 (Thr36/45). (B) CD4+ T cells (top) and B cells (bottom) from the indicated mice were labeled with CFSE and then stimulated for 48 hours as described earlier. The cells were then analyzed by flow cytometry to detect pS6 (S240/244). Red numbers in the plots indicate the percentages of pS6-positive cells. Data are representative of four experiments. (C) Cell growth at 24 hours was measured by flow cytometric analysis of FSC of CD4+ T cells (left) and B cells (right) from the indicated mice, and the decrease in size compared to that of control (stimulated flox/flox) cells was measured for each experiment as described in Fig 1B. mTOR-Δ: mTOR knockout; mTOR-KI: mTOR kinase-inactive; Raptor-Δ: Raptor knockout. For B cell–specific genetic deletion experiments, Cd19Cre heterozygotes were used as appropriate controls. (D) Groups of three to four mice of the indicated genotypes were injected with 100 μg of SEB i.p. Twenty-four hours later, their spleens were harvested and the increase in the size of CD4+Vβ8+ cells was determined by flow cytometric analysis of FSC (left). Early activation status at twenty-four hours post SEB injection was also measured by the cell surface marker CD69 on CD4+Vβ8+ cells (middle). Cell proliferation at 48 hours after injection was measured by counting the total number of splenocytes and multiplying it by the percentage of CD4+Vβ8+ cells in each sample (right). Results are representative of at least three independent experiments. Where indicated, data are means ± SEM of three to eleven experiments. *P < 0.05; **P < 0.01; ***P < 0.001, by repeated-measures analysis of variance, measured versus the control genotype.
These results were extended to an in vivo antigen-specific system in which, within 24 hours of treatment with SEB, control mice exhibited substantial SEB-induced CD4+ T cell growth within the Vβ8+ subset (Fig. 3D). After 48 hours, we observed an increase in absolute numbers of Vβ8+ CD4+ T cells in phosphate-buffered saline (PBS)-treated control mice, indicating cell proliferation (Fig. 3D). Both growth and proliferation were blunted equivalently in lymphocytes from mTOR-ΔT or mTOR-KI mice (Fig. 3D), despite their normal abundance of the early activation marker CD69 (Fig. 3D). Raptor-ΔT mice showed similar defects (fig. S3E). Comparable results were obtained from experiments in which T cells were activated with SEB in vitro (fig. S3F).
Lymphocytes have increased amounts of 4E-BP2, which is distinct from 4E-BP1 in its rapamycin-sensitivity
The 4E-BP–eIF4E axis plays a central role in lymphomagenesis (32) and regulatory T cell differentiation (33) through specific translation of mRNAs; however, little is known about its role in normal lymphocyte blastogenesis. 4E-BP proteins (4E-BP1, 4E-BP2, and 4E-BP3) are direct kinase substrates of mTORC1, which when dephosphorylated, bind to eIF4E and displace the scaffolding protein eIF4G, thereby prevents the assembly of an active eIF4F translation initiation complex (9, 34). The phosphorylation of 4E-BP by mTORC1 on several residues triggers the release of 4E-BPs from eIF4E and promotes cap-dependent translation. Phosphorylation of the key residues Thr37 and Thr46 (collectively referred to as Thr37/46) on 4E-BP1 is reduced by rapamycin only weakly in most cell types (15–18), which may explain why modulation of 4E-BP–eIF4E activity has not been considered as a likely mechanism of action for rapamycin in lymphocytes.
We assessed the phosphorylation state of 4E-BP with a phosphospecific antibody that detects Thr36/45 (the sites on mouse and rat 4E-BP1 that correspond to human Thr37/46) as well as Thr37/46 on 4E-BP2 (Fig. 4). This analysis confirmed that in activated B cells, the TOR-KI compound MLN0128, but not rapamycin, suppressed the phosphorylation of 4E-BP1 on Thr36/45 (Fig. 4A). Note that rapamycin reduced the phosphorylation of the 4E-BP2 isoform on the equivalent sites (Fig. 4A). Western blotting analysis of 4E-BP1 and 4E-BP2 singly deficient lymphocytes confirmed the phosphorylated band to be 4E-BP2 (Fig. 4A). Extensive analysis of the five conserved phosphorylation sites on mouse 4E-BP1 showed that Ser65 (equivalent to Ser64 in human 4E-BP1) contributes substantially to the SDS-PAGE migration pattern of 4E-BP1 (35). To test whether the faster gel-migration pattern of 4E-BP1 from rapamycin-treated B cells resulted from dephosphorylation of the Ser65 site that is rapamycin-sensitive in T cells (36), as well as in human embryonic kidney (HEK) 293 cells and fibroblasts (15), we analyzed activated B cells by Western blotting with an antibody specific for 4E-BP1 phosphorylated on Ser65. We observed equivalent effects of rapamycin and MLN0128 in inhibiting the phosphorylation of Ser65 on 4E-BP1 (Fig. 4B), potentially explaining the shift in migration of the band corresponding to 4E-BP1 (Fig. 4A).
Fig. 4. The phosphorylation of 4E-BP2 at Thr37/46 is rapamycin-sensitive.
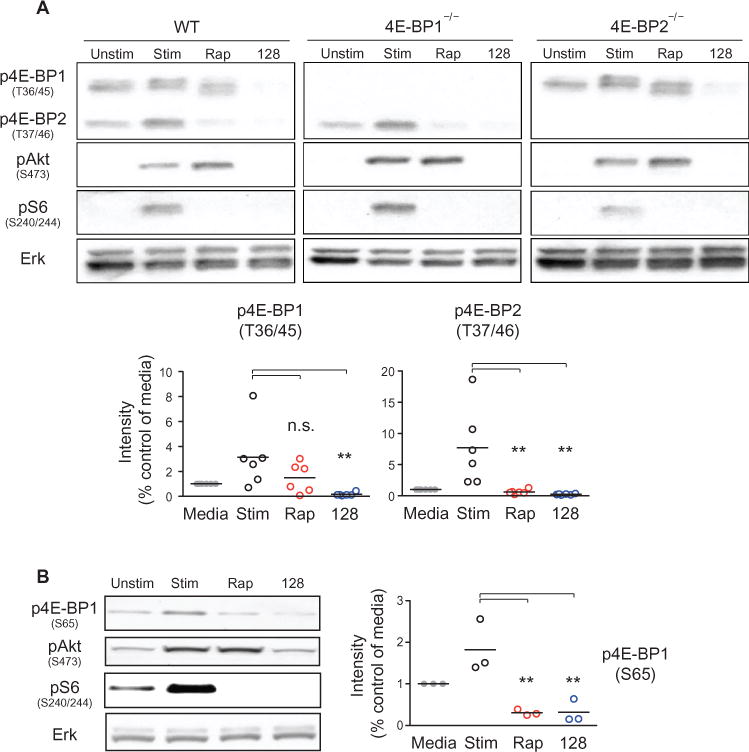
(A) Top: B cells isolated from WT, 4E-BP1 knockout (4E-BP1−/−), and 4E-BP2 knockout (4E-BP2−/−) mice were left unstimulated or were stimulated for 2 hours with anti-IgM antibody and IL-4 in the absence or presence of 20 nM rapamycin (Rap) or 50 nM MLN0128 (128). Samples were then analyzed by Western blotting with antibodies specific for the indicated proteins. Western blots are representative of two to six experiments. Bottom: Densitometric analysis of the intensities of the indicated bands normalized to that of the unstimulated condition. Data are means ± SEM of six independent experiments. (B) B cells from WT mice were left unstimulated or were stimulated as described earlier in the absence or presence of the indicated inhibitors. Cells were then analyzed by Western blotting with antibodies against the indicated proteins. Right: Densitometric analysis of the intensities of the band corresponding to p4EBP1 (S65) normalized to that of unstimulated condition. Data are means ± SEM of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001, by repeated-measures analysis of variance, measured versus the vehicle-treated control.
To check whether the differential sensitivity of 4E-BP2 was unique to primary lymphocytes, we used the diffuse large B cell lymphoma (DLBCL) cell line VAL, which had 4E-BP2, but not 4E-BP1 (Fig. 5A), as we reported previously (37). Intracellular staining showed that the p4E-BP2 signal was sensitive to rapamycin in VAL cells (Fig. 5B). In OCI-Ly1 cells, a DLBCL cell line that has high amounts of 4E-BP1 (Fig. 5A), the p4E-BP signal was rapamycin-insensitive, but MLN0128-sensitive (Fig. 5B). These results suggest that the reported rapamycin-resistance of 4E-BP phosphorylation at Thr37/46 applies more to 4E-BP1 than to 4E-BP2.
Fig. 5.
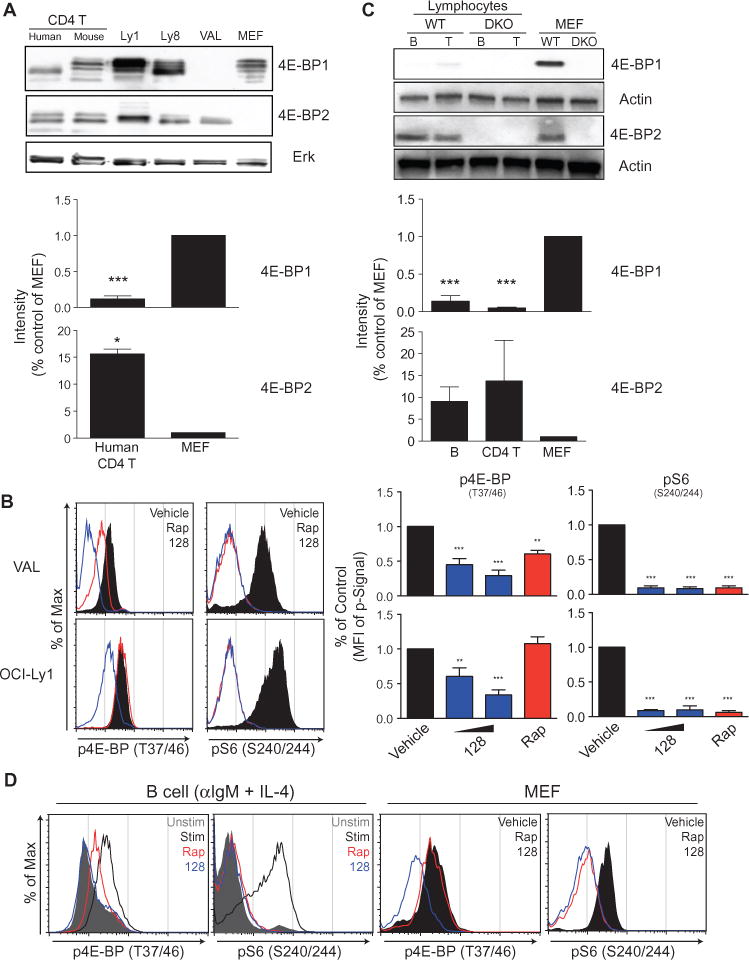
(A) CD4+ T cells purified from human PBMCs or WT mice, WT MEFs, and the DLBCL lymphoma cell lines OCI-Ly1, OCI-Ly8, and VAL were analyzed by Western blotting with antibodies against 4E-BP1 and 4E-BP2. (B) Left: The DLBCL cell lines VAL (which lacks 4E-BP1) and OCI-Ly1 were left untreated (vehicle) or were treated with the indicated inhibitors for 2 hours. The cells were then fixed, permeabilized and analyzed by flow cytometry to detect p4EBP (Thr37/46) and pS6 (Ser240/244). Right: The median fluorescent intensity (MFI) for each indicated signal was normalized to that of vehicle-treated samples. Data are means ± SEM of three to six experiments. (C) Murine CD4+ T and B cells isolated from WT splenocytes were analyzed by Western blotting with antibodies against 4E-BP1 and 4E-BP2. In the bar graphs for (A) and (C), the relative amounts of 4E-BP1 and 4E-BP2 in human CD4+ T cells (A) or murine CD4+ T cells and murine B cells (C) were compared by densitometric analysis and were normalized to those of WT MEFs. Data are means ± SEM of three or four independent experiments. (D) B cells from WT mice were left unstimulated or were stimulated for 24 hours with anti-IgM antibody and IL-4 in the absence or presence of the indicated inhibitors. MEFs from WT mice were left untreated (vehicle) or were treated for 24 hours with the indicated inhibitors. Cells were then subjected to intracellular flow cytometric analysis with antibodies specific for the indicated phosphoproteins. Data are representative of three independent experiments. *P < 0.05; **P < 0.01, ***P < 0.001, by repeated-measures analysis of variance, measured versus the vehicle-treated control.
Analysis of sequence alignments revealed a conserved glycine at position 31 of mouse 4E-BP1, a position that is filled by a polar amino acid residue (glutamine or histidine) in 4E-BP2 (fig. S4A). Mutation of Gly31 to glutamine or histidine rendered Thr37/46 in 4E-BP1 more rapamycin-sensitive (fig. S4, B and C). In this experiment, we assessed 4E-BP1 phosphorylation status by mobility shift with an antibody against total 4E-BP1, because the affinity of the phosphospecific antibody for the protein might be altered by the amino acid substitutions.
We then assessed the abundances of the 4E-BP isoforms in primary resting lymphocytes. When compared with equal amounts of protein from mouse embryo fibroblasts (MEFs), the abundance of 4E-BP1 was substantially less in T and B lymphocytes (Fig. 5C). In contrast, lymphocytes had similar or greater amounts of 4E-BP2 compared to MEFs (Fig. 5C). Purified human T cells similarly showed low amounts of 4E-BP1 and high amounts of 4E-BP2 relative to those of MEFs (Fig. 5A). A database of mRNA abundances in cells of hematopoietic lineage also showed enrichment of eif4ebp2 in mature lymphocytes (fig. S5). Consistent with a higher ratio of 4E-BP2 protein to 4E-BP1 protein in B cells, intracellular staining for p4E-BP isoforms detected reduced phosphorylation in rapamycin-treated primary B cells, but not MEFs or OCI-LY1 lymphoma cells (compare Fig. 5D with Fig. 5B).
Rapamycin disrupts eIF4F complex formation upon lymphocyte activation
The observation that rapamycin reduced the phosphorylation of 4E-BP2, which is abundant in lymphocytes, suggests that rapamycin should inhibit eIF4E function in activated lymphocytes. To test this, we assessed the interaction between eIF4G and eIF4E by 7-methylguanosine 5′-triphosphate (m7GTP) pulldown assays in lysates of B cells treated early on during activation with rapamycin. Rapamycin-treated samples showed a near complete displacement of eIF4G from eIF4E, similar to the state in resting or MLN0128-treated B cells (Fig. 6A). In contrast, rapamycin caused little or no displacement of eIF4G from eIF4E in other cell types, including MEFs that were treated with inhibitors after serum-starvation and then were restimulated with serum (fig. S6). Rapamycin and MLN0128 also had equivalent effects on protein synthesis in activated primary B cells, as detected with a puromycin incorporation assay (Fig. 6B). In MEFs, MLN0128, but not rapamycin, suppressed protein synthesis (Fig. 6C). This is consistent with MLN0128 having a greater inhibitory effect on MEF proliferation compared to that of rapamycin (fig. S7A), consistent with reported comparisons of other TOR-KIs with rapamycin (16, 17, 38).
Fig. 6. Rapamycin inhibits eIF4F formation and protein synthesis in lymphocytes.
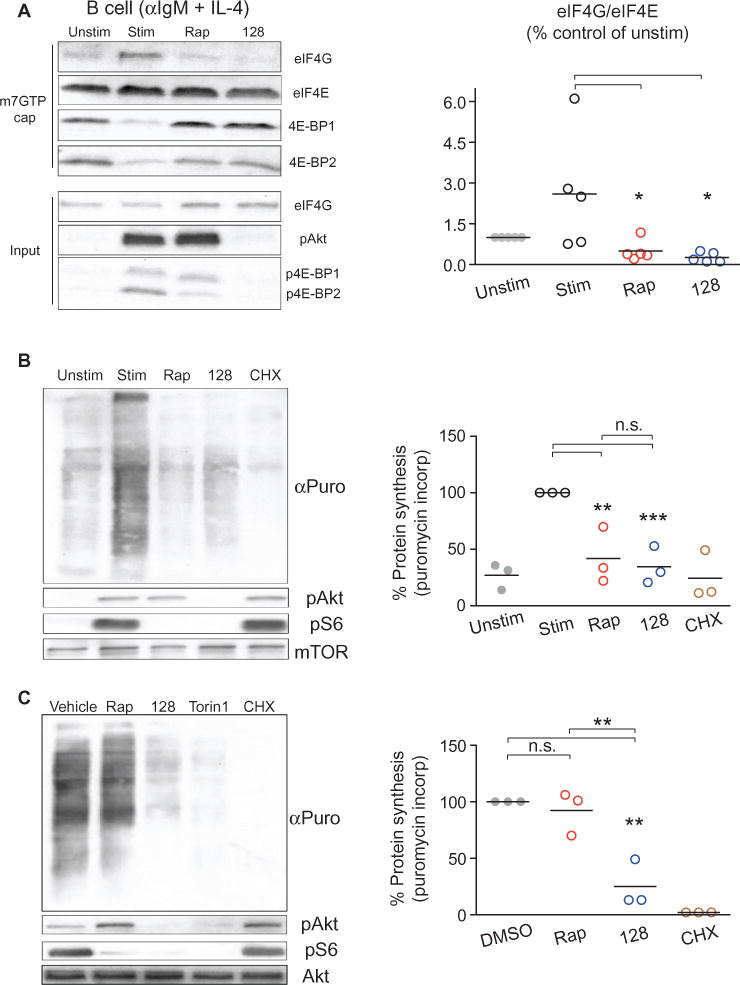
(A) Left: B cells from WT mice were left unstimulated or were stimulated for 2 hours with anti-IgM antibody and IL-4 in the absence or presence of the indicated inhibitors. The cells were then subjected to m7GTP cap pulldowns. The relative amounts of eIF4G, eIF4E, and 4E-BP1 that were bound to the cap were then measured by Western blotting analysis. Bottom: Western blotting analysis of total cell lysate (input). Right: The amount of eIF4G normalized to the amount of eIF4E pulled down from cells under the indicated conditions was quantitated over five independent experiments. Horizontal lines represent the means of each group. *P < 0.05; by repeated-measures analysis of variance, measured versus the unstimulated control. (B) Left: A puromycin incorporation assay was used to measure nascent protein synthesis in B cells that were treated as described in (A). Right: Puromycin incorporation signals for the indicated samples normalized to a loading control were quantitated for three experiments and were expressed as the percentage of protein synthesis detected. **P < 0.01; ***P < 0.001, by repeated-measures analysis of variance, measured versus the stimulated, vehicle-treated control. (C) MEFs from WT mice were left untreated or were treated for three hours with the indicated inhibitors before being subjected to puromycin incorporation assays as described in (B). Right: Puromycin incorporation signals for the indicated samples normalized to a loading control were quantitated for three experiments and were expressed as the percentage of protein synthesis detected. *P < 0.05; **P < 0.01; ***P < 0.001, by repeated-measures analysis of variance, measured versus the vehicle-treated control.
One variable that might contribute to differential rapamycin sensitivity is the presence of distinct amounts of eIF4E in different cells. We reported previously that eIF4E abundance is low in resting human T cells compared to that in T lymphoma cells (39). Here, we found that the amount of eIF4E in mouse T cells and B cells was comparable to that in MEFs, but less than that in B lymphoma cells (fig. S7B). To determine whether 4E-BPs were required for the ability of rapamycin to suppress proliferation in lymphocytes, we isolated B cells from 4E-BP1/2 double knockout (DKO) mice, and found that they were partially resistant to the anti-proliferative effects of TOR-KIs (MLN0128 and Torin1) and rapamycin (Fig. 7, A and B). This is in contrast to fibroblasts, in which deletion of 4E-BPs rescues them from the anti-proliferative effects of TOR-KIs, but not rapamycin (2).
Fig. 7. 4E-BP1/2 deficiency partially rescues lymphocyte proliferation from rapamycin treatment.
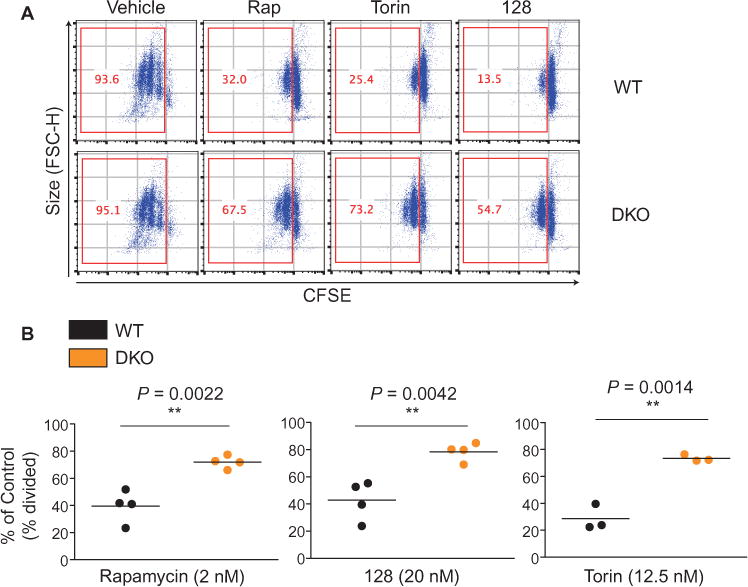
(A) B cells from WT or 4E-BP1/2 DKO mice were labeled with CFSE and were left untreated (Vehicle) or were stimulated with anti-IgM antibody and IL-4 for 72 hours in the absence or presence of the indicated inhibitors to measure cell proliferation. Cells that had divided at least once were plotted and quantified in the red boxes. Data are representative of at least three experiments. (B) For WT and DKO B cells treated with the indicated inhibitors, the percentages of cells that divided at least once (% divided) were normalized to the percentage of vehicle-treated B cells that had dividedfor each genotype. Data are means ± SEM of three to five experiments. *P < 0.05; **P < 0.01; ***P < 0.001, by unpaired two sample t-test.
Suppression of eIF4E function phenocopies the effects of mTORC1 inhibition
To address whether 4E-BPs were sufficient to mediate the effects of mTORC1 inhibition, we used transgenic mice that expressed a constitutively active mutant of 4E-BP1 (4E-BP1M), which could be expressed in a doxycycline (Dox)-inducible manner (32). These mice were crossed to Rosa26-rtTA transgenic mice, which display widespread expression of the reverse tetracycline transactivator (rtTA) protein. Upon incubation of purified lymphocytes with Dox for 6 hours, we observed substantial amounts of the 4E-BP1M mutant protein, which is characterized by a higher molecular mass because of the FLAG-tag (fig. S8A). Furthermore, there was no perturbation in the phosphorylation of upstream regulators of mTORC1 (Akt) or of downstream effectors (S6) when 4E-BP1M was expressed, suggesting that 4E-BP1M induction does not perturb overall mTOR signaling during lymphocyte activation (fig. S8, B and C).
First, we assessed the effect of 4E-BP1M on the interaction between eIF4G and eIF4E using the m7GTP pulldown assay. Resting naïve lymphocytes showed minimal binding of eIF4G to eIF4E, which was instead bound to 4E-BP1 (Fig. 8A). In B cells activated for 2 hours, the prominent increase in the extent of the interaction between eIF4G and eIF4E was abolished in the presence of 4E-BP1M (Fig. 8A). When activated for 24 hours, the 4E-BP1M-expressing lymphocytes showed a substantial defect in growth that correlated with the concentration of Dox that was added for 6 hours before the cells were activated (Fig. 8B). Note that 4E-BP1M-expressing lymphocytes closely phenocopied the growth and proliferation defect seen in Raptor-KO lymphocytes (Fig. 8, B and C); however, the ectopic expression of 4E-BP1M blocked cellular growth and proliferation independently of mTORC1 status, whereas Raptor-KO lymphocytes also showed greatly reduced amounts of pS6 (Fig. 8C). The effects of 4E-BP1M expression were similar to those of pharmacological mTORC1 inhibition by rapamycin or MLN0128 (fig. S8D). Furthermore, an adoptive transfer experiment (diagrammed in fig. S9A) showed that 4E-BP1M expression blocked the SEB-stimulated growth and proliferation of T cells in vivo without inhibiting the appearance of an early activation marker (Fig. 8D and fig. S9B). These results demonstrate that specific blockade of the 4E-BP–eIF4E signaling node downstream of mTORC1 is sufficient to block both lymphocyte growth and proliferation.
Fig. 8. A constitutively active 4E-BP1 mutant is sufficient to phenocopy mTORC1 deficiency in lymphocytes.
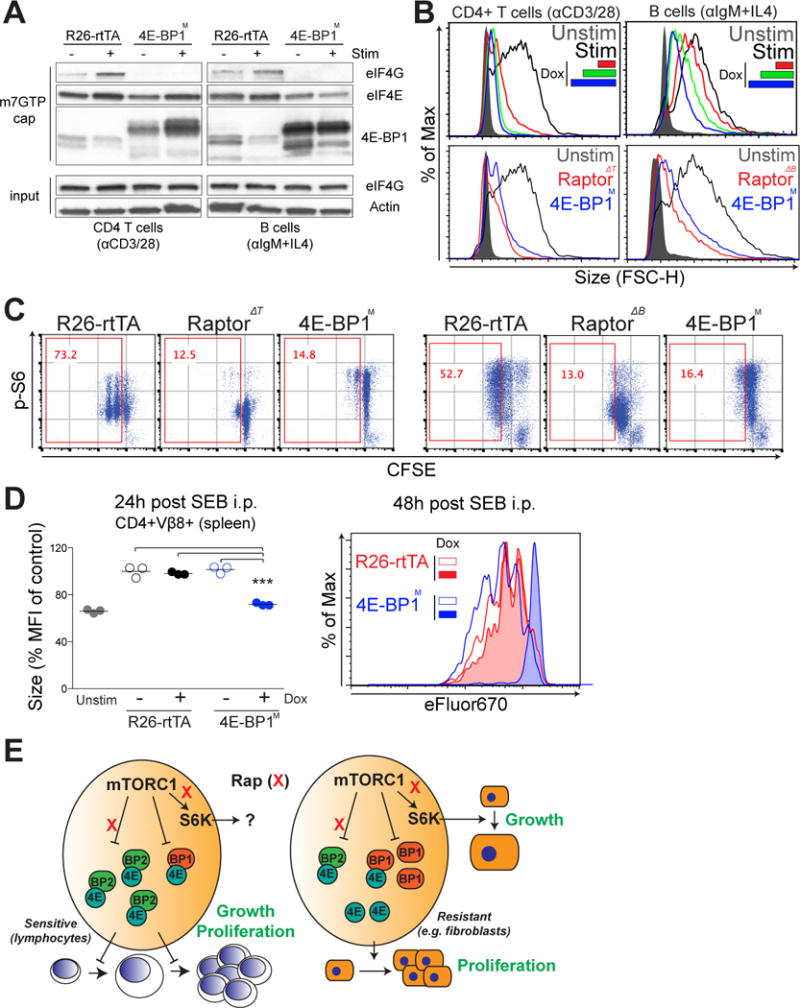
(A) CD4+ T cells and B cells from control (R26-rtTA) mice and 4E-BP1M expressing mice were left untreated or were stimulated as described earlier for 12 hours and subjected to m7GTP cap pulldown. The relative amounts of eIF4G, eIF4E, and 4E-BP1 bound to the cap were measured by Western blotting. Data are representative of two experiments. (B) Top: The growth of the indicated 4E-BP1M–expressing lymphocytes was determined by flow cytometric analysis of FSC. Different color-coding represents the different amounts of doxycycline (dox) that were added for 6 hours before cell stimulation to induce 4E-BP1M expression. Bottom: The growth of 4E-BP1M-expressing lymphocytes was compared to that of Raptor-Δ lymphocytes. Data are representative of three experiments. (C) The proliferation of the indicated 4E-BP1M-expressing lymphocytes was compared with that of Raptor-Δ lymphocytes, and mTORC1 activity was assessed by the flow cytometric measurement of pS6 abundance as described in Fig 1A. Data are representative of three experiments. (D) Syngeneic C57/B6 host mice were fed Dox in their drinking water ad libitum for 24 hours. Purified CD4+ T cells from either control (R26-rtTA) or 4E-BP1M mice were labeled with eFluor 450 and injected intravenously into syngeneic host mice. After 24 hours, mice were injected intraperitoneally with 100 μg of SEB, and spleens were analyzed at 24 and 48 hours after injection to determine the size (left) and proliferation (right) of CD4+Vβ8+ cells. Data are means ± SEM of three mice of each genotype. *P < 0.05; **P < 0.01; ***P < 0.001, by repeated-measures analysis of variance, measured versus the media control. (E) Proposed model: Cell growth and proliferation are coupled through the 4E-BP–eIF4E pathway in primary lymphocytes, which distinguishes these cells from cells, such as fibroblasts, in which both processes are regulated in a largely independent manner by S6Ks and 4E-BP–eIF4E signaling. This coordination of cell growth and proliferation through a common 4E-BP–eIF4E node renders primary lymphocytes particularly sensitive to rapamycin, which potently blocks the phosphorylation of 4E-BP2, an isoform that is highly abundant in these cells.
The effect of 4E-BP1M on cell growth was specific for primary T cells and B cells. In 3T3 fibroblasts and the OCI-Ly1 lymphoma cell line, induced expression of 4E-BP1M largely suppressed proliferation but not size or protein content (fig. S10, A and B). These results are consistent with our previous observation that the 4EBP–eIF4E axis specifically regulates cell cycle, but not size, in fibroblasts (2). Thus, lymphocytes uniquely use the 4E-BP–eIF4E axis to control both growth and proliferation.
Discussion
The coordination of cell growth and proliferation in mammals is a fundamental question in biology (10, 40). The central role of mTOR in the growth and proliferation of mammalian cells has largely been attributed to its regulation of two distinct downstream effectors: S6Ks and 4EBPs (41, 42). Here, we showed that during primary lymphocyte activation, cell growth and proliferation are coordinated through mTORC1-dependent control of the 4E-BP–eIF4E axis. Whereas inactivating S6Ks had no effect, blocking eIF4E function with an inducible 4E-BP mutant was sufficient to abrogate lymphocyte growth and proliferation both in vitro and in vivo. Conversely, deleting endogenous 4E-BPs substantially reduced the antiproliferative effect of mTOR inhibition. The ability of rapamycin to selectively inhibit lymphocyte proliferation correlates with disruption of the eIF4E translation initiation complex.
It is surprising that despite there being a conserved role for S6Ks in cell size regulation (3, 22, 23), these kinases were dispensable for T cell or B cell growth, as well as proliferation. The robust activation of S6Ks downstream of antigen receptor signaling might be more important in other cell fate decisions, such as T helper cell differentiation (43, 44).
On the contrary, both gain-of-function and loss-of-function experiments support the conclusion that 4E-BPs are key mTORC1 effectors that control growth and proliferation in activated T and B cells. Through an inducible system, we showed that the expression of constitutively active 4E-BP1M (thus inhibiting eIF4E) was sufficient to block lymphocyte growth and proliferation. Conversely, deletion of 4E-BPs partially rescued B cells from the effects of mTOR inhibition. Of note, 4E-BP1/2 DKO B cells were only partially protected from the antiproliferative effects of rapamycin and TOR-KIs. The cells that did proliferate underwent only one round of division, which could be because of either a delay in entering the cell cycle or the abortive proliferation of committed cells. It is possible that 4E-BP1/2 DKO lymphocytes have a developmental compensation that increases their dependency on other mTORC1 substrates. The 4E-BP3 isoform might also compensate.
Our analysis of the mTORC1–4E-BP–eIF4E axis in lymphocytes broadens our knowledge of the distinctions between 4E-BP isoforms. 4E-BP1 and 4E-BP2 have distinct expression patterns (45), and 4E-BP2 is enriched in the brain and regulates neuronal function (46); however, 4E-BP1 has been the main isoform studied in the context of mTOR inhibitors. Rapamycin is effective at blocking the mTORC1-mediated phosphorylation of Ser65 on 4E-BP1, but the Thr37/46 sites are rapamycin-resistant in most cell types studied to date (15, 47). Our data demonstrate that primary lymphocytes express relatively greater amounts of 4E-BP2 (both at the mRNA and protein levels) than of 4E-BP1 when compared to other cell types. In addition, the Thr37/46 sites on 4E-BP2 are more rapamycin-sensitive than are the homologous sites on 4E-BP1. This difference in rapamycin sensitivity is encoded in part by distinct amino acids at position –5 relative to the Thr37 site.
More than 15 years after rapamycin gained regulatory approval as an immunosuppressant, the mechanistic basis for its immune cell selectivity has remained elusive. Rapamycin is exquisitely selective for its target mTOR, a protein that is expressed ubiquitously. In contrast to its effects in lymphocytes, rapamycin delays, but does not block, cell cycle progression in fibroblasts and many cancer cell lines, and has generally weaker cytostatic effects than do the TOR-KIs (16–18). Our data showed that in lymphocytes, unlike in other cell types, rapamycin was equally effective as TOR-KIs at disrupting formation of the eIF4F complex and reducing protein synthesis. Displacement of eIF4G from eIF4E correlated with the increased abundance of the 4E-BP2 isoform in lymphocytes, which we found to be more rapamycin-sensitive than was 4E-BP1. Note that resting lymphocytes have less eIF4E than do lymphoma cells, which would enable the displacement of eIF4G by low amounts of dephosphorylated 4E-BPs. Conversely, the high abundance of eIF4E in VAL lymphoma cells could preserve eIF4G binding after treatment with rapamycin, despite the presence of dephosphorylated 4E-BP2. MEFs have relatively low amounts of eIF4E, similar to lymphocytes, but have barely detectable amounts of 4E-BP2. In summary, we propose that a high ratio of rapamycin-sensitive 4E-BP2 to eIF4E together with the coupling of growth and division through eIF4E can explain why rapamycin profoundly suppresses lymphocyte blastogenesis and clonal expansion (Fig. 8E). Consistent with this model, expression of 4EBP1M suppressed cell growth and division in a concentration-dependent manner.
An increase in global protein synthesis has long been observed as an early step in lymphocyte activation (48). Hence, it is surprising that translational control in lymphocyte activation is not well studied. Our results suggest that mTORC1 inhibition suppresses the increase in protein synthesis in a 4E-BP–dependent manner after antigen receptor stimulation. In non-lymphoid cells, translation of specific mRNA subsets is sensitive to the activity and amount of eIF4E (49–51). Thus, it is likely that regulated translation of specific mRNAs is necessary for increased global translation. mRNAs encoding ribosomal proteins are sensitive to eIF4E activity in fibroblasts and to rapamycin in activated human T cells (13, 51). If ribosome number is limiting in resting lymphocytes, cap-dependent translation of those mRNAs encoding ribosomal proteins might be an essential eIF4E-dependent step to increase the capacity for global protein synthesis during the first phase of lymphocyte activation.
There is growing evidence that regulated mRNA translation modulates many aspects of immune function (52). For example, regulatory T cells have a distinct translational signature compared to that of conventional T cells (33). Modulation of the translational landscape by the mTORC1–4E-BP–eIF4E axis provides a previously unappreciated mechanism to drive blastogenesis (growth) and clonal expansion (proliferation) that is distinct from well-characterized transcriptome changes (52–54). eIF4E and regulated mRNA translation might play important roles in other mTORC1-dependent phenotypes in activated lymphocytes, such as metabolic reprogramming (55–57), T helper cell differentiation (12, 20, 58, 59), and effector and memory CD8+ T cell differentiation (60, 61). Thus, it will be of interest to study changes in the translational landscape upon lymphocyte activation, an under-studied area in the field. In addition, identifying those mRNAs whose translation is eIF4E-dependent and rapamycin-sensitive in activated lymphocytes might lead to new targets for immunosuppression.
Materials and Methods
Mouse strains and reagents
C57BL6 mice were bred at the University of California-Irvine (UCI) and used when they were between 6 and 12 weeks of age. All animals were studied in compliance with protocols approved by the Institutional Animal Care and Use Committees of UCI. Raptorfl/fl mice on a C57BL6 background were obtained from Jackson Laboratories (stock number: 013138) and have been described previously (11). Mice harboring one allele encoding a kinase-inactive (KI) mTOR mutant were previously described (62) and were rederived at UCI after retrieving embryonic stem (ES) cells from Lexicon Genetics. These mice were backcrossed to the C57BL6 background at least 8 to 10 times during the course of this study. No apparent differences were seen in cellular assays from early mTORKI mice compared to later stage mice upon multiple rounds of backcrossing to the C57BL6 background. CD4Cre mice (Model #4196) and CD19Cre mice (stock number:006785) were from Taconic and Jackson Laboratories, respectively. The analog-sensitive kinase allele (ASKA) knockin mouse for Rps6k1 was developed by Taconic and purchased (Revenue ID: RDBMBFASKA0040). Mice deficient in S6K2 (S6K2−/−) were a generous gift from Dr. Sara Kozma (IDIBELL, Spain). 4E-BP1/2–deficient mice (4E-BP1/2 DKO) were previously described (2). Mice harboring a transgenic allele encoding a constitutively active form of 4EBP1 (4EBP1M) under a tetracycline responsive element (TRE) were previously described (63). These mice were crossed to a strain harboring an optimized form of reverse tetracycline controlled transactivator (rtTA-M2) inserted downstream of the ROSA26 promoter, which was purchased from Jackson Laboratories (stock number: 006965). The active site mTORC1/2 inhibitor INK128 was purchased from Cayman Chemical. The mTOR allosteric inhibitor, rapamycin, was purchased either from LC Labs or Cell Signaling Technologies.
Cell surface staining
Cells for immunophenotyping were isolated either from the spleen, lymph nodes, or thymus. Except for lymph nodes, red blood cells (RBC) were lysed with ACK lysis buffer and cells were resuspended in FACS buffer [0.5% bovine serum albumin (BSA), 0.02% sodium azide, in PBS] with Fc block (eBioscience) for 30 min on ice. Without the Fc block being removed, the cells were stained for surface markers with the following fluorophore-conjugated antibodies: CD3-FITC, B220-APC, CD4-PE, CD69-PE, Vβ8-PE, or Vβ8-APC (all from eBioscience). Samples were stained for at least 30 min on ice before undergoing flow cytometric analysis.
Lymphocyte cell culture
B cells or CD4+ T cells were purified from murine splenocytes by negative selection with the MagniSort B cell or CD4+ T cell enrichment kit (eBioscience). For proliferation assays, purified cells were labeled with 2.5 μM carboxyfluorescein succinimidyl ester (CFSE, Life Technologies) in PBS for 5 min at room temperature and were washed once with lymphocyte culture media [LCM: RPMI medium supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin (100 U/ml), 2 mM L-glutamine, 5 mM HEPES buffer, and 50 μM 2-ME] before undergoing functional assays. CFSE-labeled B cells were activated with functional grade anti-mouse immunoglobulin M (IgM) F(ab’)2 (10 μg/ml, eBioscience) with interleukin-4 (IL-4, 10 ng/ml, R&D Technologies). CD4+ T cells were activated by plate-bound anti-CD3 antibody (2C11: 5 μg/ml) and plate-bound anti-CD28 antibody (2.5 μg/ml).
Cell culture
Immortalized primary MEFs (from WT and 4E-BP1/2 DKO mice) were from the p53−/− background and have been described previously (2). MEFs and NIH/3T3 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS and penicillin-streptomycin (100 U/ml). NIH/3T3 cells were further engineered to express a doxycycline-inducible, constitutively active form of 4E-BP1 in which five phosphorylation sites were mutated to alanines: 5A. For 4E-BP1 mutant experiments, immortalized MEFs were first stably transduced with the pMA2640 vector that expresses the reverse tetracycline transactivator (rtTA) protein (Addgene plasmid #25434) and selected with puromycin (2 μg/ml). Selected cells were transduced again with pLVX-puro (Clontech) encoding the appropriate 4E-BP mutants under the control of a tetracycline-responsive element (TRE) for doxycycline-dependent induction, and selected with blastocidin (8 μg/ml). Cell proliferation was assessed by cell counting under a hemocytometer upon trypan blue exclusion. The human diffuse-large B cell lymphoma (DLBCL) cell lines OCI-Ly1 and VAL were described previously (37). DLBCL lines were cultured in RPMI medium with 10% FBS and penicillin-streptomycin (100 U/ml).
Cell growth measurement
The growth of activated B or CD4+ T cells at the times indicated in the figure legends was measured by the forward scatter (FSC) parameter with a BD FACSCalibur flow cytometer.
Intracellular staining
For intracellular staining of lymphocytes, cells were fixed and permeabilized with BD cytofix/cytoperm solution at the times indicated in the figure legends. Cells were washed twice with wash buffer (0.1% Tween-20 in PBS) and stained with antibodies against the appropriate proteins in wash buffer for 1 hour. Cells were washed once in wash buffer before being analyzed by flow cytometry. For cells other than lymphocytes, cells were fixed with 2% paraformaldehyde (PFA) for 15 min and permeabilized with ice-cold methanol. After washing with FACS buffer once, cells were stained with the appropriate antibodies for 1 hour in FACS buffer. Cells were subsequently stained with goat anti-rabbit secondary antibodies conjugated to different fluorophores for 30 min and washed once with FACS buffer before being analyzed. Alexa Fluor 647–conjugated antibody against pS6 (S240/244) (#5044) was from Cell Signaling Technologies. An Alexa Fluor 647–conjugated donkey anti-rabbit IgG was from BioLegend. For two-step staining of intracellular proteins, primary antibodies used for Western blotting were used.
Western blotting
Cells were lysed in RIPA buffer (Cell Signaling Technologies) supplemented with cOmplete protease inhibitor (Roche) and phosphatase inhibitor cocktail 2 and 3 (Sigma) for 30 min on ice with occasional mixing by vortex. Lysates were cleared by centrifugation at 4°C at max speed for 15 min. Protein amount was quantified with the Bradford Assay (Bio-Rad), and equal amounts of protein were loaded on 8, 12, or 4 to 15% TGX pre-case gels (Bio-Rad) for SDS-PAGE. Proteins were transferred onto nitrocellulose membranes with a tank blotting system (Bio-Rad) and incubated with antibodies specific for following proteins of interest: pS6 (S240/244) (#4858), p4EBP1 (T37/46) (#2855), 4EBP1 (#9644), 4EBP2 (#2845), eIF4G (#2469), Akt (#4691), pAkt (S473) (#4060), and mTOR (#2983) (all from Cell Signaling Technologies). Antibody against eIF4E (#610269) was from BD Transduction Laboratories, anti-puromycin antibody (12D10 clone) was from Millipore, and antibody against β-actin was from Sigma.
m7GTP cap assay
Cells were lysed in buffer A ([1% NP-40, 10 mM Tris-HCl (pH 7.6), 140 mM KCl, 4 mM MgCl2, 1 mM DTT, 1 mM EDTA, cOmplete protease inhibitor, and phosphatase inhibitor cocktail], and lysates were incubated with 30 μl of the mRNA cap analog m7GTP-agarose beads (Jena Bioscience) in buffer A (with 0.5% NP-40) under constant and gentle agitation for 1 hour at room temperature. Samples with beads were washed three times with buffer A (with 0.5% NP-40), and the eIF4E-associated complex was resolved by SDS-PAGE and analyzed by Western blotting.
Puromycin incorporation (SUnSET) assay
Puromycin was added to cultured cells (at a final concentration of 10 μg/ml) and the cells were pulsed for 10 min. Cells treated with cycloheximide (100 μg/ml) served as a positive control, whereas cells that were not exposed to puromycin served as a negative control in all experiments. Cells were harvested and subjected to Western blotting analysis. Puromycin incorporation (a measure of nascent protein synthesis) was assessed with an anti-puromycin antibody.
Activation of lymphocytes with SEB
To activate T cells with SEB in vitro, total splenocytes depleted of RBCs were labeled with 2.5 μM CFSE before the addition of SEB (200 ng/ml). After 96 hours, the cells were stained with anti-CD4 antibody to specifically gate on CD4+ T cells to analyze their proliferation. For in vivo T cell activation, mice (n = 3 for each group) were injected with 100 μg of SEB i.p., and cells were analyzed 24 and 48 hours later. For analysis, total splenocytes depleted of RBCs were stained with antibodies against the surface markers CD4, CD69, and Vβ8 to gate on the CD4+Vβ8+ population for growth, proliferation, and activation analysis by flow cytometry. At 48 hours after injection, total splenocytes were counted and back-calculated to the flow cytometric frequency of CD4+Vβ8+ T cells for absolute cell counts. Adoptive transfer experiments were performed by first isolating CD4+ T cells from either wild-type (WT) or 4E-BP1M donor mice. Purified CD4+ T cells were labeled with 2.5μM eFluor 670 (eBioscience), a dye that tracks cell division, and were injected into WT mice i.v. After 24 hours, these mice were injected with 100 μg of SEB, and the growth and proliferation of their CD4+Vβ8+ T cells were analyzed at 24 and 48 hours, respectively. A portion of the cells was also analyzed by flow cytometry to assess the cell-surface expression of CD69.
Sequence alignment
The ClustalW2 software provided by EMBL-EBI (www.ebi.ac.uk) was used to align the sequences for eIF4EBP1 and eIF4EBP2 across different species. The accession number for each species used is provided in table S1.
Gene expression profiling
The absolute expression profile of eIF4EBP1 and eIF4EBP2 was extracted from the Stanford Gene Expression Commons website (gexc.stanford.edu).
Statistical analysis
In experiments in which more than two conditions were compared, we used one-way ANOVA with Tukey’s post test for multiple comparisons. In experiments comparing two conditions, we used a two-tailed, two-sample unpaired t-test.
Supplementary Material
Fig. S1. Acute inhibition of S6K activity is dispensable for lymphocyte growth and proliferation.
Fig. S2. Chemical genetic inhibition shows that S6K activity is dispensable for lymphocyte growth and proliferation.
Fig. S3. Specific inactivation of mTOR kinase activity in lymphocytes phenocopies complete mTOR or mTORC1 loss in lymphocytes.
Fig. S4. Amino acid sequence surrounding the Thr37 phosphoacceptor site on 4E-BP1 and 4E-BP2 is distinct and conserved.
Fig. S5. Gene expression patterns of eif4ebp1 and eif4ebp2 are distinct specifically in mature lymphocytes.
Fig. S6. Rapamycin partially inhibits eIF4F complex formation in lymphoma cells and MEFs.
Fig. S7. Rapamycin has a lesser effect on the proliferation of MEFs than that of MLN0128.
Fig. S8. 4E-BP1M can be inducibly expressed in naïve lymphocytes and is sufficient to block growth and proliferation equivalent to rapamycin or TOR-KIs.
Fig. S9. 4E-BP1M blocks lymphocyte growth and proliferation in vivo.
Fig. S10. 4E-BP1M inhibits only proliferation without affecting size in lymphoma cells.
Table S1. Accession numbers for species analyzed.
References and Notes
- 1.Conlon IJ, Dunn GA, Mudge AW, Raff MC. Extracellular control of cell size. Nat Cell Biol. 2001;3:918–921. doi: 10.1038/ncb1001-918. [DOI] [PubMed] [Google Scholar]
- 2.Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj A, Liu Y, Kozma SC, Thomas G, Sonenberg N. mTORC1-Mediated Cell Proliferation, But Not Cell Growth, Controlled by the 4E-BPs. Science (80-) 2010;328:1172–1176. doi: 10.1126/science.1187532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohanna M, Sobering AK, Lapointe T, Lorenzo L, Praud C, Petroulakis E, Sonenberg N, Kelly PA, Sotiropoulos A, Pende M. Atrophy of S6K1(−/−) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat Cell Biol. 2005;7:286–294. doi: 10.1038/ncb1231. [DOI] [PubMed] [Google Scholar]
- 4.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poulin F, Gingras AC, Olsen H, Chevalier S, Sonenberg N. 4E-BP3, a new member of the eukaryotic initiation factor 4E-binding protein family. J Biol Chem. 1998;273:14002–14007. doi: 10.1074/jbc.273.22.14002. [DOI] [PubMed] [Google Scholar]
- 6.Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci U S A. 1998;95:1432–7. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J. 2012;441:1–21. doi: 10.1042/BJ20110892. [DOI] [PubMed] [Google Scholar]
- 8.Laplante M, Sabatini DM. mTOR Signaling in Growth Control and Disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pelletier J, Graff J, Ruggero D, Sonenberg N. Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res. 2015;75:250–63. doi: 10.1158/0008-5472.CAN-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lloyd AC. The regulation of cell size. Cell. 2013;154:1194–205. doi: 10.1016/j.cell.2013.08.053. [DOI] [PubMed] [Google Scholar]
- 11.Limon JJ, So L, Jellbauer S, Chiu H, Corado J, Sykes SM, Raffatellu M, Fruman DA. mTOR kinase inhibitors promote antibody class switching via mTORC2 inhibition. Proc Natl Acad Sci U S A. 2014;111:E5076–85. doi: 10.1073/pnas.1407104111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, Guertin DA, Lamb RF, Chi H. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity. 2013;39:1043–1056. doi: 10.1016/j.immuni.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terada N, Takase K, Papst P, Nairn AC, Gelfand EW. Rapamycin inhibits ribosomal protein synthesis and induces G1 prolongation in mitogen-activated T lymphocytes. J Immunol. 1995;155:3418–26. [PubMed] [Google Scholar]
- 14.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB, Sabatini DM. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science (80-) 2013;341:1236566. doi: 10.1126/science.1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, Konopleva M, Martin MB, Ren P, Liu Y, Rommel C, Fruman DA. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abraham RT, Wiederrecht GJ. Immunopharmacology of rapamycin. Annu Rev Immunol. 1996;14:483–510. doi: 10.1146/annurev.immunol.14.1.483. [DOI] [PubMed] [Google Scholar]
- 20.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang S, Pruitt M, Tran D, Du Bois W, Zhang K, Patel R, Hoover S, Simpson RM, Simmons J, Gary J, Snapper CM, Casellas R, Mock BA. B Cell-Specific Deficiencies in mTOR Limit Humoral Immune Responses. J Immunol. 2013;191:1692–1703. doi: 10.4049/jimmunol.1201767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. Embo J. 1998;17:6649–6659. doi: 10.1093/emboj/17.22.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: a regulator of cell size. Science (80-) 1999;285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- 24.Espeillac C, Mitchell C, Celton-Morizur S, Chauvin C, Koka V, Gillet C, Albrecht JH, Desdouets C, Pende M. S6 kinase 1 is required for rapamycin-sensitive liver proliferation after mouse hepatectomy. J Clin Invest. 2011;121:2821–2832. doi: 10.1172/JCI44203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pollizzi KN, Waickman AT, Patel CH, Sun IH, Powell JD. Cellular size as a means of tracking mTOR activity and cell fate of CD4+ T cells upon antigen recognition. PLoS One. 2015;10:e0121710. doi: 10.1371/journal.pone.0121710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuo CJ, Chung J, Fiorentino DF, Flanagan WM, Blenis J, Crabtree GR. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature. 1992;358:70–73. doi: 10.1038/358070a0. [DOI] [PubMed] [Google Scholar]
- 27.Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G. S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–3124. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tolcher A, Goldman J, Patnaik A, Papadopoulos KP, Westwood P, Kelly CS, Bumgardner W, Sams L, Geeganage S, Wang T, Capen AR, Huang J, Joseph S, Miller J, Benhadji KA, Brail LH, Rosen LS. A phase I trial of LY2584702 tosylate, a p70 S6 kinase inhibitor, in patients with advanced solid tumours. Eur J Cancer. 2014;50:867–875. doi: 10.1016/j.ejca.2013.11.039. [DOI] [PubMed] [Google Scholar]
- 29.Salmond RJ, Brownlie RJ, Meyuhas O, Zamoyska R. Mechanistic Target of Rapamycin Complex 1/S6 Kinase 1 Signals Influence T Cell Activation Independently of Ribosomal Protein S6 Phosphorylation. J Immunol. 2015 doi: 10.4049/jimmunol.1501473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ge Y, Wu AL, Warnes C, Liu J, Zhang C, Kawasome H, Terada N, Boppart MD, Schoenherr CJ, Chen J. mTOR regulates skeletal muscle regeneration in vivo through kinase-dependent and kinase-independent mechanisms. Am J Physiol Cell Physiol. 2009;297:C1434–44. doi: 10.1152/ajpcell.00248.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yip CK, Murata K, Walz T, Sabatini DM, Kang SA. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell. 2010;38:768–774. doi: 10.1016/j.molcel.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, Meyuhas O, Shokat KM, Ruggero D. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17:249–261. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bjur E, Larsson O, Yurchenko E, Zheng L, Gandin V, Topisirovic I, Li S, Wagner CR, Sonenberg N, Piccirillo CA. Distinct translational control in CD4+ T cell subsets. PLoS Genet. 2013;9:e1003494. doi: 10.1371/journal.pgen.1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mothe-satney I, Yang D, Fadden P, Haystead TaJ, CL J, Jr, Lawrence JC. Multiple Mechanisms Control Phosphorylation of PHAS-I in Five (S/T) P Sites That Govern Translational Repression. 2000 doi: 10.1128/mcb.20.10.3558-3567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, Panteleyev AA, Okkenhaug K, Cantrell DA. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012;209:2441–53. doi: 10.1084/jem.20112607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mallya S, Fitch BA, Lee JS, So L, Janes MR, Fruman DA. Resistance to mTOR kinase inhibitors in lymphoma cells lacking 4EBP1. PLoS One. 2014;9:e88865. doi: 10.1371/journal.pone.0088865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-Martinez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, Alessi DR. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR) Biochem J. 2009;421:29–42. doi: 10.1042/BJ20090489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mao X, Green JM, Safer B, Lindsten T, Frederickson RM, Miyamoto S, Sonenberg N, Thompson CB. Regulation of translation initiation factor gene expression during human T cell activation. J Biol Chem. 1992;267:20444–50. [PubMed] [Google Scholar]
- 40.Ginzberg MB, Kafri R, Kirschner M. On being the right (cell) size. Science (80-) 2015;348:1245075–1245075. doi: 10.1126/science.1245075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24:200–16. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–87. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurebayashi Y, Nagai S, Ikejiri A, Ohtani M, Ichiyama K, Baba Y, Yamada T, Egami S, Hoshii T, Hirao A, Matsuda S, Koyasu S. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORgamma. Cell Rep. 2012;1:360–373. doi: 10.1016/j.celrep.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Park Y, Jin HS, Lopez J, Elly C, Kim G, Murai M, Kronenberg M, Liu YC. TSC1 regulates the balance between effector and regulatory T cells. J Clin Invest. 2013;123:5165–78. doi: 10.1172/JCI69751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsukiyama-Kohara K, Poulin F, Kohara M, DeMaria CT, Cheng A, Wu Z, Gingras AC, Katsume A, Elchebly M, Spiegelman BM, Harper ME, Tremblay ML, Sonenberg N. Adipose tissue reduction in mice lacking the translational inhibitor 4E-BP1. Nat Med. 2001;7:1128–32. doi: 10.1038/nm1001-1128. [DOI] [PubMed] [Google Scholar]
- 46.Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, Major F, Lasko P, Ruggero D, Nader K, Lacaille JC, Sonenberg N. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2012;493:371–377. doi: 10.1038/nature11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoon SO, Roux PP. Rapamycin resistance: mTORC1 substrates hold some of the answers. Curr Biol. 2013;23:R880–3. doi: 10.1016/j.cub.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 48.Ahern T, Sampson J, Kay JE. Initiation of protein synthesis during lymphocyte stimulation. Nature. 1974;248:519–21. doi: 10.1038/248519a0. [DOI] [PubMed] [Google Scholar]
- 49.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, Wang S, Ren P, Martin M, Jessen K, Feldman ME, Weissman JS, Shokat KM, Rommel C, Ruggero D. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, Seo Y, Barna M, Ruggero D. Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell. 2015;162:59–71. doi: 10.1016/j.cell.2015.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mamane Y, Petroulakis E, Martineau Y, Sato TA, Larsson O, Rajasekhar VK, Sonenberg N. Epigenetic activation of a subset of mRNAs by eIF4E explains its effects on cell proliferation. PLoS One. 2007;2:e242. doi: 10.1371/journal.pone.0000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piccirillo CA, Bjur E, Topisirovic I, Sonenberg N, Larsson O. Translational control of immune responses: from transcripts to translatomes. Nat Immunol. 2014;15:503–511. doi: 10.1038/ni.2891. [DOI] [PubMed] [Google Scholar]
- 53.Glynne R, Ghandour G, Rayner J, Mack DH, Goodnow CC. B-lymphocyte quiescence, tolerance and activation as viewed by global gene expression profiling on microarrays. Immunol Rev. 2000;176:216–46. doi: 10.1034/j.1600-065x.2000.00614.x. [DOI] [PubMed] [Google Scholar]
- 54.Kanno Y, Vahedi G, Hirahara K, Singleton K, O’Shea JJ. Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu Rev Immunol. 2012;30:707–31. doi: 10.1146/annurev-immunol-020711-075058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev. 2012;249:43–58. doi: 10.1111/j.1600-065X.2012.01152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol. 2012;30:39–68. doi: 10.1146/annurev-immunol-020711-075024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeng H, Chi H. mTOR and lymphocyte metabolism. Curr Opin Immunol. 2013;25:347–355. doi: 10.1016/j.coi.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T-cell function. Nature. 2013 doi: 10.1038/nature12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol. 2011;11:109–117. doi: 10.1038/nri2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Araki K, Youngblood B, Ahmed R. The role of mTOR in memory CD8 T-cell differentiation. Immunol Rev. 2010;235:234–243. doi: 10.1111/j.0105-2896.2010.00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shor B, Cavender D, Harris C. A kinase-dead knock-in mutation in mTOR leads to early embryonic lethality and is dispensable for the immune system in heterozygous mice. BMC Immunol. 2009;10:28. doi: 10.1186/1471-2172-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Acute inhibition of S6K activity is dispensable for lymphocyte growth and proliferation.
Fig. S2. Chemical genetic inhibition shows that S6K activity is dispensable for lymphocyte growth and proliferation.
Fig. S3. Specific inactivation of mTOR kinase activity in lymphocytes phenocopies complete mTOR or mTORC1 loss in lymphocytes.
Fig. S4. Amino acid sequence surrounding the Thr37 phosphoacceptor site on 4E-BP1 and 4E-BP2 is distinct and conserved.
Fig. S5. Gene expression patterns of eif4ebp1 and eif4ebp2 are distinct specifically in mature lymphocytes.
Fig. S6. Rapamycin partially inhibits eIF4F complex formation in lymphoma cells and MEFs.
Fig. S7. Rapamycin has a lesser effect on the proliferation of MEFs than that of MLN0128.
Fig. S8. 4E-BP1M can be inducibly expressed in naïve lymphocytes and is sufficient to block growth and proliferation equivalent to rapamycin or TOR-KIs.
Fig. S9. 4E-BP1M blocks lymphocyte growth and proliferation in vivo.
Fig. S10. 4E-BP1M inhibits only proliferation without affecting size in lymphoma cells.
Table S1. Accession numbers for species analyzed.