

Abstract
We previously reported a systemic hyperinflammatory response to bacterial lipopolysaccharide (LPS) in children with localized aggressive periodontitis (LAP). Additionally, different levels of this response were observed within the LAP group. It is unknown whether this hyperinflammatory response influences the clinical response to periodontal treatment in these children. Therefore, the goal of this study was to evaluate the influence of LPS responsiveness present prior to treatment on the clinical response to treatment within the LAP cohort. Prior to treatment, peripheral blood was collected from 60 African American participants aged 5 to 21 y, free of systemic diseases, and diagnosed with LAP. Blood was stimulated with ultrapure LPS from Escherichia coli, and Luminex assays were performed to quantify 14 cytokine/chemokine levels. Principal component and cluster analyses were used to find patterns of cytokine/chemokine expression among participants and subdivide them into clusters. Three distinct clusters emerged among LAP participants: a high responder group (high level of response for INFg, IL6, and IL12p40), a mixed responder group (low for some and high for other cytokines/chemokines), and a low responder group (low overall cytokine/chemokine response). Periodontal clinical parameters were compared among these groups prior to and 3, 6, and 12 mo following treatment with mechanical debridement and systemic antibiotics. High responders presented the lowest reductions in clinical parameters after treatment, whereas the low responders presented the highest reductions. In our LAP participants, distinct patterns of LPS response were significantly predictive of changes in clinical parameters after treatment. Future studies are needed to evaluate the underlying mechanisms predicting the heterogeneity of LAP activity, severity, and response to treatment (ClinicalTrials.gov NCT01330719).
Keywords: inflammation, cytokines, chemokines, lipopolysaccharides, periodontal diseases, therapy
Introduction
Aggressive periodontitis (AgP) is a severe form of periodontitis characterized by a rapid progression of periodontal tissue breakdown in young individuals who are otherwise clinically healthy (Stabholz et al. 1998; Armitage 1999). The prevalence of aggressive periodontitis varies among ages and ethnic groups, ranging from 0.04% to 3.81% in North America (Albandar and Tinoco 2002). Localized aggressive periodontitis (LAP) is a type of aggressive periodontitis that primarily affects first molars and incisors in individuals around circumpubertal age (Lang et al. 1999). In addition, patients diagnosed with LAP seem to possess hyperactive inflammatory response to bacterial lipopolysaccharide (LPS; Shaddox et al. 2010) and a higher ratio of bone destruction to plaque than that of chronic periodontitis (Lang et al. 1999).
LAP destruction occurs when cell wall LPSs of gram-negative bacteria interact with Toll-like receptors (TLRs), such as TLR2 and TLR4, present on the surface of host immune cells, stimulating the production of various cytokines/chemokines and growth factors. TLRs are an essential aspect of the body’s innate immunity. However, due to their potential to overreact in particular individuals, they can also act as antagonists, exacerbating disease processes. In LAP, TLRs seem to be activated abnormally, leading to elevated levels of proinflammatory mediators that may result in the advanced destruction of the host’s periodontal tissues observed in this disease (Mahanonda and Pichyangkul 2007; Shaddox et al. 2010; Shaddox et al. 2011).
Prevalence of LAP found in related individuals has indicated a possible genetic role in this disease (Marazita et al. 1994; Schenkein and Van Dyke 1994). It has also been shown that African Americans have a higher incidence of LAP (Albandar and Tinoco 2002; Susin et al. 2014). In a previous study, it was discovered that African American children diagnosed with LAP present elevated levels of cytokines/chemokines IL1α, IL1β, IL2, IL12p40, IFNγ, TNFα GM-CSF, and IL6 in response to LPS stimulation when compared with unrelated healthy control individuals and that siblings have an intermediary response, presenting lower levels than those of LAP but higher than those of healthy unrelated controls (Shaddox et al. 2010). Additionally, it was observed that after successful periodontal treatment, these cytokine/chemokine responses were significantly modulated, and correlations were found between these responses and clinical parameters of disease (Shaddox et al. 2013). However, interestingly, the levels of inflammatory response to LPS vary greatly among LAP individuals, and although multiple factors may be involved in this disease, the mechanism and clinical significance of this hyperinflammatory response are still not well understood. Studies by Trombelli and colleagues (2006) found that periodontitis-free individuals with ongoing experimental gingivitis respond differently to similar levels of plaque accumulation and that gingival crevicular fluid volume is higher in aggressive periodontitis patients when compared with both high and low responders within the periodontitis-free patients. These findings corroborate the idea that individual susceptibility to bacteria may indeed play a role on the clinical presentation of disease and, possibly, the response to treatment. Furthermore, Nibali and colleagues (2013) showed differential IL6 concentrations in the gingival crevicular fluid associated with IL6 haplotypes in aggressive periodontitis patients. This could further indicate a genetic predisposition to a specific inflammatory response, which in turn could influence differential patterns of treatment response. Therefore, the objective of the present study was to determine if baseline levels of LPS responsiveness in LAP individuals prior to clinical intervention are significantly associated with the clinical response to periodontal treatment. Specifically, we hypothesized that there would be significant differences in the clinical response to treatment among groups of LAP patients based on their baseline levels of cytokine/chemokine release following LPS stimulation.
Materials and Methods
Participant Population
Participants were recruited from the Leon and Duval county health departments in Tallahassee and Jacksonville, Florida, respectively, and from the dental clinics at University of Florida, Gainesville, between February 2007 and January 2012. All participants (or their legal representatives) signed an informed consent form approved by the University of Florida Institutional Review Board. This study is registered at ClinicalTrials.gov (NCT01330719).
Medical and dental histories were obtained and reviewed. Participants were included if they met the following inclusion criteria: African American children aged 5 to 21 y and diagnosed with LAP, as defined by presentation of ≥2 teeth per site (first molar and/or incisor but not more than 2 teeth other than first molar and incisor), presenting pocket depth (PD) >4 mm with bleeding upon probing, and concomitant attachment loss ≥2 mm with visible radiographic bone loss. Participants were excluded if they presented a systemic disease/condition or were taking any medications that could affect their periodontal health, if they had taken antibiotics within the past 3 mo, if they were pregnant or lactating, or if they smoked >10 cigarettes a day for over 1 y.
Clinical Measurements
At baseline and at 3, 6, and 12 mo after periodontal treatment, the following clinical parameters were measured: PD, clinical attachment levels (CALs), bleeding on probing, and visible plaque. CAL was determined by the sum of PD and gingival margin position. When the gingival margin was located coronal to the cementum-enamel junction, it was given as a negative number. All measurements were performed by the use of a UNC-15 periodontal probe at 6 sites per tooth and recorded with the Florida Probe software program (Florida Probe Corporation, Gainesville, FL, USA).
Periodontal Treatment Protocol
The periodontal treatment protocol used in this study has been described (Shaddox et al. 2013). Briefly, at the baseline appointment, participants received ultrasonic full-mouth debridement (Cavitron Jet Plus; Dentsply, York, PA, USA) and site-specific scaling and root planing. Immediately after treatment, they were prescribed a regimen of 250 mg of metronidazole and 500 mg of amoxicillin (dosage was adjusted for children, <40 kg) 3 times per day for 7 d. Participants were reexamined at 3, 6, and 12 mo, and they received additional mechanical therapy for sites with pockets >4 mm, as needed, along with oral hygiene reinstructions.
LPS Stimulation and Detection of Cytokines/Chemokines
Approximately 4 mL of peripheral blood was collected by venipuncture at baseline, prior to treatment, and processed as described previously (Shaddox et al. 2010). Briefly, blood was diluted 1:4 in RPMI1640 (Invitrogen, Carlsbad, CA, USA), then stimulated with 1 µg/mL of ultrapure Escherichia coli (InvivoGen, San Diego, CA, USA). After 24 h, 14 cytokines/chemokines—eotaxin, GM-CSF, IFNγ, IL1β, IL6, IL8, IL10, IL2, IL12p40, IL12p70, IP10, MIP1α, G-CSF, and TNFα—were detected and quantified in the supernatant with multiplex fluorescence detection kits (Millipore, St. Charles, MO, USA). Culture supernatants and cytokine capture bead cocktails were incubated for 2 h. The samples were then incubated for 1.5 h with biotin-labeled anticytokine and for 30 min in a 1:12.5 dilution of streptavidin-phycoerythrin (data were obtained by Luminex 200 and analyzed with Milliplex Analyst software; Millipore) with 5 parameter logistics and standard curves.
Data and Statistical Analysis
A priori power calculation suggested that a sample size of 17 patients per group would enable a detection of a difference in PD of 0.8 mm with an SD of 0.8 mm with 80% power. Data and statistical analysis was performed with SAS 9.2 and SPSS 22 (IBM, Chicago, IL, USA). An alpha level of 0.05 was considered statistically significant for all analyses. The percentage of sites with PD ≥4 mm and the percentage of affected sites defined as having PD >4 mm and concomitant attachment loss (percentage of affected sites) were determined for each participant at each time point. Mean PD and CAL were calculated for affected sites identified at the baseline appointment, and the clinical parameters listed above, in addition to percentage bleeding on probing, at 3, 6, and 12 mo were used for the statistical analysis.
Data reduction: principal component analysis
Principal component analysis was conducted to identify principal components of immune response with an oblique rotation. Components with eigenvalues >1 were retained for interpretation and confirmed by visual inspection of the screen plot. Once the components were determined, cytokine index scores were computed as Z scores from the factor scores for each component. All Z scores were determined such that positive scores indicated greater immune response to LPS and negative scores indicated lower immune response to LPS. Pearson product-moment correlation coefficients were used to document the associations between the cytokine/chemokine components. Analysis of variance with Tukey’s post hoc test was used to compare clusters within each principal component.
Classification of participants: cluster analysis
Whereas principal component analysis uses interrelations among variables to form a smaller number of components, cluster analysis is used to assign people to groups, or “clusters” that share certain similarities. A 2-step cluster analysis was performed to organize observations into ≥2 mutually exclusive groups, where members of the groups shared properties in common. We implemented a precluster sequential clustering approach, followed by hierarchical cluster analysis using Ward’s clustering method with squared Euclidean distances. The optimal number of clusters was based on the percentage change between clusters and visual inspection of the cluster dendrogram. The baseline cytokine index scores were entered as clustering variables, and the number of clusters was not predetermined. Internal cluster validation was performed with analysis of covariance for the individual cytokines/chemokines, with 2-tailed tests and an alpha level of 0.05. We did not correct for multiple comparisons, since the aim of this analysis was to inspect the clusters to determine whether they differed significantly among the variables used to originally create them. This is a first step in determining the validity of a cluster solution (Aldenderfer and Blashfield 1984).
Differences across clusters on demographic and clinical measures
To characterize the resulting clusters, differences in sample age and baseline clinical characteristics were compared according to cluster membership via multivariate analysis of covariance for continuous variables as above and χ2 tests for categorical variables.
To examine differences in clinical measures over time, we conducted a repeated measures or doubly multivariate analysis of covariance. Given the unequal cluster sample sizes, Box’s M test was conducted to check the homogeneity of variance-covariance assumption. We tested the multivariate main effects of “time” and “cluster,” as well as the “time × cluster” interaction. If interaction effects were significant, analyses of covariance were run with all 5 dependent variables but only at the 3-mo postintervention time point to reduce the number of statistical tests performed. This time point was based on our previous work showing that LPS responsiveness decreased significantly at this time point (Shaddox et al. 2013). Sex, age, and baseline PD and CAL were included as covariates in all analyses. No further multiple comparison adjustments were performed.
Results
Study Participants
Sixty LAP patients were included in the study. Their mean age was 14 ± 4 y old, and 46 of 60 were females.
Principal Component Analyses of Cytokine Variables
Principal component analyses were performed on the cytokines/chemokines with an oblique rotation. Kaiser-Meyer-Olkin measures of sampling adequacy, as well as individual measures of sampling adequacy, were >0.70 indicating adequate matrix factorability. Four principal components resulted and are presented in Table 1. The total percentage of variance explained equaled 74.4% (Table 1). The following 4 components emerged:
Table 1.
Principal Component Analysis: Component Loadings and Eigenvalues of Cytokines.
| Cytokine/Chemokine | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| INF-γ | 0.909 | 0.107 | <0.100 | −0.238 |
| IL-6 | 0.845 | <0.100 | 0.176 | 0.249 |
| IL-12p40 | 0.798 | 0.245 | <0.100 | −0.188 |
| Eotaxin | <0.100 | 0.927 | <0.100 | <0.100 |
| IP-10 | 0.204 | 0.879 | <0.100 | −0.139 |
| IL-10 | <0.100 | <0.100 | 0.843 | <0.100 |
| IL-1β | <0.100 | <0.100 | 0.780 | <0.100 |
| TNF-α | 0.475 | 0.155 | 0.659 | <0.100 |
| IL-8 | <0.100 | <0.100 | <0.100 | 0.898 |
| MP-1α | <0.100 | −0.139 | 0.256 | 0.621 |
| MCP-1 | <0.100 | 0.559 | −0.229 | 0.579 |
| Variance, %a | 27.6 | 18.5 | 15.7 | 12.6 |
| Cumulative variance, %b | 27.6 | 46.1 | 61.8 | 74.4 |
Values in bold indicate the group of cytokines/chemokines that are mostly associated with that specific component as evidenced by the strength of the loadings or correlations.
Percentage variance indicates the amount of variability in the sample accounted for by each component.
Cumulative percentage variance is the total amount of variability accounted by each of the component’s variance.
Component 1: INF, IL-6, and IL-12p40, with loadings from 0.798 to 0.909.
Component 2: eotaxin and IP-10, with loadings of 0.927 and 0.879, respectively.
Component 3: IL-10, IL-1β, and TNF-α with loadings from 0.659 to 0.843.
Component 4: IL-8, MP-1, and MCP-1 with loadings from 0.579 to 0.898.
There were nonsignificant correlations across most Z scores, except for a modest correlation between component 1 and component 3 (r = 0.37, P < 0.05).
Cluster Analysis of Cytokine Index Scores
The cytokine index scores were then subjected to cluster analysis to identify patterns of immune response across several cytokines simultaneously, thus creating immune response profiles. The clustering procedure resulted in 3 clusters, without exclusion of cases. The clusters were characterized by 1) a group of individuals with significantly high scores on component 1 (high INF-G, IL-6, and IL-12p40; n = 18); 2) a cluster with a mixed profile of cytokine responses (n = 21); and 3) a group of individuals with the lowest scores on all cytokine responses (n = 37; Fig. 1). To assess the internal validity of the cluster solution, cluster profiles were compared on all of the raw values (e.g., nontransformed values) for each cytokine/chemokine variable. As expected, the clusters significantly differed across all of the individual cytokine variables (P<0.05; Table 2).
Figure 1.
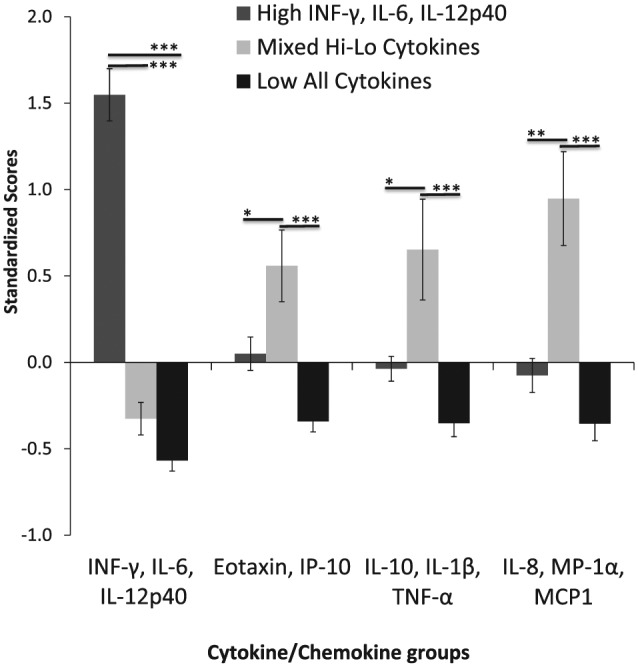
Cytokine index scores subjected to cluster analysis created 3 distinct immune response profiles (clusters): 1) individuals with significantly high scores on component 1 (INF-γ, IL-6, and IL-12p40; n=18), 2) individuals with a mixed profile of cytokine responses (n=21), and 3) individuals with the lowest scores on all cytokine responses (n=37). *P < 0.05, **P < 0.001, ***P < 0.0001, comparing clusters within components by analysis of variance with Tukey’s post hoc test.
Table 2.
Cytokine Release upon Lipopolysaccharide Stimulation across the 3 Clusters.
| Cytokine / Chemokine | Cluster 1 (n = 18) | Cluster 2 (n = 21) | Cluster 3 (n = 37) | P Value |
|---|---|---|---|---|
| INF-γ | 4,158.6 (1,344.8) | 176.5 (329.1) | 84.6 (373.0) | <0.0001 |
| IL-6 | 4,657.7 (1,321.8) | 2,553.0 (1,546.1) | 1,338.6 (1,210.2) | <0.0001 |
| IL-12p40 | 4,139.0 (2,753.8) | 704.9 (983.8) | 195.8 (682.5) | <0.0001 |
| Eotaxin | 149.4 (52.2) | 203.3 (448.9) | 9.9 (29.4) | 0.012 |
| IP-10 | 11,442.4 (9,465.6) | 10,503.9 (16,242.5) | 834.1 (959.7) | 0.006 |
| IL-10 | 2,649.1 (1,518.0) | 4,697.1 (5,117.5) | 1,159.4 (2,739.8) | 0.002 |
| IL-1β | 406.1 (269.1) | 985.3 (1,745.6) | 207.7 (273.7) | 0.017 |
| TNF-α | 1,535.5 (259.5) | 1,387.7 (1,119.4) | 428.4 (455.0) | <0.0001 |
| IL-8 | 1,635.7 (362.4) | 4,204.7 (2,485.7) | 1,951.9 (1,283.8) | <0.0001 |
| MP-1α | 2,265.5 (1,283.8) | 3,567.1 (2,035.4) | 2,121.5 (1,551.6) | 0.014 |
| MCP-1 | 1,940.7 (751.6) | 4,372.4 (2,596.6) | 1,711.2 (2,132.3) | <0.0001 |
Values presented in mean (SD), pg/mL.
Differences across Clusters on Demographic and Clinical Measures
The clusters had similar age and clinical measures at baseline (P > 0.05; see Table 3 and Fig. 2). To examine differences in the 5 clinical measures (i.e., percentage of pockets with PD >4 mm, bleeding on probing, percentage of affected sites, mean PD, and mean CAL) over time between participants across the 3 clusters, we conducted a doubly multivariate analysis of variance. Since the sample sizes were unequal across the clusters, the homogeneity of variance-covariance assumption was checked and met. Significant multivariate effects were found for the main effects of “time” (F = 3.10, P = 0.002) and “cluster” (F = 2.53, P = 0.039) as well as the “time × cluster interaction” (F = 1.98, P = 0.038). This interaction effect indicates that the difference between the clusters on the combination of the 5 variables is different at baseline from the other time points. To reduce the number of comparisons made and as guided by our previous findings, we focused our post hoc analysis on the difference among the clusters at the 3-mo postintervention time point. All subsequent analyses of covariance on the 5 clinical measures supported significant differences among the clusters (P < 0.05). Specifically, individuals in cluster 1 (i.e., high INF-G, IL-6, IL-12p40) had significantly greater percentage of sites with PD >4 mm, bleeding on probing, and percentage of affected sites at 3 mo posttreatment compared with the other 2 clusters (P’s < 0.05). However, individuals in cluster 3 (i.e., low all cytokines) had significantly lower mean PD at 3 mo posttreatment compared with clusters 1 and 2 (P’s < 0.05). Similarly, cluster 3 (i.e., low all cytokines) had lower mean CAL at 3 mo posttreatment compared with clusters 1 and 2 (P’s < 0.05; see Fig. 2).
Table 3.
Demographic and Baseline Clinical Data of Patients Distributed in the 3 Clusters.
| Cluster 1 (n = 18) | Cluster 2 (n = 21) | Cluster 3 (n = 37) | |
|---|---|---|---|
| Age, y | 13.7 (4.5) | 13.0 (4.4) | 13.6 (3.3) |
| % PD >4 mm | 16.6 (4.5) | 15.1 (4.4) | 16.0 (3.3) |
| % BoP | 15.3 (11.6) | 14.4 (9.6) | 16.3 (13.9) |
| % Plaque | 51.4 (20.0) | 42.1 (28.8) | 43.1 (26.6) |
| Mean PD, mm | 4.9 (0.8) | 5.3 (0.9) | 5.3 (0.6) |
| Mean CAL, mm | 3.2 (1.3) | 3.5 (1.5) | 3.2 (1.1) |
| % Affected sites | 14.8 (9.2) | 11.0 (8.6) | 12.2 (9.4) |
% affected sites, percentage of sites with PD >4 mm + CAL >2 mm; % BoP, percentage of sites with bleeding on probing; % PD >4 mm, percentage of pockets with probing depth >4 mm; % plaque, percentage of sites with visible plaque; CAL, clinical attachment level; PD, pocket depth.
Values presented in mean (SD). No differences were detected among the clusters for any of the parameters here (P > 0.05).
Figure 2.

Clinical parameters at baseline and after treatment (3, 6, and 12 mo) for patients subdivided in the 3 clusters: high lipopolysaccharide responders (high levels of INFg, IL6, and IL12p40), mixed (low for first component and high on others markers), and low all (low for all markers tested in the model). Different covariance models examined adjusted for age and baseline clinical parameters showed statistically significant changes among clusters at specific time points and within clusters overall. Cluster 1 (high INF-g, IL-6, IL-12p40) had greater percentage of sites with pocket depth >4 mm (%PD >4 mm), percentage bleeding on probing (BoP), and percentage of affected sites at 3 mo posttreatment versus the other 2 clusters (P < 0.05). Yet, individuals in cluster 3 (i.e., low for all cytokines) had lower mean pocket depth (PD) at 3, 6, and 12 mo posttreatment versus other clusters (P < 0.05) and lower mean clinical attachment levels (CALs) at 3 and 6 mo posttreatment versus other clusters (P < 0.05). *P < 0.05 versus other groups.
Discussion
Patients exhibiting periodontitis present different levels of success when given equal periodontal treatment (Cobb 1996). Periodontitis is considered a complex multifactorial disease, where genetic, pathogenic, and environmental factors have been suggested to play a role in the susceptibility and healing processes of different individuals. In determining the specific factors involved, predictions of treatment success and stability of disease can be made.
We previously reported E. coli and Porphyromonas gingivalis LPS-stimulated hyperinflammatory cytokine/chemokine response profiles for patients diagnosed with LAP when compared with those of healthy unrelated controls and healthy siblings, where LAP individuals presented elevated LPS responsiveness measured by exacerbated expression of IL1α, IL1β, IL2, IL12p40, IFNγ, TNFα GM-CSF, and IL6 (Shaddox et al. 2010). We further correlated these LPS-induced cytokine/chemokine levels with several clinical parameters at baseline and after the course of treatment (Shaddox et al. 2013). This discovery led us to question whether the magnitude of this cytokine/chemokine response to LPS stimulation prior to treatment would have an effect on treatment results.
We present here findings that LAP patients with similar baseline clinical parameters but varying initial response to LPS, as measured by levels of different cytokines/chemokines, exhibited differences in response to treatment. We identified 3 response groups through cluster analysis, and the ones exhibiting relatively lower LPS-stimulated levels of all inflammatory markers tested in the model at baseline (low responders) presented overall greater reductions of clinical parameters after treatment as compared with those who presented relatively high LPS response levels of some of these cytokines/chemokines at baseline (high and mixed responder groups; P < 0.05).
Baseline response levels of many cytokines/chemokines were strongly associated with clinical parameters in the short term after treatment (3 mo), although mean PD was lower in the low responder cluster at all time points after treatment. It is possible that clinical reductions are frequently higher at the initial phase after treatment for the low responder group due to an initial low inflammatory burden on these patients, which may allow the host to show a quicker healing response. However, the high inflammatory response clusters (high and mixed responders) eventually show decreases in clinical parameters after treatment, and this may be due to a reduction in the cytokine/chemokine LPS response after treatment, as observed in a previous study (Shaddox et al. 2013). This previous study observed that the LPS responsiveness decreased significantly at 3 mo after initial treatment. This reduction may have allowed the high responder clusters to be able to mount a healing response to treatment after that time point. Although this previous study also showed a rebound in LPS inflammatory response by 6 to 12 mo, this apparently did not influence the ability of these high responders to eventually show reductions in the clinical parameters of disease, possibly due to an LPS tolerance in these patients (Nahid et al. 2011).
Additionally, different cytokines/chemokines are known to cross-regulate one another. Interestingly, most proinflammatory markers were highly correlated with one another in the different components found here, such as component 1 (INFγ, IL6, and IL12p40), high in cluster 1 patients, who showed the worse initial response to treatment when compared with the individuals presenting low LPS response for all inflammatory markers (cluster 3). Indeed, INFγ has been well associated with bone loss in periodontal disease in animal models (Teng et al. 2005; Garlet et al. 2008). Interestingly, in our sample, INFγ was significantly associated with IL12 and, together with IL6, characterized one of the high response groups. Although IL12 role’s in periodontal disease is controversial, it can be considered a Th1 response driver, and it has also been associated with bone loss (Garlet 2010). Similarly, cluster 2—which was low in this first component but high in several other proinflammatory markers, such as TNFα, IL1β, IL8, and MIP1α—seemed to follow the same pattern of cluster 1 for lower reductions in mean PD and CAL. Interestingly, MIP1α, along with IL8 and IL1β, was recently associated with initiation of attachment loss in young individuals (Fine et al. 2014). The initial high levels of these markers, followed by their eventual reduction after treatment, as shown previously (Thunell et al. 2010; Shaddox et al. 2013), could reinforce the current observation of a less robust/delayed response to treatment in these individuals of clusters 1 and 2.
It is important to note that the levels of cytokines/chemokines presented in this study were obtained after in vitro stimulation of whole blood cells with E. coli LPS. Thus, the present study showed how these levels, represented as the result of the host response to gram-negative bacterial stimuli, could influence response to treatment. Further studies need to be conducted to evaluate the influence of the actual nonstimulated local and systemic levels of these cytokines/chemokines in the response to treatment, as well as the mechanisms behind the heterogeneity of the immune response in this population to further elucidate their roles in disease response, progression, and stability. In addition, there is always the possibility that the results observed may be due to “regression to the mean,” where any group defined on the basis of extreme values at baseline tends to regress toward the mean even in the absence of any treatment. We aimed to decrease this effect by using different measures postbaseline (i.e., clinical measures), which were not included in the baseline grouping of participants, and by accounting for the baseline clinical measures (i.e., covariates). Future clinical trials may be designed with a control group to help in minimizing these effects. Finally, we were not able to statistically compare the clusters across all time points measured due to the large number of comparisons increasing the chances of a type I error. Future randomized controlled studies with larger sample sizes are needed to address this issue and fully understand the longitudinal effects of baseline immune responses.
Despite these limitations, the present study shows that an initial specific pattern of cytokine/chemokine LPS responsiveness was significantly associated with different responses to treatment in African Americans diagnosed with localized aggressive periodontitis. Further understanding the impact of this inflammatory response in the subsequent clinical response to treatment could affect clinical care. Specifically, it may enable clinicians to better predict clinical responses to treatment—as well as preselect different treatment approaches targeted to these hyperresponsive patients, such as the use of specific anti-inflammatory agents or antibiotics—or even establish longer follow-up of clinical responses to nonsurgical treatments prior to surgical intervention.
Author Contributions
N. Allin, Y. Cruz-Almeida, contributed to data analysis, drafted the manuscript; I. Velsko, contributed to data analysis, drafted and critically revised the manuscript; A. Vovk, contributed to design, data analysis, and interpretation, critically revised the manuscript; N. Hovemcamp, contributed to data acquisition, critically revised the manuscript; P. Harrison, contributed to design, data acquisition, and interpretation, critically revised the manuscript; H. Huang, contributed to data analysis, critically revised the manuscript; I. Aukhil, contributed to conception, design, data acquisition, and interpretation, critically revised the manuscript; S.M. Wallet, contributed to conception, design, data analysis, and interpretation, drafted and critically revised the manuscript; L.M. Shaddox, contributed to conception, design, data acquisition, analysis, and interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
This study is supported by the National Institutes of Health / National Institute of Dental and Craniofacial Research (R01DE 019456) and is registered in Clinicaltrials.gov (registration NCT01330719).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Albandar JM, Tinoco EM. 2002. Global epidemiology of periodontal diseases in children and young persons. Periodontol 2000. 29(1):153–176. [DOI] [PubMed] [Google Scholar]
- Aldenderfer MS, Blashfield RK. 1984. Cluster analysis. Newbury Park (CA): Sage Publications. [Google Scholar]
- Armitage GC. 1999. Development of a classification system for periodontal diseases and conditions. Ann Periodontol. 4(1):1–6. [DOI] [PubMed] [Google Scholar]
- Cobb CM. 1996. Non-surgical pocket therapy: mechanical. Ann Periodontol. 1(1):443–90. [DOI] [PubMed] [Google Scholar]
- Fine DH, Markowitz K, Fairlie K, Tischio-Bereski D, Ferrandiz J, Godboley D, Furgang D, Gunsolley J, Best A. 2014. Macrophage inflammatory protein-1alpha shows predictive value as a risk marker for subjects and sites vulnerable to bone loss in a longitudinal model of aggressive periodontitis. PLoS One. 9(6):e98541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlet GP. 2010. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J Dent Res. 89(12):1349–1363. [DOI] [PubMed] [Google Scholar]
- Garlet GP, Cardoso CR, Campanelli AP, Garlet TP, Avila-Campos MJ, Cunha FQ, Silva JS. 2008. The essential role of IFN-gamma in the control of lethal Aggregatibacter actinomycetemcomitans infection in mice. Microbes Infect. 10 (5):489–496. [DOI] [PubMed] [Google Scholar]
- Lang N, Bartold PM, Cullinan M, Jeffcoat M, Mombelli A, Murakami S, Page R, Papapanou P, Tonetti M, Van Dyke T. 1999. Consensus report: aggressive periodontitis. Ann Periodontol. 4(1):53. [Google Scholar]
- Mahanonda R, Pichyangkul S. 2007. Toll-like receptors and their role in periodontal health and disease. Periodontol 2000. 43:41–55. [DOI] [PubMed] [Google Scholar]
- Marazita ML, Burmeister JA, Gunsolley JC, Koertge TE, Lake K, Schenkein HA. 1994. Evidence for autosomal dominant inheritance and race-specific heterogeneity in early-onset periodontitis. J Periodontol. 65(6):623–630. [DOI] [PubMed] [Google Scholar]
- Nahid MA, Satoh M, Chan EK. 2011. MicroRNA in TLR signaling and endotoxin tolerance. Cell Mol Immunol. 8(5):388–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibali L, Pelekos G, D’Aiuto F, Chaudhary N, Habeeb R, Ready D, Parkar M, Donos N. 2013. Influence of IL-6 haplotypes on clinical and inflammatory response in aggressive periodontitis. Clin Oral Investig. 17(4):1235–1242. [DOI] [PubMed] [Google Scholar]
- Schenkein HA, Van Dyke TE. 1994. Early-onset periodontitis: systemic aspects of etiology and pathogenesis. Periodontology 2000. 6:7–25. [DOI] [PubMed] [Google Scholar]
- Shaddox LM, Gonçalves PF, Vovk A, Allin N, Huang H, Hou W, Aukhil I, Wallet SM. 2013. LPS-induced inflammatory response after therapy of aggressive periodontitis. J Dent Res. 92(8):702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaddox LM, Wiedey J, Bimstein E, Magnuson I, Clare-Salzler M, Aukhil I, Wallet SM. 2010. Hyper-responsive phenotype in localized aggressive periodontitis. J Dent Res. 89(2):143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaddox LM, Wiedey J, Calderon NL, Magnusson I, Bimstein E, Bidwell JA, Zapert EF, Aukhil I, Wallet SM. 2011. Local inflammatory markers and systemic endotoxin in aggressive periodontitis. J Dent Res. 90(9):1140–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabholz A, Mann J, Agmon S, Soskolne WA. 1998. The description of a unique population with a very high prevalence of localized juvenile periodontitis. J Clin Periodontol. 25(11, pt 1):872–878. [DOI] [PubMed] [Google Scholar]
- Susin C, Haas AN, Albandar JM. 2014. Epidemiology and demographics of aggressive periodontitis. Periodontology 2000. 65(1):27–45. [DOI] [PubMed] [Google Scholar]
- Teng YT, Mahamed D, Singh B. 2005. Gamma interferon positively modulates Actinobacillus actinomycetemcomitans-specific RANKL+ CD4+ Th-cell-mediated alveolar bone destruction in vivo. Infect Immun. 73(6):3453–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thunell DH, Tymkiw KD, Johnson GK, Joly S, Burnell KK, Cavanaugh JE, Brogden KA, Guthmiller JM. 2010. A multiplex immunoassay demonstrates reductions in gingival crevicular fluid cytokines following initial periodontal therapy. J Periodontal Res. 45(1):148–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombelli L, Scapoli C, Calura G, Tatakis DN. 2006. Time as a factor in the identification of subjects with different susceptibility to plaque-induced gingivitis. J Clin Periodontol. 33(5):324–328. [DOI] [PubMed] [Google Scholar]