

Abstract
Nonsense-mediated mRNA decay (NMD) is an RNA surveillance mechanism that requires upframeshift protein 1 (UPF1). This study demonstrates that human UPF1 exerts protective effects in a rat paralysis model based on the amyotrophic lateral sclerosis (ALS)-associated protein, TDP-43 (transactive response DNA-binding protein 43 kDa). An adeno-associated virus vector (AAV9) was used to express TDP-43 throughout the spinal cord of rats, inducing reproducible limb paralysis, to recapitulate the paralysis in ALS. We selected UPF1 for therapeutic testing based on a genetic screen in yeast. The expression of human TDP-43 or human UPF1 in the spinal cord was titrated to less than twofold over the respective endogenous level. AAV9 human mycUPF1 clearly improved overall motor scores in rats also expressing TDP-43. The gene therapy effect of mycUPF1 was specific and reproducible compared with groups receiving either empty vector or green fluorescent protein vector controls. The gene therapy maintained forelimb motor function in rats that would otherwise become quadriplegic. This work helps validate UPF1 as a novel therapeutic for ALS and other TDP-43-related diseases and may implicate UPF1 and NMD involvement in the underlying disease mechanisms.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a deadly neurodegenerative disease involving progressive paralysis. There are no highly efficacious strategies to treat ALS despite great effort by doctors and scientists. Successful treatments in mouse models, most of which are based on rare familial mutations in the ALS gene SOD1, have so far had little impact on modifying the disease in humans. Novel models based on transactive response DNA-binding protein 43 kDa (TDP-43) may offer a more predictive test system given that the vast majority of ALS cases harbor TDP-43 pathology in their neurons and glia.1–3 Abnormal TDP-43 aggregates are also prominent in the class of diseases known as frontotemporal lobar degeneration (FTLD-TDP).4 TDP-43 is an RNA-binding protein that is normally found predominantly in the nucleus. In FTLD-TDP and the majority of ALS, abnormal TDP-43 accumulation occurs in the cytoplasm in the form of hyperphosphorylated and ubiquitinated pathological protein aggregates, and thus serves as a postmortem diagnostic marker.1–4 One of the ways by which TDP-43 has been studied in animals is by gene delivery, which has proven to be sufficiently reproducible to allow the discrimination of genotype–phenotype differences among TDP-43 isoforms in our previous work.5–7 This reproducibility and the ability to experimentally control the onset and severity of the disease state offer advantages for modeling, given that TDP-43 overexpression is highly toxic to cells.8 Here we use TDP-43 gene transfer to induce motor paralysis in rats to study limb symptomatology that is germane to ALS as a platform for gene therapy. Overexpression of TDP-43 causes progressive paresis to paralysis of the limbs in a highly reproducible manner,6,7 offering an assay for therapeutic efficacy such as gene therapy. Gene therapy is worth considering for this disease given that ALS is fatal and irreversible. In this report, recombinant TDP-43 expression was titrated to a low level for a partial disease state retaining restorative capacity.
Refinement of TDP-43 animal models continues to be an important goal in the field.9 Reports of experimental treatments that slow or block TDP-43-mediated toxicity are beginning to emerge, either by genetic or pharmacological interventions in several TDP-43 models.10–14 Here we report behavioral outcomes from testing an empirically chosen therapeutic target, cDNA coding for human upframeshift protein 1 (UPF1), in a rat model of ALS-like paralysis based on TDP-43.
UPF1 is best known for its role in nonsense-mediated mRNA decay (NMD), a surveillance mechanism that degrades mRNAs containing a premature termination codon, which can be generated, for example, through alternative splicing. NMD prevents the production of truncated proteins that could harm the cell. NMD is also involved in the regulation of the expression of ~ 10% of normal physiologic transcripts in the cell, and is essential in mice.15–17 We pursued the possibility that UPF1 could ameliorate ALS-like symptoms based on the work carried out in yeast and neuronal cultures.18,19 In a genetic screen of several thousands of proteins, a yeast homolog of hUPF1, and then the human gene itself, was found to prevent FUS- and TDP-43-mediated toxicity in yeast,19 Ju et al., unpublished. Barmada et al.18 have advanced this approach, demonstrating that UPF1 protects primary neuronal cultures from TDP-43 cytotoxicity, possibly by upregulating NMD, as inhibitors of NMD blocked the protective effect.18 The fact that expressing UPF1 blocks the toxic actions of TDP-43 in yeast cells and cultured neurons is consistent with the hypothesis that TDP-43-induced toxicity involves inhibition of UPF1 function, because TDP-43 toxicity can be suppressed by adding back UPF1 to restore NMD.
The main purpose of this study was to evaluate the expression of human mycUPF1 (i.e. human UPF1 with an N-terminal myc epitope tag) as a protection against TDP-43-induced limb paralysis in rats. MycUPF1 was tested in parallel with several different types of control treatments, all confirming that mycUPF1 elicits a specific therapeutic effect. We also evaluated whether the expression of recombinant TDP-43 or mycUPF1 would affect either each others recombinant gene expression or the expression of endogenous rat TDP-43 or UPF1. The data demonstrate that augmenting the cellular abundance of UPF1 provides a useful means of abrogating the devastating paralysis induced by TDP-43 overexpression.
RESULTS
Quantifying exogenous TDP-43 and mycUPF1 in rat tissues Exogenous TDP-43 and green fluorescent protein (GFP) expression levels were purposefully set relatively low compared with the previous studies to test a rat model with a partial lesion and restorative capacity. This titration was advantageous to observe a therapeutic effect, but the low expression levels rendered detection of the transgene products inefficient. Nevertheless, previous work demonstrated that intravenous adeno–associated virus vector (AAV9) TDP-43 gene transfer specifically induces hindlimb paralysis even when the resulting level of exogenous TDP-43 is only faintly detectable.6 We chose the intravenous AAV9 method because it produces widespread central nervous system (CNS) expression, leading to marked expression in spinal motor neurons, dorsal root ganglia (DRG) neurons and cerebellar Purkinje neurons,6,20 with only a small fold overexpression of the encoded protein, for example, less than twofold overexpression relative to the corresponding endogenous protein as estimated in the spinal cord in Dayton et al.7
For studying the effect of mycUPF1 expression, we harvested DRG neurons because this tissue provides a relatively high percentage of transduced cells in the nervous system, allowing for detection of transgene product. By comparison, the spinal cord and cerebellar samples include a greater percentage of non-transduced cells. We used antibodies for total TDP-43 or total UPF1 that detect both the endogenous rat plus exogenous human TDP-43 or UPF1. In DRG, the increase in total TDP-43 expression in AAV9 TDP-43/Empty vs uninjected animals was 2.4-fold (t-test, P<0.02, N = 3 per group), whereas for total UPF1, we estimated the increase to be 1.6-fold in AAV9 mycUPF1 vs uninjected subjects (Figure 1). The fold increases were relatively lower in the spinal cord (Figure 1) and cerebellum (not shown), as expected: the estimated ratio in the spinal cord and cerebellum was 1.4- and 1.2-fold for AAV9 TDP-43/Empty vs uninjected subjects and 1.1- and 1.1-fold for AAV9 mycUPF1 vs uninjected subjects (N = 3 per group). Although fold overexpression levels were small, recombinant mycUPF1 could be specifically visualized using myc antibody, which detected recombinant mycUPF1 only in subjects receiving AAV9 mycUPF1 only or AAV9 TDP-43/mycUPF1 (Figure 2).
Figure 1.
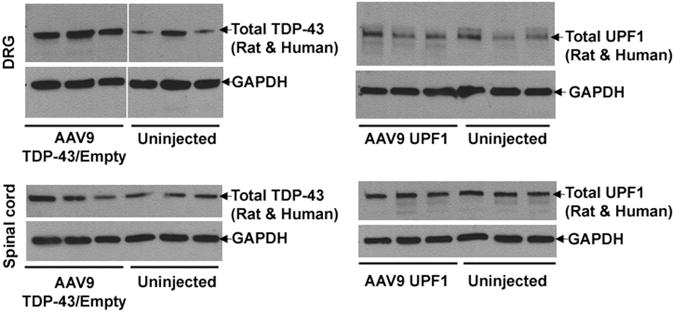
Overexpression of TDP-43 or UPF1 in the rat CNS. Protein from dissected DRG and lumbar spinal cord was analyzed by western blotting 12 weeks after intravenous injection of AAV9 expression vectors. Three animals are shown for each condition. The level of total TDP-43 (endogenous rat plus recombinant human TDP-43) was significantly increased in the DRG of the AAV9 TDP-43/Empty group compared with uninjected subjects (t-test, P<0.02, N = 3), but less so in the spinal cord or cerebellum (not shown). The expression level of human mycUPF1 compared with endogenous rat UPF1 was relatively small in all the three regions. The bands were normalized to GAPDH. See Results for details.
Figure 2.
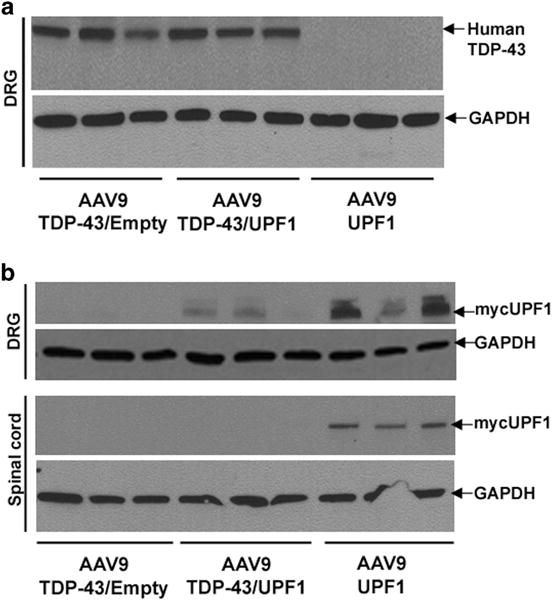
Selective detection of only recombinant human TDP-43 or mycUPF1. (a) A human-specific TDP-43 antibody detected exogenous human but not endogenous rat TDP-43 in DRGs. The level of TDP-43 expression was indistinguishable with or without mycUPF1 coexpression. (b) The level of exogenous mycUPF1 was detected with a myc antibody and only observed in rats that received AAV9 mycUPF1. In contrast to (a), MycUPF1 expression levels were reduced when AAV9 TDP-43 was coexpressed (t-test, P<0.05, N = 3 for DRG). The bands were normalized to GAPDH.
Non-reciprocal effects of TDP-43/mycUPF1 coexpression
We studied whether coexpression of TDP-43 and UPF1 would affect the expression of each protein. We initially hypothesized that UPF1 would lower TDP-43 expression based on the data in Polymenidou et al.21 However, recombinant TDP-43 protein levels were unaltered by mycUPF1 (Figure 2a; this anti-TDP-43 is specific to human TDP-43 and does not crossreact with rat TDP-43). Conversely, we found that mycUPF1 expression was consistently lowered by TDP-43 coexpression: there was less exogenous mycUPF1 in the DRG of the TDP-43/mycUPF1 coexpression group than in the mycUPF1-alone group despite an equal amount of AAV9 mycUPF1 vector being administered (t-test, P<0.05, N = 3; Figure 2b). The same effect was also observed in lumbar spinal cord, where mycUPF1 levels were below the limits of detection in the TDP-43/mycUPF1 rats but not in the mycUPF1-alone rats (Figure 2b). A similar TDP-43-mediated decrease in the level of UPF1 expression was observed in human embryonic kidney 293T cells that had been transiently transfected with human TDP-43 and mycUPF1 expression vectors when compared with cells that had been transiently transfected with the mycUPF1 expression vector and an empty expression vector so that the total amount of transfecting DNA was constant (Supplementary Figure S1; t-test, P<0.05, N = 3 per group). TDP-43 expression did not alter the expression of a vector-expressing GFP, suggesting that TDP-43 specifically decreases the level of recombinant UPF1 expression (Supplementary Figure S1). We tested whether single TDP-43 transfection would alter levels of endogenous 293T cell UPF1 in controls over the course of 2–3 days transient transfections, but TDP-43 transfection did not alter the endogenous UPF1 levels under these conditions (not shown).
PCR primers were used to specifically assay mRNAs for human TDP-43, human mycUPF1 or endogenous rat TDP-43 and endogenous UPF1. Reverse transcription coupled to real-time PCR, with normalization to the housekeeping enzyme RNA for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), revealed that the level of mycUPF1 mRNA was lowered in transduced rats upon human TDP-43 coexpression (P<0.05, N = 3 per group, t-test), whereas human TDP-43 mRNA levels were unaffected by mycUPF1 coexpression. Thus, we observed a specific lowering of mycUPF1 mRNA and protein when TDP-43 was coexpressed in cells or in rats (Supplementary Figure S2). Although we were not able to demonstrate any effects on the level of endogenous rat UPF1 mRNA (Supplementary Figure S2), our ability to assay mycUPF1 mRNA and protein using specific PCR primers and myc antibody, respectively, allowed us to selectively assay only the transduced cells without interference from non-transduced tissue.
TDP-43 or mycUPF1 transgene expression pattern
In addition to expression in the CNS, delivery of AAV9 intravenously also transduces the heart, liver and, less so, muscle in the rat.6,7 A similar pattern was found in this study at intentionally lower expression levels. In the spinal cord, human TDP-43 was observed in the nucleus of motor neurons in animals transduced with AAV9 TDP-43 (Figure 3a). The myc antibody allowed for specific visualization of the human mycUPF1 (Figure 3b). MycUPF1 immunoreactivity was not found in cell bodies in the ventral horn (not shown) but was unequivocally present in axonal processes in the dorsal horns (gray matter) and dorsal columns (white matter) of the spinal cord. An animal coinjected with AAV9 TDP-43 and AAV9 GFP demonstrated human TDP-43 and GFP expression in the same area of the ventral cerebellum on adjacent sections (Figures 3c and d). A merged image of TDP-43 and GFP immunoreactivity in Figure 3e shows examples of neurons coexpressing both transgenes. Non-transduced control tissues lacked recombinant TDP-43, mycUPF1 or GFP immunoreactivity. A low magnification image (Figure 3f) showed that the cerebellar GFP expression in this study was relatively weak compared with gene transfer using higher vector doses, as in the previous studies in which the cerebellum was thoroughly transduced (Supplementary Figure S3).6,7 In the TDP-43/GFP animal, cardiomyocytes were positive for GFP as expected (Figure 3g). In contrast, GFP expression in the skeletal muscle was not detected by either fluorescence or real-time PCR.
Figure 3.
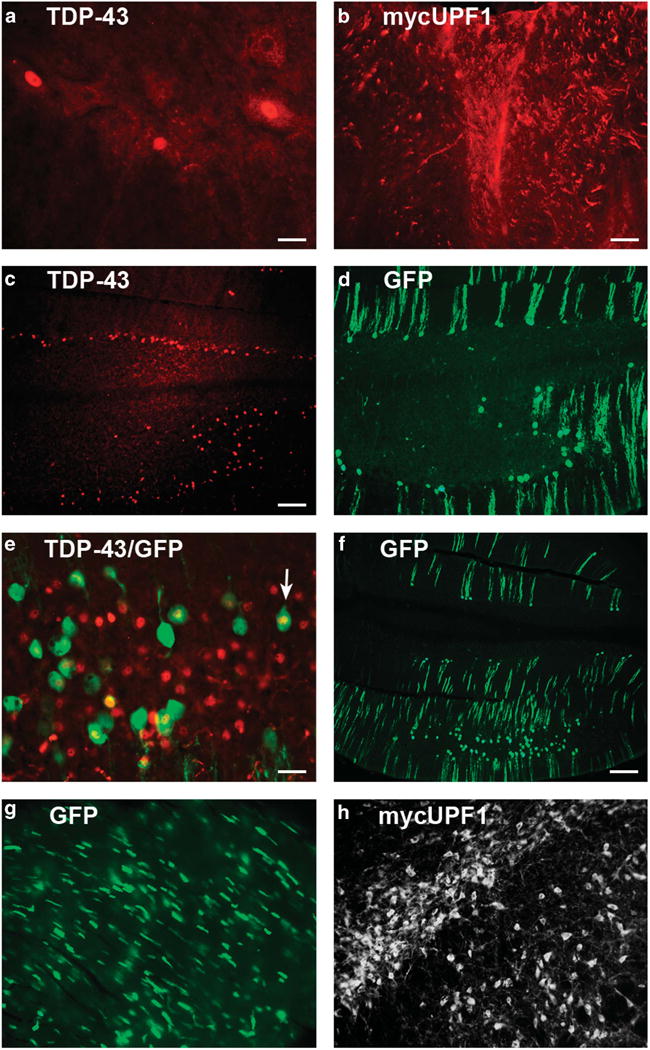
Recombinant TDP-43, mycUPF1 or GFP transduction pattern after intravenous AAV9 vector administration. (a) Human-specific TDP-43 immunoreactivity in the ventral horn of the lumbar spinal cord in a rat that received the AAV9 TDP-43 vector. Three large motor neurons are expressing the human TDP-43 in the nucleus. (b) MycUPF1 immunoreactivity in the lumbar spinal cord. Myc-positive fibers were unequivocal in the dorsal spinal cord in rats receiving the AAV9 mycUPF1. Negative control samples not receiving the respective vectors in (a and b) were blank for immunoreactivity. (c) Ventral cerebellum from an animal receiving AAV9 vectors for both TDP-43 and GFP. Recombinant TDP-43 is expressed in the nuclei of Purkinje layer cells. (d) GFP immunoreactivity from an adjacent section shows transduced cells in the same area as (a). (e) Merger of TDP-43 and GFP immunoreactivity. Most of the GFP-positive cells contain a human TDP-43-positive nucleus. However, there are TDP-43-positive cells that are negative for GFP, which may be because of technical reasons. (f) GFP in the ventral cerebellum, low magnification. (g) GFP expression in heart cells. (h) Specific labeling of mycUPF1 from the substantia nigra of an animal injected with AAV9 mycUPF1 directly into this region. (a and c–g) Interval 8 weeks; (b) interval 12 weeks; and (f) interval 2 weeks. Bar in (a) = 34 μm; bar in (b) = 67 μm, same magnification in (h); bar in (c) = 134 μm, same magnification in (d); bar in (e) = 42 μm; and bar in (f) = 268 μm, same magnification in (g).
To more efficiently demonstrate specific expression of mycUPF1 in the rat, the sample in Figure 3h was injected with AAV9 UPF1 stereotaxically into the substantia nigra, which achieves a higher level of recombinant vector per cell than does intravenous delivery. In this case, specific expression of mycUPF1 in neuronal cell bodies is unequivocal, and uninjected tissues lacked c-myc immunoreactivity under the staining conditions that were used. There was no evidence for either loss of spinal motor neurons (Supplementary Figure S4) or severe muscle atrophy (Supplementary Figure S5), as expected given the relatively low level of TDP-43 expression after intravenous gene delivery.
Positive effects on motor function conferred by mycUPF1
In this TDP-43 lesion model, we noticed marked positive effects on motor function when human mycUPF1 was expressed, relative to a control empty vector that did not express any recombinant transgene (Figures 4a–e). The effect of mycUPF1 was sufficiently pronounced and penetrant that the groups could be faithfully and readily typed by observing the subjects ambulate in their home cages. Not only did mycUPF1 prevent some of the behavioral deficits induced by TDP-43 but it also restored weight gain in male subjects (Figure 5). In terms of potential side effects induced by either AAV9 empty vector (N = 4) or AAV9 mycUPF1 (N = 4), there were no group differences compared with uninjected controls (N = 6) in assays of weight gain or specific behaviors, such as the rotarod assay (Supplementary Figure S6), which provides a sensitive measure for motor function deficits. In an additional set of control experiments, we compared the expression of mycUPF1 to the expression of GFP so as to ensure that coexpression of an unrelated protein would not mimic the effect. In comparing the TDP-43/GFP group (N = 6) vs the TDP-43/mycUPF1 group (N = 4) over 8 weeks, it was obvious that the protective effects of AA9V mycUPF1 were specifically reproducible and not recapitulated by AAV9 GFP (Figures 5f–i). Positive effects of mycUPF1 are highlighted in Figures 4 and 5. Statistical comparisons are described as follows.
Figure 4.
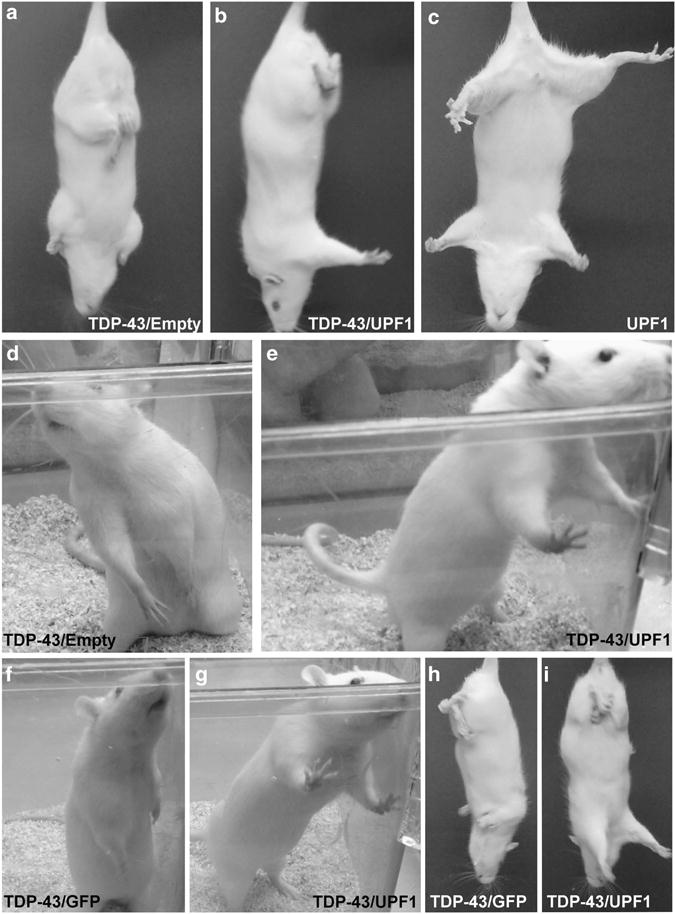
Human UPF1 protects rats from forelimb impairments induced by human TDP-43 expression. Limb impairments were assessed by assays of the escape reflex and by observing rearing behavior in home cages. (a–c) Rats injected with AAV9 TDP-43 show clasping of their hindlimbs, whereas rats injected with AAV9 mycUPF1 alone show a normal escape reflex. When AAV9 TDP-43 was coinjected with AAV9 empty vector, the rats show clasping of both hindlimbs and forelimbs (a). However, when AAV9 TDP-43 was coinjected with AAV9 mycUPF1, normal forelimb extension was maintained in the escape reflex (b). (d) Rats injected with AAV9 TDP-43 and AAV9 empty vector showed abnormal rearing with both forelimbs lowered. (e) The TDP-43/mycUPF1 group manifested normal rearing posture with both forelimbs extended. (f–i) Separate groups were analyzed in parallel with an AAV9 GFP control. When TDP-43 was coexpressed with GFP, the rats showed abnormal rearing posture with both forelimbs lowered (f) and clasping of both hind- and forelimbs in the escape reflex (h). TDP-43/mycUPF1 rats displayed normal forelimb function (g and i). (a–e) Interval 12 weeks; (f–i) interval 8 weeks.
Figure 5.
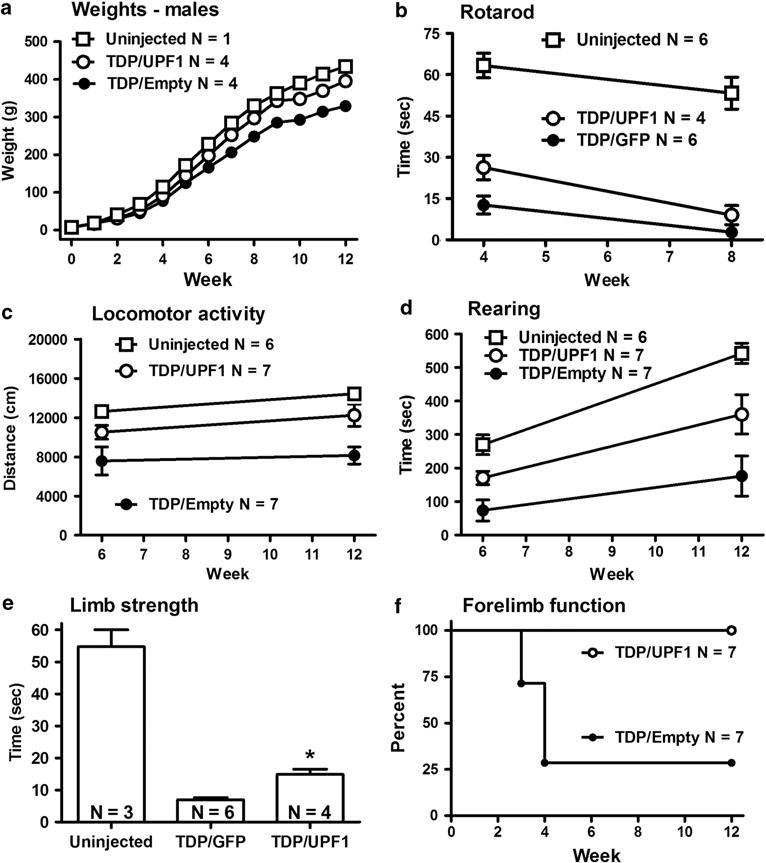
Positive outcomes exerted by AAV9 mycUPF1 compared with control vectors. (a) Weight gain curves. (b) Motor performance was assessed using rotarod assays. (c) Locomotor activity: distance traveled in 30-min test sessions. (d) Time spent rearing in 30-min test sessions. (e) Grip strength as measured by a hang test. (f) Percent of subjects with normally functional forelimbs in the escape reflex test. N values are indicated for each panel. Vector group differences between mycUPF1-treated and control groups were found in (a–f) by repeated-measures analysis of variance (ANOVA), t-test or log-rank survival analysis (P<0.05 to 0.001). See Results for details.
Although we previously observed weight loss and mortality in rats expressing a higher dose of AAV9 TDP-43,6 there was no mortality in the current studies. Human mycUPF1 did partially block the morbidity of weight loss in this study (Figure 5a). Comparisons of the two male TDP-43 vector groups with or without mycUPF1 coexpression revealed a specific effect of vector treatment on weight gain (i.e. TDP-43/mycUPF1 vs TDP-43/Empty; F1,72 = 37.53; P<0.001) as well as an effect of weight gain over time as expected (F12,72 = 2942; P<0.0001), and a significant interaction between vector treatment group and time (F12,72 = 26.01; P<0.0001), demonstrating a specific effect of the vector treatment over time. In Bonferroni posttests, there were weight differences between the two vector groups at all time points from 6 to 12 weeks (P<0.01). However, in TDP-43-expressing female groups, we did not observe differences in weight gain compared with uninjected female groups (N = 5) nor a difference in weight gain between the two female TDP-43 vector groups with or without mycUPF1 (N = 3 per group). We believe that the TDP-43-related morbidity of slightly lower weights was more readily discernible in males because males weigh more, and not because of gender differences in gene transfer efficiency, which was not observed on western blots or by immunhistochemistry throughout the study.
Rotarod performance is an index of overall motor function and coordination (Figure 5b). There was consistent motor impairment in all groups that received AAV9 TDP-43. However, in repeated-measures analysis of variance of rats administered AAV9 TDP-43/Empty compared with rats administered AAV9 TDP-43/mycUPF1 from 4 to 12 weeks, there was a significant effect of both vector group (F1,24 = 6.60, P<0.05) and time (F2,24 = 7.57, P<0.005), as well as a significant interaction (F2,24 = 5.76, P<0.01). Bonferroni posttests showed a difference between the groups at 4 weeks (P<0.001) but not 8 or 12 weeks. With the separate TDP-43/GFP and TDP-43/mycUPF1 subjects (Figure 5b), there was a significant effect of vector (F1,8 = 7.68, P<0.05) and time (F1,8 = 34.29, P<0.0005) but no interaction. Again the Bonferroni posttests showed a significant difference at 4 (P<0.05) but not 8 weeks.
Locomotor activity was measured at 6 and 12 weeks for the AAV9 TDP-43/Empty and AAV9 TDP-43/mycUPF1 groups (Figure 5c). There was a significant effect of vector group (F1,12 = 8.64; P<0.05) but no effect of time nor interaction. In the posttests, the TDP-43/Empty group had lower locomotor scores at 12 weeks (P<0.05). Rearing is a typical behavior that depends on hindlimb function (Figure 5d). As an index of rearing behavior, we analyzed the time spent rearing during 30 min sessions. There was a significant effect of vector group (F1,12 = 6.27, P<0.05) and time interval (F1,12 = 19.95, P<0.001), but no interaction. In the posttests, the TDP-43/Empty group had reduced rearing at 12 weeks (P<0.05). The separate TDP-43/GFP and TDP-43/mycUPF1 groups were analyzed for locomotor and rearing behavior at 6 weeks. No significant differences were found by t-tests, despite trends of increased behaviors in the rats receiving mycUPF1. However, this was not surprising given there were no differences in the posttests for the TDP-43/Empty and TDP-43/mycUPF1 comparisons at 6 weeks.
For an index of grip strength, a hang test was analyzed for the TDP-43/GFP and TDP-43/mycUPF1 groups at 4 weeks (Figure 5e). The animals receiving mycUPF1 with TDP-43 performed better than animals receiving TDP-43 without the mycUPF1 (t-test, P<0.01).
Overall forelimb use was observed during normal rearing in the home cages (Figure 4). In the escape reflex test, we recorded abnormal clasping of the hindlimbs and forelimbs (Figures 4 and 5f). All animals that received AAV9 TDP-43 developed hindlimb defects that were not present in any of the other types of controls not receiving TDP-43. Forelimb deficits and clasping also developed in some of the animals: in five of the seven TDP-43/Empty animals and in four of the six TDP-43/GFP animals. Interestingly, none of the 11 TDP-43/mycUFP1 animals developed forelimb deficits. Analysis of the survival curves for forelimb function yielded a significant difference between the TDP-43/Empty and TDP-43/mycUPF1 groups (P<0.01) and between the separate TDP-43/GFP and TDP-43/mycUPF1 groups (P = 0.05).
In summary, the benefits of mycUPF1 were evidenced by the vector group differences in weight gain, overall motor function and coordination, grip strength and survival of forelimb function.
DISCUSSION
All of the rats that were administered the AAV9 human TDP-43 manifested characteristic motor impairments as expected. Impairments were evidenced by low rotarod scores and progressive hindlimb paralysis in all of the TDP-43 rats by 4 weeks. Forelimb dysfunction occurred in most of the animals receiving AAV9 TDP-43 combined with control vector. In contrast to rats expressing human TDP-43, control groups expressing empty vector, GFP or human mycUPF1 did not develop any observable impairments.
To avoid severe atrophy and mortality, a lower AAV9 TDP-43 dose than previously assayed in rats was used so as to establish a moderate disease state.6 In some of our previous work, we have used the woodchuck hepatitis virus posttranscriptional regulatory enhancer element (WPRE) to augment transgene expression by 5–10-fold.7,22,23 It is worth noting that here we purposefully did not incorporate the woodchuck hepatitis virus posttranscriptional regulatory enhancer element as only small increases in TDP-43 expression have been proposed to induce disease based on analyses of human samples.24 In fact, our data suggest that increases in TDP-43 expression of less than twofold in the spinal cord are sufficient to induce paralysis, providing information that may be relevant to both disease and its treatment. Our data also indicate that UPF1 gene therapy could be feasible even with low gene dosages.
To evaluate the potential of hUPF1 as a gene therapy for TDP-43-associated diseases, first we compared AAV9 TDP-43 rats administered either AAV9 mycUPF1 or, as a control, AAV9 empty vector or AAV9 GFP, and we observed protective effects mediated only by mycUPF1 expression. These findings indicated that protection was not due to either a nonspecific effect or gene competition by coexpressing a foreign transgene unrelated to the specific actions of mycUPF1.
Rats coexpressing mycUPF1 manifested clear improvements in their functional outcome. They performed better on rotarod at the early 4-week interval, displayed greater locomotor activity and rearing and displayed better grip strength relative to rats receiving either type of control treatment. The improvements were readily apparent by observing the mycUPF1 rats rearing in their home cages and in the escape reflex: all of the mycUPF1 rats retained relevant function of their forelimbs as evidenced during both normal rearing and escape reflex. If an analogous treatment could be achieved in human ALS patients, then forelimb function could be preserved and prolonged so to improve the quality of life. However, mycUPF1 did not appear to protect the hindlimbs. In both our prior work and in other rodent models of ALS, the hindlimbs are affected earlier and more severely than the forelimbs.6,7,25 Thus, the differential effect of mycUPF1 on the forelimbs relative to the hindlimbs may be related to the forelimbs retaining more restorative capacity than the hindlimbs. Further refinement of this therapeutic strategy will be necessary to extend protection to the hindlimbs. As the protection of motor function was only partial, strategies that could potentially augment the effect are either expressing UPF1 before TDP-43 or achieving greater coexpression of the two transgenes in the same neurons than we observed here.
There are several potential TDP-43 splice variants in mice, rats and humans, and at least one of them is a putative substrate for NMD.26–28 In a cell culture experiment that blocked UPF1 expression, TDP-43 isoform 3 expression was increased, suggesting that UPF1 may regulate TDP-43 levels, as it does for ~ 10% of the normal transcriptome.21 We thus thought exogenous UPF1 might exert its protection by lowering TDP-43 levels. However, we found no indication of changes in either rat or human TDP-43 mRNA or protein level with mycUPF1 coexpression. Another potential protective mechanism could involve UPF1-mediated inhibition of TDP-43 toxicity via a direct or indirect interaction of the two proteins. However, studies in yeast and neurons have thus far not found evidence for colocalization of cytosolic TDP-43 and UPF1 proteins (Ju et al., unpublished).18
We found decreases in the expression of recombinant mycUPF1 RNA and protein when TDP-43 was coexpressed, which was unexpected yet consistent in both the rat and in transfected cells. A transcriptional regulation of the recombinant promoter and recombinant mycUPF1 cDNA in the vector by TDP-43 is not likely as TDP-43 coexpression did not exert a similar reduction on the control GFP expression in cells as it did on mycUPF1. The data are more consistent with posttranscriptional effects of TDP-43 on mycUPF1 mRNA stability or on mycUPF1 protein degradation.29 We could not detect an effect of TDP-43 on endogenous rat UPF1 levels, perhaps because of the high background levels of rat UPF1 in non-transduced cells not expressing exogenous TDP-43. For example, although we were able to detect the recombinant mycUPF1 protein, the estimated upregulations were not significant; there was a high fraction of non-transduced tissue in our samples. The changes in recombinant UPF1 expression support further study of potential dysregulation of endogenous UPF1 in this animal model or in ALS. We hypothesize that TDP-43 inhibits UPF1 function and that expressing exogenous UPF1 compensates for this inhibition by at least partially restoring UPF1 function.
The clinically relevant protection of forelimb function by AAV9-produced human UPF1 in our rat model of ALS provides reasons for further research and development. We know that TDP-43 expression in the spinal cord is essential to observe TDP-43-induced paralysis.7 However, we cannot completely rule out contributing comorbidities from highly transduced tissues such as the liver and the heart. In terms of researching underlying causes in ALS, it would be beneficial to tightly restrict TDP-43 expression to neurons vs glia or neurons vs muscle in future investigations. Further therapeutic development will test if UPF1 is protective in a more severe late-stage model, how long after the disease state is initiated can UPF1 counteract TDP-43, that is, a clinical sequence, and if protection of forelimb function can be extended to protection of hindlimb function by higher levels of UPF1 expression or alternative routes of administration. Although connections between UPF1 function in NMD and UPF1-mediated protection from TDP-43-induced toxicity have yet to be understood, it is worth noting that overexpression of UPF1 in yeast overcomes the normal requirement of UPF2 and UPF3 NMD factors.30 Thus, UPF1 gene delivery could be an important tool to manipulate NMD in vivo.
CONCLUSIONS
This work expands studies demonstrating suppression of human TDP-43 cytotoxicity by coexpressing human UPF1 and TDP-43 in yeast and neuronal cultures18 and helps to further establish and develop the human UPF1 gene as a therapeutic target by validating efficacy against clinically relevant limb paralysis induced by TDP-43 in rodents. We show that an intravenous AAV9 gene transfer approach in rats is sufficiently reproducible to provide a preclinical test for possible ALS gene therapies using highly relevant assays of motor function and coordination. Further studies will be needed to establish optimal dosage and delivery methods, and to determine if AAV9 human UPF1 gene therapy is safe and effective in other models of ALS.
MATERIALS AND METHODS
DNA and AAVs
The transgene expression cassette included AAV2 terminal repeats, the hybrid cytomegalovirus/chicken β-actin promoter and the bovine growth hormone polyadenylation sequence.5 This study used separate constructs with DNA encoding either GFP, human wild-type TDP-435 or human UPF1 with an N-terminal myc epitope tag (mycUPF1).31 The addition of the myc epitope tag does not diminish UPF1’s function in a cellular assay of NMD.32 Additionally, a vector-only construct, that is, empty vector, was used to control for viral particles. DNAs were packaged into recombinant AAV9 as described previously.5 The AAV9 serotype was first described by Gao et al.33 and has enhanced gene transfer to the CNS.6,9,20,34–37 Helper and AAV9 capsid plasmids used to generate AAV9 were from the University of Pennsylvania (Philadelphia, PA, USA).33 Viral stocks were sterilized using Millipore (Billerica, MA, USA) Millex-GV syringe filter, aliquoted and frozen. Viral genome copies were titered using dot-blot assay, and equal titer doses were obtained by diluting stocks in lactated Ringer’s solution (Baxter Healthcare, Deerfield, IL, USA). Through the course of this study, two batches of AAV9 TDP-43 and three batches of AAV9 mycUPF1 were used, and consistent results were obtained across the batches.
Animals and treatments
Five litters of Sprague–Dawley rats (Harlan, Indianapolis, IN, USA) totaling 39 animals of both genders were injected with AAV9 on postnatal day 1 and studied for up to 12 weeks. Animals received a fixed amount of AAV9 TDP-43 (1 × 1012 vector genomes, vg). To test for a protection from TDP-43-induced paralysis by UPF1, AAV9 mycUPF1 (7.9 × 1011 vg) or an equivalent dose of a transgene-less empty AAV9 vector was administered with AAV9 TDP-43 to generate the TDP-43/mycUPF1 group (N = 7) or the TDP-43/Empty group (N = 7). These groups were evaluated for a total of 12 weeks, with testing for motor function at several intervals. Additional control groups included AAV9 mycUPF1 only (dose = 7.9 × 1011 vg; N = 4), AAV9 empty vector-only (dose = 7.9 × 1011 vg; N = 4) and uninjected subjects (N = 6). In a second comparison to test for a protective effect of the UPF1 relative to when an unrelated recombinant control protein is expressed, we compared AAV9 mycUPF1 against an equivalent dose of an AAV9 GFP of 7.9 × 1011 vg. For this set, there was a TDP-43/mycUPF1 group (N = 4) and a TDP-43/GFP group (N = 6) tested for motor function over 8 weeks.
For the intravenous injections, 100 μl of each AAV9 in lactated Ringer’s solution was loaded into a 1 ml syringe attached to a 30 G needle. The AAV9 solution was injected into the temporal vein as described previously, which leads to highly consistent gene transfer results.6,7 The paws of the animals were inked (Spaulding Color, Voorheesville, NY, USA) for identification. After injection, rats were returned to the mother’s cage, and weaning occurred at 3 weeks of age. In addition, one adult female rat underwent a stereotaxic injection into the substantia nigra by described methods.22 AAV9 mycUPF1 was injected at a dose of 1 × 1010 vg and the tissue was analyzed 2 weeks later. All animal procedures followed protocols approved by the Institutional Animal Care and Use Committee and the NIH Guide for Care and Use of Laboratory Animals. The methods for intravenous delivery of AAV9 derived from earlier work in mice.20,35.
Motor function tests
Animals were assessed for motor-related abilities that included hindlimb escape reflex, rotarod (time spent on a spinning wheel before falling), open field (distance traveled and time spent rearing) and a grip-strength hang test. To observe the hindlimb escape reflex, rats were gently lifted by their tail for up to 10 s. A normal escape reflex, which consists of the outward extension of the limbs, was tested weekly from 1 to 12 weeks. Three trials were conducted per week on the day of observation, and an abnormal reflex was noted only if present during all three trials. Rotarod (Rotarod/RS; Letica Scientific Instruments, Barcelona, Spain) testing was conducted during weeks 4, 8 and 12 using an accelerating (4–40 r.p.m) rotarod over a 2-min time period. The time spent on the wheel before falling was averaged over three trials. Open field testing was conducted using a photobeam activity monitoring system (Truscan 2.0; Coulbourn Instruments, Whitehall, PA, USA) to observe locomotor and rearing behaviors at 6 and 12 weeks. The trials were conducted for 30 min in a dark room. An index of the animal’s grip strength was measured by the hang test. Animals were placed on a wire mesh cage cover above the home cage. The cage cover was raised to a perpendicular angle, and resting on the cage and the time it took for the animal to drop down a few inches into the bedding of the home cage was measured. Three trials, each capped at 60 s, were undertaken over a 30 min period, and results were averaged.
Real-time RT-PCR
Dissected tissues were frozen on dry ice and stored in 1 ml of RNAlater (Ambion, Austin, TX, USA) overnight at 4 °C. The tissues were homogenized in 1 ml RNA STAT-60 (Tel-Test Inc., Friendswood, TX, USA), and RNA was extracted using chloroform/isopropanol, washed with ethanol and dissolved in RNase-free water (Ambion). RNA was then purified with the RNeasy MinElute Cleanup Kit (Qiagen, Valencia, CA, USA) and stored at − 80 °C. RNA integrity was assessed by electrophoresis using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). The RNA was converted to cDNA using the iScript cDNA Synthesis Kit (Invitrogen, Carlsbad, CA, USA) and stored at − 20 °C. PCR was performed using the Bio-Rad C1000 Thermocycler with the Bio-Rad CFX96 Real-Time System (Bio-Rad, Hercules, CA, USA). Primers used were human TDP-43 sense (5′-TAATAACCAAAACCAAGG-3′) and antisense (5′-AGAAGACTTAGAATCCAT-3′), targeting the coding region of human TDP-43 and the polyadenylation signal sequence of the recombinant vector backbone; rat TDP-43 sense (5′-CACTTGTCTTCCTTCATAT-3′) and antisense (5′-AGTATTCCTATGGCAGAA-3′), targeting sequences found in rat but not human TDP-43, rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) sense (5′-CATTCTTCCACCTTTGAT-3′) and antisense (5′-CTGTAGCCATATTCATTGT-3′); exogenous mycUPF1 sense (5′-TGGAGCAGAAGCTGATCTCA-3′) and antisense (5′-GTCTGCGAGCTGGGCCCGTA-3′), targeting the sequence encoding the N-terminal c-myc epitope tag; and rat UPF1 sense (5′-CAGACTACCCTTCCCAACAG-3′) and antisense (5′-AGGGCTGGCTCATGGACACA-3′), targeting sequences found in the rat but not the human UPF1 coding regions (Eurofins MWG Operon, Huntsville, AL, USA).
Western blotting
Tissues were dissected from the animals and frozen on dry ice. The samples were Dounce-homogenized in RIPA buffer (1% nonidet-P40/0.5% sodium deoxycholate/0.1% sodium dodecyl sulfate/phosphate-buffered saline) with protease inhibitors (Halt Protease Inhibitor Cocktail Kit; from Pierce, Rockford, IL, USA) and then centrifuged. Protein content was determined by Bio-Rad Protein Assay Dye. Samples were normalized for protein content and electrophoresed in 12% polyacrylamide containing sodium dodecyl sulfate (Bio-Rad). Primary antibodies used to probe blots were anti-TDP-43 (Abnova, Taipei City, Taiwan), anti-c-myc (9E10; Santa Cruz Biotechnology, Dallas, TX, USA), anti-UPF138,39 and anti-GAPDH (Ambion). Secondary antibodies and ECL reagents were from Amersham (Buckinghamshire, UK). Primary antibody dilutions were 1:1500, and secondary antibody dilutions were 1:10 000.
Immunohistochemistry
Animals were anesthetized using a cocktail of xylazine (20 mg ml−1; Butler, Columbus, OH, USA), ketamine (100 mg ml−1; Fort Dodge Animal Health, Fort Dodge, IA, USA) and acepromazine (10 mg ml−1; Boerhinger Ingelheim, St Joseph, MO, USA) in a 3:3:1 fluid ratio. Animals were administered the cocktail intramuscularly at a dose of 1 ml kg−1 and perfused with phosphate-buffered saline followed by cold 4% paraformaldehyde in phosphate-buffered saline. Tissues were removed and immersed in 4% paraformaldehyde overnight at 4 °C. Fifty-micrometer sections were cut on a sliding microtome with a freezing stage. Primary antibodies used were anti-TDP-43 (Abnova), anti-c-myc (Santa Cruz Biotechnology) and anti-GFP (Invitrogen) at dilutions of 1:500 and 1:1000. Secondary antibodies include biotinylated antibodies from DAKO Cytomation (Carpinteria, CA, USA; 1:2000) and Alexa Fluor 488-(Invitrogen) or Cy3-conjugated antibodies (Jackson ImmunoResearch, West Grove, PA, USA) at 1:300. 4′,6-Diamidino-2-phenylindole (Sigma, St Louis, MO, USA) counterstaining and Nissl staining followed standard methods.
Cell transfections
Human embryonic kidney 293T cells (6 cm dish) were transfected with several plasmid DNA combinations (TDP-43, mycUPF1, GFP and Empty) using a calcium-phosphate method and harvested for western blotting 2 days later. Equal amounts of DNA totaling 4 μg were transfected in each dish, for example, 2 μg of TDP-43 DNA and 2 μg of mycUPF1 DNA. Transfections were performed in triplicate.
Statistics
Results are presented as the mean ± s.e.m. Statistical tests included repeated-measures analysis of variance with Bonferroni posttests, log-rank test and Student’s t-test as indicated.
Supplementary Material
Acknowledgments
We thank Sami Barmada and Stacie Weninger for advice and insightful discussion, Ellie Hall and J Steven Alexander for technical advice and Mychal Grames, Adam Richard, Sarah Lopez, Christopher Jackson and Isaac Hardman for technical assistance. This study was supported by the Fidelity Biosciences Research Initiative (RLK), Karyopharm Therapeutics Inc. (RLK) and NIH R01 GM059614 (LEM).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on Gene Therapy website (http://www.nature.com/gt)
References
- 1.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 2.Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 3.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 4.Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6:211–220. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tatom JB, Wang DB, Dayton RD, Skalli O, Hutton ML, Dickson DW, et al. Mimicking aspects of frontotemporal lobar degeneration and Lou Gehrig’s disease in rats via TDP-43 overexpression. Mol Ther. 2009;17:607–613. doi: 10.1038/mt.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang DB, Dayton RD, Henning PP, Cain CD, Zhao LR, Schrott LM, et al. Expansive gene transfer to the rat CNS and amyotrophic lateral sclerosis relevant sequelae when TDP-43 is overexpressed. Mol Ther. 2010;18:2064–2074. doi: 10.1038/mt.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dayton RD, Gitcho MA, Orchard EA, Wilson JD, Wang DB, Cain CD, et al. Selective forelimb impairment in rats expressing a pathological TDP-43 25 kDa C-terminal fragment to mimic amyotrophic lateral sclerosis. Mol Ther. 2013;21:1324–1334. doi: 10.1038/mt.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 2010;30:639–649. doi: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang DB, Gitcho MA, Kraemer BC, Klein RL. Genetic strategies to study TDP-43 in rodents and to develop preclinical therapeutics for amyotrophic lateral sclerosis. Eur J Neurosci. 2011;34:1179–1188. doi: 10.1111/j.1460-9568.2011.07803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armakola M, Higgins MJ, Figley MD, Barmada SJ, Scarborough EA, Diaz Z, et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet. 2012;44:1302–1309. doi: 10.1038/ng.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hebron M, Chen W, Miessau MJ, Lonskava I, Moussa CE. Parkin reverses TDP-43-induced cell death and failure of amino acid homeostasis. J Neurochem. 2013;129:305–361. doi: 10.1111/jnc.12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HJ, Raphael AR, Ladow ES, McGurk L, Weber RA, Trojanowski JQ, et al. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2013;46:152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaccaro A, Patten SA, Ciura S, Maios C, Therrien M, Drapeau P, et al. Methylene blue protects against TDP-43 and FUS neuronal toxicity in C. elegans and D. rerio. PLoS One. 2012;7:e42117. doi: 10.1371/journal.pone.0042117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, et al. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci USA. 2012;109:15024–15029. doi: 10.1073/pnas.1206362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karam R, Wengrod J, Gardner LB, Wilkinson MF. Regulation of nonsense-mediated mRNA decay: implications for physiology and disease. Biochim Biophys Acta. 2013;1829:624–633. doi: 10.1016/j.bbagrm.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Popp MW, Maquat LE. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu Rev Genet. 2013;47:139–165. doi: 10.1146/annurev-genet-111212-133424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen LS, Wilkinson MF, Gecz J. Nonsense-mediated mRNA decay: Inter-individual variability and human disease. Neurosci Biobehav Rev. 2013;S0149-7634:270–274. doi: 10.1016/j.neubiorev.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barmada S, Ju S, Arjun A, Batarse A, Qu H, Huang EE, et al. RNA helicases ameliorate ALS- and FTD related neurotoxicity. Ann Neurol. 2013;74:S93. [Google Scholar]
- 19.Ju S, Tardiff DF, Han H, Divya K, Zhong Q, Maquat LE, et al. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 2011;9:e1001052. doi: 10.1371/journal.pbio.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klein RL, Hamby ME, Hirko AC, Gong Y, Wang S, Hughes JA, et al. Dose and promoter effects of adeno-associated viral vector for green fluorescent protein expression in the rat brain. Exp Neurol. 2002;176:66–74. doi: 10.1006/exnr.2002.7942. [DOI] [PubMed] [Google Scholar]
- 23.Loeb JE, Cordier WS, Harris ME, Weitzman MD, Hope TJ. Enhanced expression of transgenes from adeno-associated virus vectors with the woodchuck hepatitis virus posttranscriptional regulatory element: implications for gene therapy. Hum Gene Ther. 1995;10:2295–2305. doi: 10.1089/10430349950016942. [DOI] [PubMed] [Google Scholar]
- 24.Gitcho MA, Bigio EH, Mishra M, Johnson N, Weintraub S, Mesulam M, et al. TARDBP 3′-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol. 2009;118:633–645. doi: 10.1007/s00401-009-0571-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA. 2010;107:3858–3863. doi: 10.1073/pnas.0912417107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang HY, Wang IF, Bose J, Shen CK. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004;83:130–139. doi: 10.1016/s0888-7543(03)00214-3. [DOI] [PubMed] [Google Scholar]
- 27.Avendaño-Vázquez SE, Dhir A, Bembich S, Buratti E, Proudfood N, Baralle FE. Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 2012;26:1679–1684. doi: 10.1101/gad.194829.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ayala YM, De Conti L, Avendaño-Vázquez SE, Dhir A, Romano M, D’Ambrogio A, et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011;30:277–288. doi: 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wengrod J, Martin L, Wang D, Frischmeyer-Guerrerio P, Dietz HC, Gardner LB. Inhibition of nonsense-mediated RNA decay activates autophagy. Mol Cell Biol. 2013;33:2128–2135. doi: 10.1128/MCB.00174-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isken O, Kim YK, Hosoda N, Mayeur GL, Hershey JW, Maquat LE. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell. 2008;133:314–327. doi: 10.1016/j.cell.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurosaki T, Li W, Hoque M, Popp MW-L, Ermolenko DN, Tian B, Maquat LE. A post-translational regulatory switch on UPF1 controls targeted mRNA degradation. Genes Dev. 2014;28:1900–1916. doi: 10.1101/gad.245506.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dayton RD, Wang DB, Klein RL. The advent of AAV9 expands applications for brain and spinal cord gene delivery. Expert Opin Biol Ther. 2012;12:757–766. doi: 10.1517/14712598.2012.681463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duque S, Joussemet B, Riviere C, Marais T, Dubreil L, Douar AM, et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu H, Dirosario J, Killedar S, Zaraspe K, McCarty DM. Correction of neurological disease of mucopolysaccharidosis IIIB in adult mice by rAAV9 trans-blood–brain barrier gene delivery. Mol Ther. 2011;19:1025–1033. doi: 10.1038/mt.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinberg MS, Samulski RJ, McCown TJ. Adeno-associated virus (AAV) gene therapy for neurological disease. Neuropharmacology. 2013;69:82–88. doi: 10.1016/j.neuropharm.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gong C, Kim YK, Woeller CF, Tang Y, Maquat LE. SMD and NMD are competitive pathways that contribute to myogenesis: effects on PAX3 and myogenin mRNAs. Genes Dev. 2009;23:54–66. doi: 10.1101/gad.1717309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurosaki T, Maquat LE. Rules that govern UPF1 binding to mRNA 3’UTRs. Proc Natl Acad Sci USA. 2013;110:3357–3362. doi: 10.1073/pnas.1219908110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.