

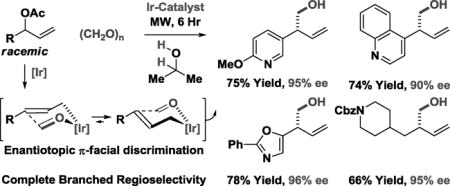
Abstract
Chiral iridium complexes modified by SEGPHOS catalyze the 2-propanol mediated reductive coupling of branched allylic acetates 1a–1o with formaldehyde to form primary homoallylic alcohols 2a–2o with excellent control of regio- and enantioselectivity. These processes, which rely on enantiotopic π-facial discrimination of σ-allyliridium intermediates, represent the first examples of enantioselective formaldehyde C-C coupling beyond aldol addition.
Graphical abstract
Enantiotopic facial discrimination of electrophilic allylmetal species is well established in asymmetric allylic alkylation (W,1a Pd,1b Mo,1c Ir1d) (Scheme 1, eq. 1). However, despite nearly four decades of work on enantioselective carbonyl allylation,2–8 this mode of enantioselection is unknown in the context of nucleophilic allylmetal species, where work has focused exclusively on asymmetric additions to aldehydes or ketones to form chiral α-stereogenic secondary or tertiary alcohols, respectively (Scheme 1, eq. 2). Nucleophilic allylations of formaldehyde to form chiral β-stereogenic primary homoallylic alcohols have not been described. Indeed, the only catalytic enantioselective C-C couplings of formaldehyde reported, to date, involve asymmetric aldol addition.9,10
Scheme 1.
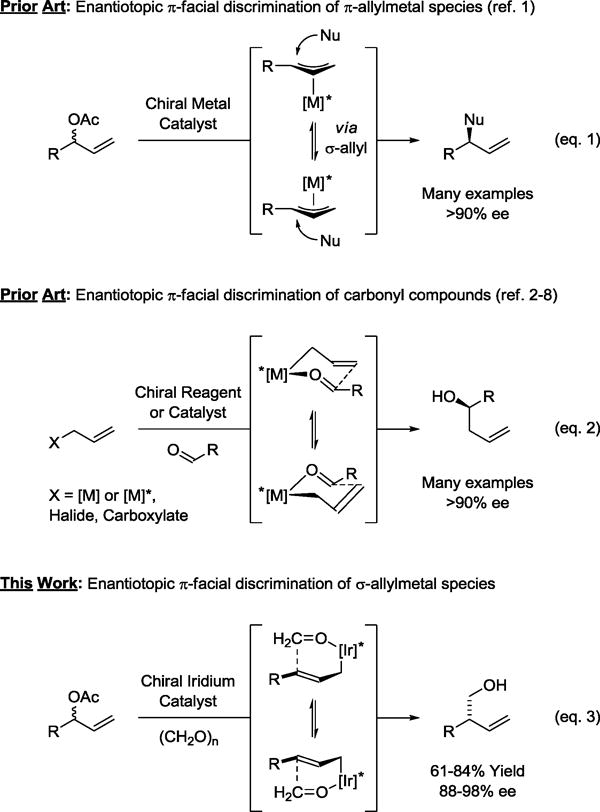
Enantiotopic π-facial discrimination in electrophilic and nucleophilic allylation.
Merging the chemistry of transfer hydrogenation and carbonyl addition,7 formaldehyde (or methanol11b) mediated hydrohydroxymethylations of diverse π-unsaturated reactants (allenes,11a,b,c dienes11d,e and alkynes11f) were developed.11,12,13 While these processes are efficient and regioselective in contexts where carbonylation/hydroformylation is not, enantioselective variants have not been established, nor has the hydroxymethylation of allylic acetates been explored. Here, we apply a chiral iridium catalyst to the enantioselective reductive coupling of allylic acetates with formaldehyde to form chiral β-stereogenic primary homoallylic alcohols via enantiotopic π-facial discrimination of σ-allyliridium intermediates (Scheme 1, eq. 3). These processes constitute the first examples of enantioselective formaldehyde C-C coupling beyond aldol addition.
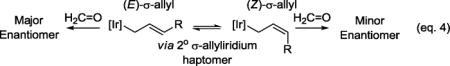
The use of paraformaldehyde as an electrophilic partner in asymmetric nucleophilic allylation poses several challenges. First, high levels of enantioselectivity require intervention of a single geometrical isomer of the σ-allylmetal species in the carbonyl addition event. Our collective data are consistent with a catalytic mechanism wherein carbonyl addition occurs by way of a closed chair-like transition structure.7,8 Second, as established in studies on the stereochemistry of mechanistically related aldol additions,14 competition between chair-like vs boat-like transition structures increases with decreasing steric demand of the reactants, which inverts the enantiotopic nucleophile π-face undergoing addition. Finally, in the presence of group 9 metals, paraformaldehyde will transform to synthesis gas,15 which can promote a variety of side reactions and act as a catalyst poison.
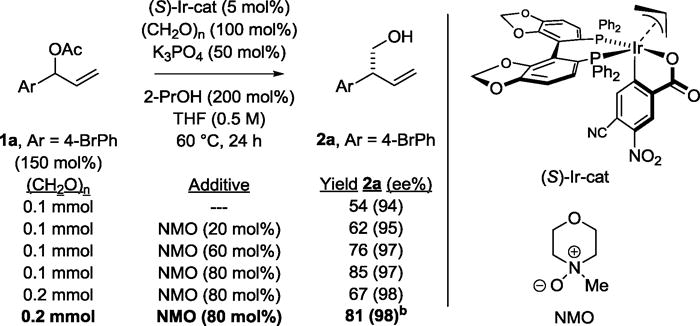
Cognizant of these potential obstacles, initial optimization experiments were undertaken. Gratifyingly, it was found that the indicated π-allyliridium C,O-benzoate complex modified by (S)-SEGPHOS promotes the reductive coupling of the 4-bromophenyl substituted allylic acetate 1a (150 mol%) with paraformaldehyde (100 mol%) to provide the desired product, the primary homoallylic alcohol 2a, in 54% yield and 94% ee as a single regioisomer (Table 1). It was postulated that synthesis gas generated upon decomposition of paraformaldehyde15 might contribute to the formation of catalytically inactive iridium carbonyl complexes. This hypothesis was corroborated by the observance of absorptions at 2125 cm−1 in IR spectra taken from aliquots of crude reaction mixtures.16 Hence, N-methyl morpholine oxide (NMO), which is commonly used for the oxidative removal of carbonyl ligands,17 was employed as an additive. As hoped, the addition of NMO proved to be beneficial, allowing homoallylic alcohol 2a to be isolated in 85% yield and 97% ee. However, this yield could not be reproduced on larger scale due to solubility issues involving both paraformaldehyde and NMO. Fortunately, it was found that microwave heating largely restored the isolated yield of 2a with the advantage of significantly shortened reaction times.
Table 1.
Selected optimization experiments in the enantioselective iridium catalyzed reductive coupling of allylic acetate 1a with formaldehyde illustrating the importance of NMO.a
|
Yields are of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details
Microwave heating, 6h.
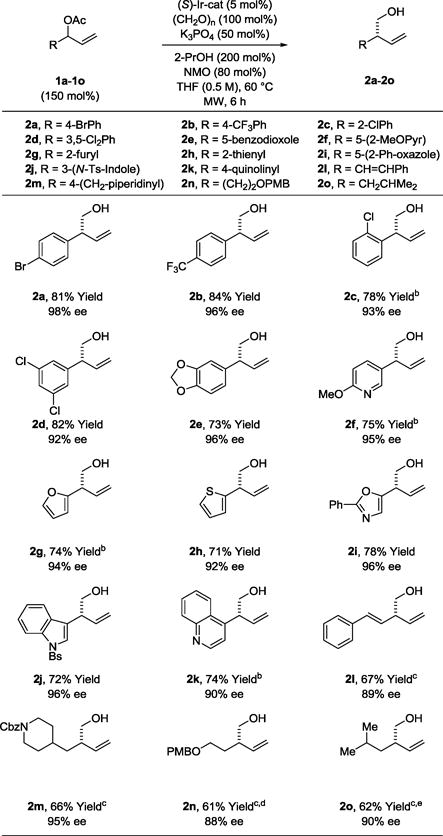
To assess scope, optimized conditions were applied to the reductive coupling of paraformaldehyde with diverse branched allylic acetates 1a–1o (Table 2). As illustrated in the formation of primary homoallylic alcohols 2a–2e, a variety of substituted aryl groups are tolerated, including ortho-substituted aryl moieties (2c). The formation of compounds 2f–2k establish the tolerance of these conditions toward N-, S- and O-bearing heterocycles. In each case, aryl- and heteroaryl-substituted substituted products 2a–2k are formed in good yield with uniformly high levels of enantioselectivity, consistent with good partitioning of (E)- and (Z)-σ-allyliridium isomers in the carbonyl addition event (eq. 4). Even in the case of branched
 |
(eq. 4) |
allylic acetates substituted by styryl groups (1l) or alkyl chains (1m–1o), high levels of enantioselectivity are retained. In all cases, complete levels of branch-regioselectivity are observed. Attempted redox-neutral hydroxymethylations wherein methanol serves dually as reductant and formaldehyde precursor were inefficient due to transesterification and transfer hydrogenation of the terminal olefin of the allylic acetate.
Table 2.
Enantioselective iridium catalyzed reductive coupling of branched allylic acetates 1a–1o with formaldehyde to form primary homoallylic alcohols 2a–2o.a
|
Yields are of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. 2k, 8 h. 2l, 10 h. See Supporting Information for further experimental details.
80 °C.
70 °C.
The catalyst modified by (S)-DM-SEGPHOS was used.
Isolated as the 4-nitrobenzoate due to volatility.
The products of reductive coupling serve as useful building blocks in chemical synthesis (Scheme 2). For example, rhodium catalyzed hydroformylation18 of adduct 2a provides a lactol, which upon treatment with pyridinium chlorochromate (PCC) is converted to the enantiomerically enriched δ-lactone 3a (eq. 5). The absolute stereochemistry of 3a was determined by single crystal X-ray diffraction and is the basis for the stereochemical assignments of compounds 2a–2o. Conversion of alcohol 2a to the corresponding acrylic ester, followed by ring-closing metathesis, delivers the α,β-unsaturated δ-lactone 4a (eq. 6). Amination of the alcohol moiety of 2a by way of the p-toluene sulfonate was challenging due to competing elimination. However, treating the p-toluene sulfonate derived from 2a with sodium azide in DMF delivered the primary azide in good yield. Reduction of the azide provided the amine, which was isolated as the N-Boc amide 5a (eq. 7).
Scheme 2.
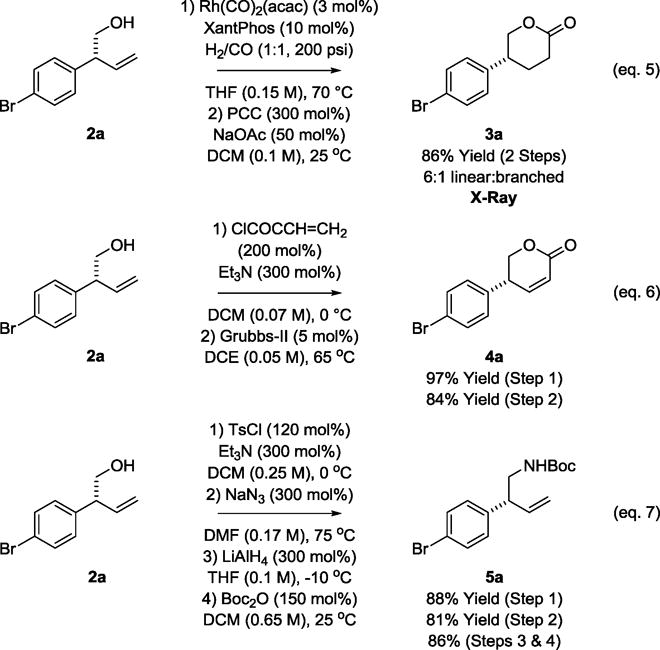
Elaboration of adduct 2a and assignment of absolute stereochemistry.a
aYields are of material isolated by silica gel chromatography.
A catalytic mechanism and stereochemical have been proposed (Scheme 3). Entry into the catalytic cycle occurs via protonolytic cleavage of the π-allyliridium C,O-benzoate complex mediated by 2-propanol to furnish the indicated iridium 2-propoxide complex. β-Hydride elimination with loss of acetone generates an iridium hydride, which upon deprotonation forms an anionic iridium(I) species. Ionization of the allylic acetate delivers the mono-substituted π-allyliridium complex. Complete levels of branch-regioselectivity accompanied by high levels of enantioselectivity suggest formaldehyde addition occurs predominantly by way of the primary (E)-σ-allyliridium haptomer through a closed 6-centered transition structure.7,8 Protonolytic cleavage of the resulting homoallylic alkoxide mediated by 2-propanol regenerates the iridium 2-propoxide complex to close the catalytic cycle. The absolute stereochemical course of the carbonyl addition event is consistent with our previously proposed stereochemical model.8b Allylic acetate recovered from the reaction is racemic, indicating kinetic resolution does not occur in the ionization event.
Scheme 3.
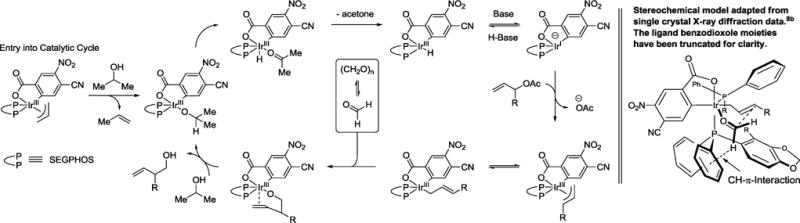
General catalytic mechanism and stereochemical model.a
In summary, we report the first enantioselective formaldehyde C-C couplings beyond aldol addition. Specifically, chiral iridium complexes modified by SEGPHOS catalyze the 2-propanol mediated reductive coupling of branched allylic acetates 1a–1o with formaldehyde to form primary homoallylic alcohols 2a–2o with complete control of regioselectivity and uniformly high levels of enantioselectivity. This process provides access to products of hydroxyalkylation that are inaccessible using classical carbonylative methods11,12 and support the feasibility of related transformations, including the non-carbonylative aminomethylation19 of branched allylic acetates.
Supplementary Material
Acknowledgments
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) are acknowledged for partial support of this research. Chinh Ngo, Zhicheng Zhang and Nicole Behnke are acknowledged for technical assistance.
Footnotes
Supporting Information Available: Experimental procedures and spectral data. HPLC traces corresponding to racemic and enantiomerically enriched samples. Single crystal X-ray diffraction data for compound 3a. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For seminal examples of enantiotopic facial discrimination in electrophilic π-allyl complexes based on W, Pd, Mo and Ir, respectively, see:; (a) Lloyd-Jones GC, Pfaltz A. Angew Chem Int Ed Engl. 1995;34:462. [Google Scholar]; (b) Trost BM, Krische MJ, Radinov R, Zanoni G. J Am Chem Soc. 1996;118:6297. [Google Scholar]; (c) Janssen JP, Helmchen G. Tetrahedron Lett. 1997;38:8025. [Google Scholar]; (d) Trost BM, Hachiya I. J Am Chem Soc. 1998;120:1104. [Google Scholar]
- 2.For selected reviews on enantioselective carbonyl allylation, see:; (a) Ramachandran PV. Aldrichim Acta. 2002;35:23. [Google Scholar]; (b) Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h. [DOI] [PubMed] [Google Scholar]; (c) Kennedy JWJ, Hall DG. Angew Chem, Int Ed. 2003;42:473. [Google Scholar]; (d) Yu C-M, Youn J, Jung H-K. Bull Korean Chem Soc. 2006;27:462. [Google Scholar]; (e) Marek I, Sklute G. Chem Commun. 2007;1683 doi: 10.1039/b615042j. [DOI] [PubMed] [Google Scholar]; (f) Hall DG. Synlett. 2007;1644 [Google Scholar]; (g) Lachance H, Hall DG. Org React. 2008;73:1. [Google Scholar]; (h) Yus M, González-Gómez JC, Foubelo F. Chem Rev. 2011;111:7774. doi: 10.1021/cr1004474. [DOI] [PubMed] [Google Scholar]
- 3.For selected examples of enantioselective carbonyl allylation using chirally modified allylmetal reagents, see:; (a) Herold T, Hoffmann RW. Angew Chem Int Ed. 1978;17:768. [Google Scholar]; (b) Hoffmann RW, Herold T. Chem Ber. 1981;114:375. [Google Scholar]; (c) Hayashi T, Konishi M, Kumada M. J Am Chem Soc. 1982;104:4963. [Google Scholar]; (d) Brown HC, Jadhav PK. J Am Chem Soc. 1983;105:2092. [Google Scholar]; (e) Roush WR, Walts AE, Hoong LK. J Am Chem Soc. 1985;107:8186. [Google Scholar]; (f) Reetz MT. Pure Appl Chem. 1988;60:1607. [Google Scholar]; (g) Short RP, Masamune S. J Am Chem Soc. 1989;111:1892. [Google Scholar]; (h) Corey EJ, Yu C-M, Kim SS. J Am Chem Soc. 1989;111:5495. [Google Scholar]; (i) Seebach D, Beck AK, Imwinkelzied R, Roggo S, Wonnacott A. Helv Chim Acta. 1987;70:954. [Google Scholar]; (j) Riediker M, Duthaler RO. Angew Chem, Int Ed. 1989;28:494. [Google Scholar]; (k) Panek JS, Yang M. J Am Chem Soc. 1991;113:6594. [Google Scholar]; (l) Kinnaird JWA, Ng PY, Kubota K, Wang X, Leighton JL. J Am Chem Soc. 2002;124:7920. doi: 10.1021/ja0264908. [DOI] [PubMed] [Google Scholar]; (m) Hackman BM, Lombardi PJ, Leighton JL. Org Lett. 2004;6:4375. doi: 10.1021/ol0480731. [DOI] [PubMed] [Google Scholar]; (n) Burgos CH, Canales E, Matos K, Soderquist JA. J Am Chem Soc. 2005;127:8044. doi: 10.1021/ja043612i. [DOI] [PubMed] [Google Scholar]
- 4.For selected examples of enantioselective carbonyl allylation using achiral allylmetal reagents in combination with chiral catalysts, see:; (a) Furuta K, Mouri M, Yamamoto H. Synlett. 1991;561 [Google Scholar]; (b) Costa AL, Piazza MG, Tagliavini E, Trombini C, Umani-Ronchi A. J Am Chem Soc. 1993;115:7001. [Google Scholar]; (c) Keck GE, Tar-bet KH, Geraci LS. J Am Chem Soc. 1993;115:8467. [Google Scholar]; (d) Denmark SE, Coe DM, Pratt NE, Griedel BD. J Org Chem. 1994;59:6161. doi: 10.1021/jo052202p. [DOI] [PubMed] [Google Scholar]; (e) Denmark SE, Fu J. J Am Chem Soc. 2001;123:9488. doi: 10.1021/ja016552e. [DOI] [PubMed] [Google Scholar]; (f) Lachance H, Lu X, Gravel M, Hall DG. J Am Chem Soc. 2003;125:10160. doi: 10.1021/ja036807j. [DOI] [PubMed] [Google Scholar]; (g) Wada R, Oisaki K, Kanai M, Shibasaki M. J Am Chem Soc. 2004;126:8910. doi: 10.1021/ja047200l. [DOI] [PubMed] [Google Scholar]; (h) Rauniyar V, Hall DG. Angew Chem Int Ed. 2006;45:2426. doi: 10.1002/anie.200504432. [DOI] [PubMed] [Google Scholar]; (i) Rauniyar V, Zhai H, Hall DG. J Am Chem Soc. 2008;130:8481. doi: 10.1021/ja8016076. [DOI] [PubMed] [Google Scholar]; (j) Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2006;128:12660. doi: 10.1021/ja0651308. [DOI] [PubMed] [Google Scholar]; (k) Barnett DS, Moquist PN, Schaus SE. Angew Chem, Int Ed. 2009;48:8679. doi: 10.1002/anie.200904715. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Jain P, Antilla JC. J Am Chem Soc. 2010;132:11884. doi: 10.1021/ja104956s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For selected reviews on enantioselective carbonyl allylation via Nozaki-Hiyama-Kishi coupling, see:; (a) Avalos M, Babiano R, Cintas P, Jiménez JL, Palacios JC. Chem Soc Rev. 1999;28:169. [Google Scholar]; (b) Bandini M, Cozzi PG, Umani-Ronchi A. Chem Commun. 2002;919 doi: 10.1039/b109945k. [DOI] [PubMed] [Google Scholar]; (c) Hargaden GC, Guiry PJ. Adv Synth Catal. 2007;349:2407. [Google Scholar]
- 6.For selected reviews covering carbonyl allylation via umpolung of π-allyls, see:; (a) Masuyama Y. In: Advances in Metal-Organic Chemistry. Liebeskind LS, editor. Vol. 3. JAI Press; Greenwich: 1994. p. 255. [Google Scholar]; (b) Tamaru Y. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E-i, de Meijere A., editors. Vol. 2. Wiley; New York: 2002. p. 1917. [Google Scholar]; (c) Tamaru Y. In: Perspectives in Organopalladium Chemistry for the XXI Century. Tsuji J, editor. Elsevier; Amsterdam: 1999. p. 215. [Google Scholar]; (d) Kondo T, Mitsudo T-A. Curr Org Chem. 2002;6:1163. [Google Scholar]; (e) Tamaru Y. Eur J Org Chem. 2005;13:2647. [Google Scholar]; (f) Zanoni G, Pontiroli A, Marchetti A, Vidari G. Eur J Org Chem. 2007;22:3599. [Google Scholar]
- 7.For a recent review on enantioselective carbonyl allylation via transfer hydrogenation, see:; Ketcham JM, Shin I, Montgomery TP, Krische MJ. Angew Chem Int Ed. 2014;53:9142. doi: 10.1002/anie.201403873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For selected examples of catalytic enantioselective redox-triggered carbonyl allylations employing allylic carboxylates as pronucleophiles, see:; (a) Kim IS, Ngai M-Y, Krische MJ. J Am Chem Soc. 2008;130:6340. doi: 10.1021/ja802001b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim IS, Ngai M-Y, Krische MJ. J Am Chem Soc. 2008;130:14891. doi: 10.1021/ja805722e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514. doi: 10.1021/ja808857w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gao X, Townsend IA, Krische MJ. J Org Chem. 2011;76:2350. doi: 10.1021/jo200068q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Schmitt DC, Dechert-Schmitt A-MR, Krische MJ. Org Lett. 2012;14:6302. doi: 10.1021/ol3030692. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dechert-Schmitt A-MR, Schmitt DC, Krische MJ. Angew Chem Int Ed. 2013;52:3195. doi: 10.1002/anie.201209863. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Shin I, Wang G, Krische MJ. Chem Eur J. 2014;13382 doi: 10.1002/chem.201404065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For catalytic enantioselective Mukaiyama aldol additions of formaldehyde, see:; (a) Ozasa N, Wadamoto M, Ishihara K, Yamamoto H. Synlett. 2003;2219 [Google Scholar]; (b) Manabe K, Ishikawa S, Hamada T, Kobayashi S. Tetrahedron. 2003;59:10439. [Google Scholar]; (c) Ishikawa S, Hamada T, Manabe K, Kobayashi S. J Am Chem Soc. 2004;126:12236. doi: 10.1021/ja047896i. [DOI] [PubMed] [Google Scholar]; (d) Kobayashi S, Ogino T, Shimizu H, Ishikawa S, Hamada T, Manabe K. Org Lett. 2005;7:4729. doi: 10.1021/ol051965w. [DOI] [PubMed] [Google Scholar]; (e) Kokubo M, Ogawa C, Kobayashi S. Angew Chem Int Ed. 2008;47:6909. doi: 10.1002/anie.200801849. [DOI] [PubMed] [Google Scholar]
- 10.For catalytic enantioselective direct aldol additions of formaldehyde, see:; (a) Kuwano R. Chem Commun. 1998;71 [Google Scholar]; (b) Torii H, Naka-dai M, Ishihara K, Saito S, Yamamoto H. Angew Chem Int Ed. 2004;43:1983. doi: 10.1002/anie.200352724. [DOI] [PubMed] [Google Scholar]; (c) Casas J, Sundén H, Córdova A. Tetrahedron Lett. 2004;45:6117. [Google Scholar]; (d) Fukuchi I, Hamashima Y, Sodeoka M. Adv Synth Catal. 2007;349:509. [Google Scholar]; (e) Mouri S, Chen Z, Matsunaga S, Shibasaki M. Chem Commun. 2009;5138 doi: 10.1039/b912380f. [DOI] [PubMed] [Google Scholar]; (f) Mase N, Inoue A, Nishio M, Takabe K. Bioorg Med Chem Lett. 2009;3955 doi: 10.1016/j.bmcl.2009.03.012. [DOI] [PubMed] [Google Scholar]; (g) Boeckman RK, Jr, Miller JR. Org Lett. 2009;11:4544. doi: 10.1021/ol9017479. [DOI] [PubMed] [Google Scholar]; (h) Pasternak M, Paradowska J, Rogozińska M, Mlynarski J. Tetrahedron Lett. 2010;51:4088. [Google Scholar]; (i) Shirakawa S, Ota K, Terao SJ, Maruoka K. Org Biomol Chem. 2012;10:5753. doi: 10.1039/c2ob07193b. [DOI] [PubMed] [Google Scholar]; (j) Yasui Y, Benohoud M, Sato I, Hayashi Y. Chem Lett. 2014;43:556. [Google Scholar]; (k) Meninno S, Fuoco T, Tedesco C, Lattanzi A. Org Lett. 2014;16:4746. doi: 10.1021/ol502148a. [DOI] [PubMed] [Google Scholar]
- 11.For catalytic hydrohydroxymethylation of allenes (a,b,c), dienes (d,e) and alkynes (f,g) to paraformaldehyde via C–C bond forming transfer hydrogenation, see:; (a) Ngai M-Y, Skucas E, Krische MJ. Org Lett. 2008;10:2705. doi: 10.1021/ol800836v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moran J, Preetz A, Mesch RA, Krische MJ. Nat Chem. 2011;3:287. doi: 10.1038/nchem.1001. [DOI] [PubMed] [Google Scholar]; (c) Sam B, Montgomery TP, Krische MJ. Org Lett. 2013;15:3790. doi: 10.1021/ol401771a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smejkal T, Han H, Breit B, Krische MJ. J Am Chem Soc. 2009;131:10366. doi: 10.1021/ja904124b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Köpfer A, Sam B, Breit B, Krische MJ. Chem Sci. 2013;4:1876. [Google Scholar]; (f) Bausch CC, Patman RL, Breit B, Krische MJ. Angew Chem Int Ed. 2011;50:5687. doi: 10.1002/anie.201101496. [DOI] [PubMed] [Google Scholar]
- 12.For reviews encompassing use of paraformaldehyde and methanol as C1-feedstocks in metal catalyzed C-C coupling, see:; (a) Wu L, Liu Q, Jackstell R, Beller M. Angew Chem Int Ed. 2014;53:6310. doi: 10.1002/anie.201400793. [DOI] [PubMed] [Google Scholar]; (b) Sam B, Breit B, Krische MJ. Angew Chem Int Ed. 2015;54:3267. doi: 10.1002/anie.201407888. [DOI] [PubMed] [Google Scholar]
- 13.For methanol-mediated Guerbet-type aldol reactions, see:; (a) Chan LKM, Poole DL, Shen D, Healy MP, Donohoe TJ. An-gew Chem Int Ed. 2014;53:761. doi: 10.1002/anie.201307950. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shen D, Poole DL, Shot-ton CC, Kornahrens AF, Healy MP, Donohoe TJ. Angew Chem Int Ed. 2015;54:1642. doi: 10.1002/anie.201410391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans DA, Nelson JV, Taber T. Top Stereochem. 1982;13:1. [Google Scholar]
- 15.Fuentes JA, Pittaway R, Clarke ML. Chem Eur J. 2015;21:10645. doi: 10.1002/chem.201502049. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The absorption at 2125 cm−1 in IR is consistent with an iridium(III) carbonyl complex:; (a) Vaska L, DiLuzio JW. J Am Chem Soc. 1961;83:2784. [Google Scholar]; (b) Vaska L. J Am Chem Soc. 1966;88:4100. doi: 10.1021/ja00969a043. [DOI] [PubMed] [Google Scholar]
- 17.Shen JK, Gao Y-C, Shi Q-Z, Basolo F. Coord Chem Rev. 1993;128:69. and references cited therein. [Google Scholar]
- 18.for impact of large bite-angle ligands on regioselectivity in hydro-formylation see reference (a). For seminal use of XantPhos in linear regioselective hydroformylation, see reference (b):; (a) Casey CP, Whiteker GT, Melville MG, Petrovich LM, Gavney JA, Jr, Powel DR. J Am Chem Soc. 1992;114:5535. [Google Scholar]; (b) Kranen-burg M, van der Burgt YEM, Kamer PCJ, van Leeuwen PWNM, Goubitz KG, Fraanje J. Organometallics. 1995;14:3081. [Google Scholar]
- 19.(a) Oda S, Sam B, Krische MJ. Angew Chem Int Ed. 2015;54:8525. doi: 10.1002/anie.201503250. [DOI] [PubMed] [Google Scholar]; (b) Oda S, Franke J, Krische MJ. Chem Sci. 2016;7:136. doi: 10.1039/c5sc03854e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.