

Abstract
Multiple substrate enzymes present a particular challenge when it comes to understanding their activity in a complex system. Although a single target may be easy to model, it does not always present an accurate representation of what that enzyme will do in the presence of multiple substrates simultaneously. Therefore, there is a need to find better ways to both study these enzymes in complicated systems, as well as accurately describe the interactions through kinetic parameters. This review looks at different methods for studying multiple substrate enzymes, as well as explores options on how to most accurately describe an enzyme’s activity within these multi-substrate systems. Identifying and defining this enzymatic activity should clear the way ro use in vitro systems to accurately predict the behavior of multi-substrate enzymes in vivo.
Keywords: Internal competition, Kinetics, Specificity, Selectivity, Mass spectrometry
1. Introduction
It is common for one enzyme to be able to catalyze multiple substrates or interact with multiple sites, as has been found from various in vitro enzymatic studies (for example, cytochrome P450 enzymes [1, 2], lysine acetyltransferases [3], and kinases [4]). In in vivo systems, as a consequence, all these potential substrates/sites also have the potential to act as competitors. Enzyme preference is usually revealed by different rates or affinities for substrates. The preference of an enzyme for one specific substrate is defined as the specificity, and the preference for one substrate over another is its selectivity. Given these facts, single target substrates matched with a single enzyme is the most direct and simplest system for investigating enzyme specificity in vitro (i.e., classical steady-state approach). From the kinetic parameters obtained via this straightforward approach, one can determine the specificity (i.e., specificity constant, kcat/Km) of a substrate [5, 6]. The ratio of specificity constants from two different substrates with the same enzyme may then be used to interpret the preference of that enzyme for one substrate over the other: the selectivity [7]. These enzymatic kinetic parameters seem adequate for applications for in vitro systems, but it has been shown that some predictions of these parameters fail to match up with corresponding observations from in vivo assays [8–10]. The complexity of in vivo assays may lead to the following potential factors being overlooked: protein-protein interactions [11–14], enzymatic structural/conformational changes [15–18], and internal inhibition [19–22]. Thus, recent research has utilized the method of internal competition (multiple substrates to one enzyme) to study the selectivity of an enzyme between substrates [23–26]. This experimental design can/may more closely simulate the in vivo environment. However, such assays also create difficulties in producing accurate detections for multiple targets, as signals from one target have to be independent of the others. Fortunately, advances in current technologies allow for the measurement of multi-substrates/products in a less labor-intensive and time-consuming manner.
In this review, we examine multiplexed, high throughput, and potentially even real-time methodologies applied on different enzymatic selectivity assays and we provide an overview of how to use the kinetic parameters from internal competition to interpret the potential selectivity in vivo. Using this approach, we also highlight the possible constraints of each method, such as choices of substrate concentrations, time frame selections (steady-state condition), and/or available cofactors/inhibitors.
2. Techniques for multiplexed, high throughput measurements of multiple substrates/products
Internal competition is a method that has been used to investigate the differences of an enzyme for individual mixed substrates by measuring either the consumption rate of individual substrates or the generation rate of individual products. This method has also been extensively used in studying kinetic isotope effects [27–29]. Since the concentrations of multi-substrates and/or multi-products need to be monitored, a multiplexed analytical technique is required to measure all of the concentrations of each of these components for data analysis. This section will discuss the recent analytical techniques applied to study the kinetics of internal enzyme competition.
This simplest method for these approaches is liquid chromatography (LC), which relies on the separation of multiple substrates by hydrophobicity or cation/anion exchange, depending on what is being separated. This is often a reverse phase column with the accompanying detection as UV absorption, fluorescence, or radio chemical. An example of this approach is the analysis of multiple substances in the bioremediation of polycyclic aromatic hydrocarbons (PAHs) [30, 31]. In this case LC alone can separate various forms of PAHs; in other cases additional verification is needed, such as mass/charge.
Mass changes (either cleavage or addition/removal of functional groups) of a substrate are often the result of catalytic reactions. Thus, mass spectrometry (MS) is a common analytical technology that is utilized for these types of studies [32–35]. The coupling of LC or gas chromatography to MS provides separation of multiple substrates, and therefore more accurate quantification, for multiple target analysis [36, 37]. Furthermore, tandem MS (MS/MS) can be used to acquire more spatial or structural information of analytes. For example, LC-MS/MS has been used to quantitate the substrates and/or products from enzymatic kinetic assays [13, 38–40], and a detection resolution as small as a single amino acid residue can be reached [7, 41, 42]. Recently, this kind of site-specific study has even been utilized for the investigation of non-enzymatic protein modifications [7, 14, 43].
Another technique used for these internal competition assays, nuclear magnetic resonance spectrometry (NMR), is used to determine the kinetic isotope effects between stable isotope labeled substrates and unlabeled substrates [27–29]. As more innovative methodologies have been developed to utilize this advanced technology, a very high degree of precision and accuracy can be obtained for the measurement of low abundance, stably isotope-labeled substrates [44–47]. Additionally, with the proper sample and safety controls, a method of radioactive remote labeling can be utilized to study internal competition. A scintillation detector with a multi-channel analyzer can record different radiation energy from different radioactive sources. With this technique, a high sensitivity and high accuracy of measurements can be achieved for the detection of various radioactively labeled substrates [48–50]. The study of hydrogen tunneling by using radioactive remote labeling is one example of the application of this technique [51, 52].
Internal competition assay are not limited to proteins. Substrates can also include DNA and/or RNA. Current biochemical and labeling techniques have been developed to effectively and efficiently measure the DNA/RNA kinetics of enzymatic catalysis [53–56]. For example, Goodman (et al.) used kinetic assays to investigate DNA polymerase fidelity by comparing the competition of right and wrong nucleotide incorporations [56]. As another example, substrate competition of endoribonucleases can occur in vivo because endoribonucleases can cleave multiple RNA substrates [57]. Harris (et al.) examined the internal competition between different tRNA precursors for ribonuclease P by radiolabelling substrates and directly quantifying the substrate specificity [58, 59]. Furthermore, it has been found that RNA sequence is very specific for ribonucleases (for example, RNase H) [60–62]. To understand the substrate sequence specificity and site specificity, deep sequencing methods (for review see [63–65]) were used to investigate the frequency and locations of ribonuclease L cleavage sites of viral RNAs [66, 67]. Site-specific studies have also been carried out, using primer extension reactions to characterize ribonuclease L specific cleavage sites in hepatitis C virus RNA [68] and DNA damage sites [69]. Utilizing the aforementioned analytical technologies (i.e., MS) coupled with these types of assays can be useful for the detection of not only the DNA/RNA sequences [70–72] but also the modifications on the individual nucleosides [73–75]. Additionally, MS analysis has been used to investigate the substrate selectivity of artificial restriction enzymes [76], which can be applied to manipulating RNAs for biotechnology applications.
Finally, the concept of this internal competition method was also applied to developing quantitative competitive polymerase chain reaction techniques to quantitate target DNA [77]. By competing with internal DNA segment, Tompkins (et al.) demonstrated that the reverse transcription-quantitative competitive polymerase chain reaction technique can quantitate the expression of seven cytokine mRNAs in domestic mammals [78]. This approach can be extended to barcoded samples like those demonstrated by Nguyen (et al.) where they paired unique DNA sequences to specific histone modifications in nucleosomes [79]. While this would require a separation step, it would also allow simple quantitative PCR in place of expensive sequencing or other technology. Through these types of experiments, we have seen the progress in adapting the knowledge gained from internal competition assays into the utilization in practical applications. All aforementioned methods that can be utilized to measure multiple substrates/products are summarized in Table 1.
Table 1.
Summary of published techniques for multiplexed measurements of multiple substrates/products
| Subjects studied | Techniques for measurements | Categories of studies | Reference (year) |
|---|---|---|---|
|
| |||
| Multiple PAH biodegradation | HPLC | Monod kinetics | [30] (1988); [31] (1999) |
| Glycogen synthase for adenine- and uridine-diphosphate glucose polymerization | MS | Double-reciprocal kinetic | [33] (2003) |
| Sugar nucleotidyltransferases for multiple substrates | Michaelis-Menten kinetics | [34] (2005) | |
| NodST, a GlcNAc-6-O-sulfotransferase, for four chitoologosaccharides synthesis | Internal competition kinetics | [38] (2004) | |
| Gcn5-mediated histone H3 lysine acetylation and nonenzymatic acetylation | Site specific internal competition kinetics | [7] (2013) | |
| Comparison of Rtt109-Vps75 mediated histone acetylation on different histone complexes | Site specific internal competition kinetics | [14] (2015) | |
| P300 and CBP for histone H3 and H4 acetylation | Site specific internal competition kinetics | [41] (2013) | |
| DNA methylation | Quantification of nucleosides | [73] (2008) | |
| tRNA modifications | Quantification of nucleosides | [74] (2010); [75] (2014) | |
|
| |||
| Sialidase hydrolysis of Neu5Acα2, 6LacβSPh (12C/13C and 16O/18O) | NMR | Competitive kinetic isotope effects | [28] (2010) |
| Catalysis by thymidylate synthase | Kinetic isotope effects and hydrogen tunneling | [51] (2004) | |
| Catalysis by dihydrofolate reductase | Kinetic isotope effects and hydrogen tunneling | [52] (2011) | |
|
| |||
| Mechanism of mutant and wild type T4 DNA polymerases (3H and 14C) | Radiolabeling | Internal competition kinetics | [53] (1979) |
| Steady state and pre-steady state kinetics for studying DNA polymerase fidelity (32P) | Internal competition kinetics | [56] (2010) | |
| Kinetics of ribonuclease P over 80 competing tRNA precursors (32P) | Internal competition kinetics | [58] (2013) | |
| Modeling of ribonuclease P processing 3 competing tRNA precursors (32P) | Internal competition kinetics | [59] (2014) | |
| Specificity study on ribozyme for variable substrate sequences (32P) | Individual kinetics | [60] (1988) | |
| Ribonuclease L cleavage sites in viral RNAs (32P) | Cleavage site study | [68] (2004) | |
| Serial analysis of mutation spectra (32P) | Identification of DNA damage sites | [69] (2008) | |
|
| |||
| Proofreading efficiency of wild type T4 DNA polymerase | Fluorescence | Internal competition kinetics | [54] (1994) |
| Cleavage rate study on 8–17 deoxyribozyme | Internal competition kinetics | [55] (2010) | |
|
| |||
| Ribonuclease L cleavage sites in host (HeLa cell) and viral RNAs | Deep sequencing method | Characterization of cleavage sites | [66] (2014) |
| Ribonuclease L cleavage sites in viral RNAs | Characterization of cleavage sites | [67] (2014) | |
|
| |||
| Cytokine gene expression in domestic mammals | Quantitative competitive PCR | Internal competition | [78] (1996) |
3. Analysis of enzyme kinetics for substrate selectivity
3.1 Steady-state analysis of specificity and selectivity for multiple substrates/sites in simple systems
The enzyme kinetics of one substrate under multiple turnover steady-state conditions can be described by the Michaelis-Menten equation (eq. 1). The Michaelis-Menten equation describes a hyperbolic relationship when plotting the initial rate (v) versus substrate concentrations, [S]. Where [E] is the concentration of enzyme, and kcat and Km are steady-state kinetic parameters, representing the catalytic constant and the Michaelis constant, respectively. Conceptually, kcat represents the number of turnover events occurring per unit time, and Km is a relative measure of substrate binding affinity.
| eq. 1 |
Differences in substrate specificity by a single enzyme have been studied since the 1920s [80], but it wasn’t until the 1960s that a usable definition was articulated [81]. Specificity is “… defined as a higher rate of reaction with respect to some reference substrate or reaction … to measure the special contribution of the enzyme to the catalysis, we should compare the velocity of the enzymatic reaction to the velocity of a nonenzymatic reaction” [81]. From this point Brot and Bender use the term specificity constant to refer to kcat/Km [82], but it wasn’t until 1974 that Fersht linked specificity and selectivity together, by using (v/[E])1/(v/[E])2, to show that induced fit and non-productive complexes are not represented in the specificity of an enzyme (in a simple system) [83]. From this point we can see specificity is linked to it ability to choose one substrate over another, or selectivity, and that kcat /Km, is the best description of specificity for a substrate because it will predict selectivity in a mixture of substrates in a simple system (eq. 2).
| eq. 2 |
We can apply this foundation to modern methods to understand selectivity between larger numbers of substrates. To do this we need to keep the standard steady-state assumptions, namely that total substrate concentration should remain close to the free substrate concentration, that enzyme should be much less than the substrate concentration and that less than 10% of total substrate should be consumed [6]. This gets more complicated when you have either one substrate that can produce multiple products like we have with histone acetyltransferases [7, 14, 41] or multiple different substrates. In this simple system one substrate will not impact the specificity or selectivity of another substrate or product (see the next section of complications). It is important to note that in this description we are assuming that all substrates are in equal concentrations or that we have one substrate and multiple products. We can solve for the steady-state rate (v/[E]) for one substrate in the presence of multiple substrates or products from one substrate, in eq. 3, here kx and Kx is rate and binding constant for the substrate we are monitoring, A and D are given by equations 4 and 5. Two important features come from this: 1) solving for kcat/Km we get kx/Kx [7]; and 2) this makes it obvious that if we divide the rate for any substrate in this system by any other we will return to eq. 2.
| eq. 3 |
| eq. 4 |
| eq. 5 |
If your main concern in studying an enzyme is substrate preference then measuring substrate turnover in the presence of all possible substrates has an advantage over individual kinetic measurements because they are able to determine selectivity with fewer measurements. We have shown that while the apparent kcat/Km for any one substrate measured in a complex background is the same as measured independently the apparent kcat is not. However, if we solve for the ratio of kcat(s) to compare any two substrates (eq. 6) then we get the same value as we would for the ratio of kcat/Km(s).
| eq. 6 |
This means that as long as we can be confident that substrate is at saturation then we can measure selectivity or substrate preference by comparing the ratios of the apparent kcat(s). With advancing technologies (e.g., deep sequencing and mass spectrometry, Table 1), which allow us to monitor multiple substrates in complex mixtures, this approach has potential to greatly increase our understanding of how enzymes function in a cell.
3.2 Steady-state analysis of specificity and selectivity for multiple substrates/sites in complex systems
In the previous section we assumed that all substrates are consumed in a hyperbolic dependence and the presence of once substrate would have little to no influence on the steady-state rate of another substrate. However, there are circumstances where the substrate binding can either positively or negatively influence catalytic activities [84–86], which can result in apparent cooperativity. This causes a deviation from the hyperbolic kinetics described by eq. 1. For example, the appearance of a sigmoidal curve in the plot of initial reaction velocity vs. substrate concentration indicates a potential event of positive cooperativity [84]. Thus, the Hill coefficient (nH) is used in eq. 7 to describe the kinetics of cooperativity [87]. In this case, Km is replaced by K1/2, which shows the substrate concentration where the reaction reaches half-maximal velocity. Sigmoidal kinetics might also be indicative of a slow transient conformational change seen in a monomeric single-site enzyme [88] or it could be a case of a random ordered mechanism of a two-substrate enzyme [89].
| eq. 7 |
When the competing alternative substrates are considered in the steady-state conditions for a specific enzyme, the kinetics of individual substrates has been illustrated by eq. 7. From this equation you can see that the only difference is a Hill coefficient for every substrate and equilibrium constant, and that if we rewrite eq. 2, we get eq. 8, where we have added the Hill coefficients for each substrate. In this case we can only use the ratio of the apparent kcat(s) if we know the Hill coefficients, which prevent single rate measurements.
| eq. 8 |
However, if the Hill coefficients are the same for all sites the substrate concentration still cancels out. While Cornish-Bowden orginally only consider substrate concentrations where they were beyond the inflection point of the enzyme [90], or that selectivity would not change as you move from low to high concentrations, we have seen this effect in lysine acetyltransferases p300 and CBP [41, 91].
In cases where different substrates have different Hill coefficients, understanding selectivity can be difficult. This is because at low substrate concentrations an enzyme could prefer A and at high concentrations it could prefer B; consider a enzyme where the K1/2 and kcat are the same but one has a nH of 2 and the other is not cooperative. The substrate with a Hill coefficient of 1 will be preferred at low concentrations while the substrate with the Hill coefficient will be preferred as we move to the maximum rate (Figure 1). We have proposed to use the plot of catalytic proficiency as a function of substrate concentration to simplify the understanding the enzyme specificity and how it changes with concentration. Catalytic proficiency is the second order rate constant kcat/Km divided by the non-enzymatic rate of catalysis, and is consistent with the definition of specificity as set by Bender and Kizdy [81]. This value will not change as a function of substrate concentration unless there is a Hill coefficient that is not equal to one, in which case the value will respond up (nH>1) or down (nH<1) in response to substrate. When dealing with multiple possible products like we have with p300 and CBP, converting this to a ΔΔG can make this easier to understand. In this case the selectivity between any two sites is simply the difference between the ΔΔG.
Figure 1. Use of catalytic proficiency to describe substrate dependent changes in selectivity.
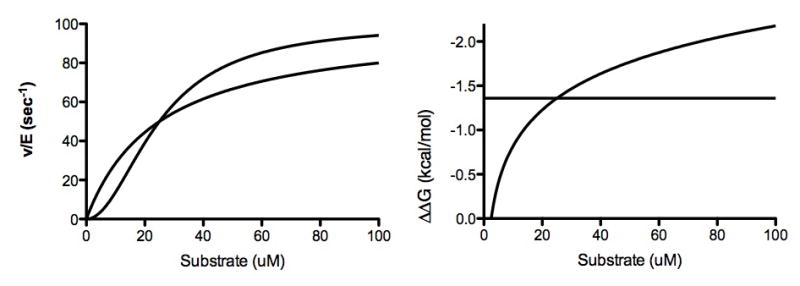
We simulated v/E vs. substrate for two substrates both with the same kcat (100/sec) and Km (25μM) values, but one with a nH of 2 and one with a nH of 1. Right: is the standard v/E vs substrate and Left: is the ΔΔG of catalytic proficiency, or −RTln([S]nH-1(k/K1/2nH)/(knE).
In cases where there are multiple substrates and one substrate can influence the activity of another it can be extremely difficult to describe the possible selectivity in a cell. Under these conditions it is best to try and limit the possible conditions using any information that can be obtained from cellular studies. It may also be possible to build initial models and refine these by comparing them measured differences in cells. In these cases chemical biology approaches [91] such as inhibitors/activators and/or isotopic labeling of substrates or products can aid in the refining of proposed models.
3.3 Progress curve assays
For practical reasons, the same concentrations for individual competing substrates may not always be achievable, especially when more than two substrates are being investigated. Thus, in eq. 4, the terms of substrate concentration cannot be canceled out, even when no cooperativity is observed. However, in the case of nH,1 = nH,2 = 1, a viable alternative is to estimate the ratio of specificity constants by eq. 9 via measuring either individual substrate concentrations ([S1] and [S2]) or individual product concentrations ([P1] and [P2]) at a specific time point [40, 59, 92]. [S1,0] and [S2,0] in eq. 9 represent the initial concentrations of S1 and S2, respectively, before the assay starts. Taken together, this approach has several advantages; for example, there is no need to provide identical concentrations for all competing substrates, and one set of progress curves from an internal competition assay can be used to estimate the ratios of the specificity constants. The parameters estimated from eq. 9 are also in accordance with the calculations from eq. 7 or 8, without requiring the substrate concentrations to be the same. However, note that this approach can only be applied to cases where no cooperativity is observed. In addition, to obtain optimal estimations while maintaining a comparably competitive system, none of any substrate should be consumed either less than 30% or more than 70%.
| eq. 9 |
4. Case studies
4.1 Lysine acetyltransferases (KAT)
The majority of HATs can acetylate more than one residue on a single histone, and often multiple histones. Acetylation can have different biological outcomes depending on which residues are acetylated. This presents a unique problem in that every substrate could result in multiple products. Almost all substrates used in KAT assays contain more than one target residue, including even short peptide substrates [7].
For this reason our group has been interested in the challenge of finding meaningful ways to describe specificity through in vitro assays which have predictive power in cellular and in vivo models. Much of the mechanistic discussion covered above became clear to us during our studies with Gcn5 and Rtt109 [7, 14]. Gcn5 acetylates only H3K14 as an initial acetylation event but after H3K14 has been acetylated, Gcn5 can then acetylate multiple residues, while Rtt109-Vps75 will initially acetylate two residues. It is the ability to observe multiple products simultaneously that has allowed us to understand how these two enzymes function. Additionally, simultaneous detection of multiple histone residues enabled us to characterize the differences in selectivity between highly conserved KATs, p300 and CBP: these results included the observation that while both p300 and CBP target similar residues, CBP has a much stronger preference for H3K18 than p300, and that p300 is more efficient than CBP at targeting H3K9 [41].
After characterizing these two enzymes, our follow-up investigation [91] involved determining how the in vitro kinetics of p300 in response to drug treatment correlated with in vivo effects of that drug. p300 is a prolific KAT, which is able to acetylate multiple residues of the histone [41]. We had previously shown that some residues were acetylated by p300 with cooperative dependence on acetyl-CoA, where other residues were hyperbolically dependent on acetyl-CoA. We hypothesized that this cooperativity was monomeric and the result of a slow conformation change between two or more conformations. This was evident by the fact that, had it followed a classically cooperative model, the result would have been suppression of non-cooperative products, which was not observed. Modeling this type of kinetic mechanism based on the model for hexokinase (see below), we could show that competitive inhibitors are capable of actually stimulating activity by altering the distribution of enzyme conformations. We tested this hypothesis by looking at the kinetics of the p300 response to the drug C646, a small molecule that competes for binding to the acetyl-CoA binding pocket of p300 [93]. One of the novel findings from this study was that while C646 was originally classified as an inhibitor of p300, in vitro assays determined that there was actually a biphasic effect, with low concentrations of the drug able to stimulate p300 activity.
To test whether this effect could be seen in vivo, cells were treated with differing amounts of C646. The acetylation patterns of the cells were compared to the kinetic data obtained from the in vitro assays. It was found that the changes in histone acetylation at individual residues that were detected in vivo correlated well with the changes in kcat(app) at those residues in vitro. Specifically, looking at the ratio of the kcat(app) (eq. 8) of histone H3 lysine 18 (H3K18) compared to H3K23 (or K18/K23) matched the increase in acetylation of K18 compared to K23 in cells.
These results help to support the usage of a multi-substrate in vitro system for determining enzyme selectivity. In the case of p300, the histone itself presents multiple potential targets for p300 binding and acetylation. As the findings of this study apply to multi-substrate enzymes in general, it would suggest that in a complex system with a multi-target enzyme, determining the kcat(app) of that enzyme using an assay which contains multiple potential substrates could serve as a strong indicator of that enzyme’s selectivity in vivo.
4.2 Hexokinase
Hexokinase, originally called glucokinase was the first enzyme in which monomeric cooperativity was described: a sigmoidal dependence in the absence of a multiple substrate model. Hexokinase was shown to have a sigmoidal dependence on glucose but not on 2-deoxyglucose [94, 95]. Early models for this type of cooperativity suggested that cooperativity is due to a slow conformational step between two enzyme conformations where only one state was active (Figure 2) [94, 95]. Decades later NMR confirmed an order-disorder transition is responsible for the observed monomeric cooperativity [18]. This type of mechanism can also lead to difficulties in understanding the enzyme’s selectivity in a complex background. The presence of one substrate can affect the rate of another substrate’s turnover, by altering the concentration of the active enzyme complex [96]. In this model, anything that can bind in the active site could influence the conformational change and result in stimulation of enzyme activity, as modeled in Figure 2. These types of considerations highlight the difficulty in predicting enzyme selectivity in vivo that can act on multiple substrates, as their concentrations can vary independently of each other In this case, in order to accurately describe the selectivity between substrates for an enzyme displaying monomeric cooperativity, it becomes necessary to either know the free concentrations of all substrates or to know that at least one substrate is saturating. Without this information, the description of specificity becomes dependent on multiple substrates concentrations. Such a situation is problematic for analysis as a small amount of one substrate could increase the other substrate’s specificity, resulting in a decrease in its own selectivity.
Figure 2. Simulations of monomeric cooperativity and the link between second substrate/inhibitor activation.
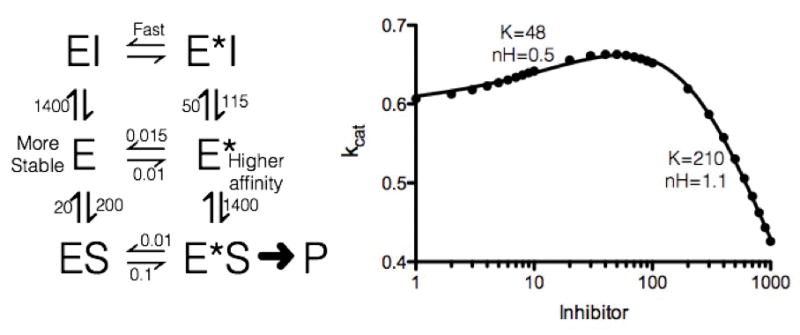
We used KinTek Explorer to simulate monomeric cooperativity based on hexokinase [97, 98]. We then added an inhibitor to the simulation that binds one state of the enzyme and facilitates the conversation of enzyme conformation. Parameters are in the figure (enzyme concentration was 0.06 E and 0.04 E*, and 100 S). This simulation was used to calculate steady-state rate under kcat conditions as a function of inhibitor concentration [I]. This data demonstrated a biphasic profile with an activation and inhibition phase. Right panel was fit using v/E=((kinitial+(kactive*[I])/KactivenH)/(1+ [I]((KactivenH+KinhibitnH)−1)) using data generated form KinTek Explorer.
5. Conclusions and outlook
Advancing technologies have facilitated the detection and quantitation of multiple substrate/product systems. Mass spectrometry, chromatography, NMR, and even DNA/RNA sequencing methods have all aided the advancement of this type of investigation. Using these systems, and carefully considering the conditions of the reactions, it is possible to obtain meaningful data from in vitro assays that can predict the behavior of enzymes in vivo. By understanding the caveats, as well as the kinetic analysis behind concepts like specificity and selectivity, we can continue to tailor multiple substrate and product experiments to more accurately model protein behavior in cellular systems.
Acknowledgments
Our work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01 GM102503 to A. J. A. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
Abbreviation
- KAT
lysine acetyltransferases
- PAH
polycyclic aromatic hydrocarbons
- LC
liquid chromatography
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- NMR
nuclear magnetic resonance spectrometry
- Gcn5
general control nonderepressible 5
- Rtt109
regulator of Ty1 transposition 109
- Vps75
vacuolar protein sorting 75
- CBP
CREB-binding protein
References
- 1.Porter TD, Coon MJ. Cytochrome P-450. Multiplicity of isoforms, substrates, and catalytic and regulatory mechanisms. J Biol Chem. 1991;266:13469–13472. [PubMed] [Google Scholar]
- 2.Wrighton SA, Schuetz EG, Thummel KE, Shen DD, Korzekwa KR, Watkins PB. The human CYP3A subfamily: practical considerations. Drug Metab Rev. 2000;32:339–361. doi: 10.1081/dmr-100102338. [DOI] [PubMed] [Google Scholar]
- 3.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allende J, Allende C. Protein kinases. 4. Protein kinase CK2: an enzyme with multiple substrates and a puzzling regulation. The FASEB Journal. 1995;9:313–323. doi: 10.1096/fasebj.9.5.7896000. [DOI] [PubMed] [Google Scholar]
- 5.Cornish-Bowden A. Fundamentals of enzyme kinetics, revised ed. Portland Press Ltd; London, U.K: 1995. [Google Scholar]
- 6.Copeland RA. Enzymes: A practical introduction to structure, mechanism, and data analysis. 2. John Wiley & Sons, Inc; New York: 2000. [Google Scholar]
- 7.Kuo YM, Andrews AJ. Quantitating the specificity and selectivity of Gcn5-mediated acetylation of histone H3. PloS one. 2013;8:e54896. doi: 10.1371/journal.pone.0054896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houston JB, Kenworthy KE. In vitro-in vivo scaling of CYP kinetic data not consistent with the classical Michaelis-Menten model. Drug Metab Disposition. 2000;28:246–254. [PubMed] [Google Scholar]
- 9.Ekins S, Stresser DM, Williams JA. In vitro and pharmacophore insights into CYP3A enzymes. Trends Pharmacol Sci. 2003;24:161–166. doi: 10.1016/s0165-6147(03)00049-x. [DOI] [PubMed] [Google Scholar]
- 10.Wienkers LC, Heath TG. Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov. 2005;4:825–833. doi: 10.1038/nrd1851. [DOI] [PubMed] [Google Scholar]
- 11.Grant PA, Eberharter A, John S, Cook RG, Turner BM, Workman JL. Expanded lysine acetylation specificity of Gcn5 in native complexes. J Biol Chem. 1999;274:5895–5900. doi: 10.1074/jbc.274.9.5895. [DOI] [PubMed] [Google Scholar]
- 12.Berndsen CE, Selleck W, McBryant SJ, Hansen JC, Tan S, Denu JM. Nucleosome recognition by the Piccolo NuA4 histone acetyltransferase complex. Biochemistry. 2007;46:2091–2099. doi: 10.1021/bi602366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abshiru N, Ippersiel K, Tang Y, Yuan H, Marmorstein R, Verreault A, Thibault P. Chaperone-mediated acetylation of histones by Rtt109 identified by quantitative proteomics. J Proteomics. 2013;81:80–90. doi: 10.1016/j.jprot.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuo YM, Henry RA, Huang L, Chen X, Stargell LA, Andrews AJ. Utilizing targeted mass spectrometry to demonstrate Asf1-dependent increases in residue specificity for Rtt109-Vps75 mediated histone acetylation. PloS one. 2015;10:e0118516. doi: 10.1371/journal.pone.0118516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rabin B. Co-operative effects in enzyme catalysis: a possible kinetic model based on substrate-induced conformation isomerization. Biochem J. 1967;102:22C–23C. doi: 10.1042/bj1020022c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herschlag D. The role of induced fit and conformational changes of enzymes in specificity and catalysis. Bioorg Chem. 1988;16:62–96. [Google Scholar]
- 17.Pezza JA, Stopa JD, Brunyak EM, Allen KN, Tolan DR. Thermodynamic analysis shows conformational coupling and dynamics confer substrate specificity in fructose-1, 6-bisphosphate aldolase. Biochemistry. 2007;46:13010–13018. doi: 10.1021/bi700713s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larion M, Salinas RK, Bruschweiler-Li L, Miller BG, Brüschweiler R. Order–disorder transitions govern kinetic cooperativity and allostery of monomeric human glucokinase. PLoS Biol. 2012;10:e1001452. doi: 10.1371/journal.pbio.1001452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards VH. The influence of high substrate concentrations on microbial kinetics. Biotechnol Bioeng. 1970;12:679–712. doi: 10.1002/bit.260120504. [DOI] [PubMed] [Google Scholar]
- 20.Hill GA, Robinson CW. Substrate inhibition kinetics: phenol degradation by Pseudomonas putida. Biotechnol Bioeng. 1975;17:1599–1615. [Google Scholar]
- 21.Tanner KG, Langer MR, Kim Y, Denu JM. Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. J Biol Chem. 2000;275:22048–22055. doi: 10.1074/jbc.M002893200. [DOI] [PubMed] [Google Scholar]
- 22.Lin Y, Lu P, Tang C, Mei Q, Sandig G, Rodrigues AD, Rushmore TH, Shou M. Substrate inhibition kinetics for cytochrome P450-catalyzed reactions. Drug Metab Disposition. 2001;29:368–374. [PubMed] [Google Scholar]
- 23.Cha S. Kinetics of enzyme reactions with competing alternative substrates. Mol Pharmacol. 1968;4:621–629. [PubMed] [Google Scholar]
- 24.Schellenberger V, Siegel RA, Rutter WJ. Analysis of enzyme specificity by multiple substrate kinetics. Biochemistry. 1993;32:4344–4348. doi: 10.1021/bi00067a025. [DOI] [PubMed] [Google Scholar]
- 25.Cleland W. The use of isotope effects to determine enzyme mechanisms. Arch Biochem Biophys. 2005;433:2–12. doi: 10.1016/j.abb.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 26.Gu H, Zhang S. Advances in kinetic isotope effect measurement techniques for enzyme mechanism study. Molecules. 2013;18:9278–9292. doi: 10.3390/molecules18089278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pascal RA, Baum MW, Wagner CK, Rodgers LR, Huang DS. Measurement of deuterium kinetic isotope effects in organic and biochemical reactions by natural abundance deuterium NMR spectroscopy. J Am Chem Soc. 1986;108:6477–6482. [Google Scholar]
- 28.Chan J, Lewis AR, Gilbert M, Karwaski MF, Bennet AJ. A direct NMR method for the measurement of competitive kinetic isotope effects. Nat Chem Biol. 2010;6:405–407. doi: 10.1038/nchembio.352. [DOI] [PubMed] [Google Scholar]
- 29.Liuni P, Olkhov-Mitsel E, Orellana A, Wilson DJ. Measuring kinetic isotope effects in enzyme reactions using time-resolved electrospray mass spectrometry. Anal Chem. 2013;85:3758–3764. doi: 10.1021/ac400191t. [DOI] [PubMed] [Google Scholar]
- 30.Bauer JE, Capone D. Effects of co-occurring aromatic hydrocarbons on degradation of individual polycyclic aromatic hydrocarbons in marine sediment slurries. Appl Environ Microbiol. 1988;54:1649–1655. doi: 10.1128/aem.54.7.1649-1655.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guha S, Peters CA, Jaffe PR. Multisubstrate biodegradation kinetics of naphthalene, phenanthrene, and pyrene mixtures. Biotechnol Bioeng. 1999;65:491–499. doi: 10.1002/(sici)1097-0290(19991205)65:5<491::aid-bit1>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 32.Zakett D, Flynn R, Cooks R. Chlorine isotope effects in mass spectrometry by multiple reaction monitoring. J Phys Chem. 1978;82:2359–2362. [Google Scholar]
- 33.Zea CJ, MacDonell SW, Pohl NL. Discovery of the archaeal chemical link between glycogen (starch) synthase families using a new mass spectrometry assay. J Am Chem Soc. 2003;125:13666–13667. doi: 10.1021/ja037298o. [DOI] [PubMed] [Google Scholar]
- 34.Ko KS, Zea CJ, Pohl NL. Strategies for the chemoenzymatic synthesis of deoxysugar nucleotides: substrate binding versus catalysis. J Org Chem. 2005;70:1919–1921. doi: 10.1021/jo048424p. [DOI] [PubMed] [Google Scholar]
- 35.Liesener A, Karst U. Monitoring enzymatic conversions by mass spectrometry: a critical review. Anal Bioanal Chem. 2005;382:1451–1464. doi: 10.1007/s00216-005-3305-2. [DOI] [PubMed] [Google Scholar]
- 36.Kline PC, Rezaee M, Lee TA. Determination of kinetic isotope effects for nucleoside hydrolases using gas chromatography/mass spectrometry. Anal Biochem. 1999;275:6–10. doi: 10.1006/abio.1999.4308. [DOI] [PubMed] [Google Scholar]
- 37.Hunt C, Gillani N, Farone A, Rezaei M, Kline PC. Kinetic isotope effects of nucleoside hydrolase from Escherichia coli. Biochim Biophys Acta: Proteins Proteomics. 2005;1751:140–149. doi: 10.1016/j.bbapap.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Pi N, Leary JA. Determination of enzyme/substrate specificity constants using a multiple substrate ESI-MS assay. J Am Soc Mass Spectrom. 2004;15:233–243. doi: 10.1016/j.jasms.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 39.Harris ME, Dai Q, Gu H, Kellerman DL, Piccirilli JA, Anderson VE. Kinetic isotope effects for RNA cleavage by 2′-O-transphosphorylation: nucleophilic activation by specific base. J Am Chem Soc. 2010;132:11613–11621. doi: 10.1021/ja103550e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu H, Zhang S, Wong KY, Radak BK, Dissanayake T, Kellerman DL, Dai Q, Miyagi M, Anderson VE, York DM. Experimental and computational analysis of the transition state for ribonuclease A-catalyzed RNA 2′-O-transphosphorylation. Proc Natl Acad Sci USA. 2013;110:13002–13007. doi: 10.1073/pnas.1215086110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henry RA, Kuo YM, Andrews AJ. Differences in specificity and selectivity between CBP and p300 acetylation of histone H3 and H3/H4. Biochemistry. 2013;52:5746–5759. doi: 10.1021/bi400684q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuo YM, Henry RA, Andrews AJ. A quantitative multiplexed mass spectrometry assay for studying the kinetic of residue-specific histone acetylation. Methods. 2014;70:127–133. doi: 10.1016/j.ymeth.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baeza J, Smallegan MJ, Denu JM. Site-specific reactivity of non-enzymatic lysine acetylation. ACS Chem Biol. 2015;10:122–128. doi: 10.1021/cb500848p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singleton DA, Thomas AA. High-precision simultaneous determination of multiple small kinetic isotope effects at natural abundance. J Am Chem Soc. 1995;117:9357–9358. [Google Scholar]
- 45.Chan J, Tang A, Bennet AJ. A stepwise solvent-promoted SNi reaction of α-D-glucopyranosyl fluoride: Mechanistic implications for retaining glycosyltransferases. J Am Chem Soc. 2011;134:1212–1220. doi: 10.1021/ja209339j. [DOI] [PubMed] [Google Scholar]
- 46.Pabis A, Kamiński R, Ciepielowski G, Jankowski S, Paneth P. Measurements of heavy-atom isotope effects using 1H NMR spectroscopy. J Org Chem. 2011;76:8033–8035. doi: 10.1021/jo201226g. [DOI] [PubMed] [Google Scholar]
- 47.Manning KA, Sathyamoorthy B, Eletsky A, Szyperski T, Murkin AS. Highly precise measurement of kinetic isotope effects using 1H-detected 2D [13C, 1H]-HSQC NMR spectroscopy. J Am Chem Soc. 2012;134:20589–20592. doi: 10.1021/ja310353c. [DOI] [PubMed] [Google Scholar]
- 48.Horenstein BA, Parkin DW, Estupinan B, Schramm VL. Transition-state analysis of nucleoside hydrolase from Crithidia fasciculata. Biochemistry. 1991;30:10788–10795. doi: 10.1021/bi00108a026. [DOI] [PubMed] [Google Scholar]
- 49.Knapp MJ, Klinman JP. Environmentally coupled hydrogen tunneling. Eur J Biochem. 2002;269:3113–3121. doi: 10.1046/j.1432-1033.2002.03022.x. [DOI] [PubMed] [Google Scholar]
- 50.Schwartz PA, Vetticatt MJ, Schramm VL. Transition state analysis of the arsenolytic depyrimidination of thymidine by human thymidine phosphorylase. Biochemistry. 2011;50:1412–1420. doi: 10.1021/bi101900b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Agrawal N, Hong B, Mihai C, Kohen A. Vibrationally enhanced hydrogen tunneling in the Escherichia coli thymidylate synthase catalyzed reaction. Biochemistry. 2004;43:1998–2006. doi: 10.1021/bi036124g. [DOI] [PubMed] [Google Scholar]
- 52.Sen A, Yahashiri A, Kohen A. Triple isotopic labeling and kinetic isotope effects: Exposing H-transfer steps in enzymatic systems. Biochemistry. 2011;50:6462–6468. doi: 10.1021/bi2003873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clayton LK, Goodman MF, Branscomb EW, Galas DJ. Error induction and correction by mutant and wild type T4 DNA polymerases. Kinetic error discrimination mechanisms. J Biol Chem. 1979;254:1902–1912. [PubMed] [Google Scholar]
- 54.Bloom LB, Otto MR, Eritja R, Reha-Krantz LJ, Goodman MF, Beechem JM. Pre-steady-state kinetic analysis of sequence-dependent nucleotide excision by the 3′-exonuclease activity of bacteriophage T4 DNA polymerase. Biochemistry. 1994;33:7576–7586. doi: 10.1021/bi00190a010. [DOI] [PubMed] [Google Scholar]
- 55.KiáLee N, RanáKoh H, YoungáHan K, KeunáKim S. Single-molecule, real-time measurement of enzyme kinetics by alternating-laser excitation fluorescence resonance energy transfer. Chem Commun. 2010;46:4683–4685. doi: 10.1039/c002666b. [DOI] [PubMed] [Google Scholar]
- 56.Bertram JG, Oertell K, Petruska J, Goodman MF. DNA polymerase fidelity: comparing direct competition of right and wrong dNTP substrates with steady state and pre-steady state kinetics. Biochemistry. 2010;49:20–28. doi: 10.1021/bi901653g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang W. Nucleases: diversity of structure, function and mechanism. Q Rev Biophys. 2011;44:1–93. doi: 10.1017/S0033583510000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yandek LE, Lin HC, Harris ME. Alternative substrate kinetics of Escherichia coli ribonuclease P. Determination of relative rate constants by internal competition. J Biol Chem. 2013;288:8342–8354. doi: 10.1074/jbc.M112.435420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin HC, Yandek LE, Gjermeni I, Harris ME. Determination of relative rate constants for in vitro RNA processing reactions by internal competition. Anal Biochem. 2014;467:54–61. doi: 10.1016/j.ab.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zaug AJ, Grosshans CA, Cech TR. Sequence-specific endoribonuclease activity of the Tetrahymena ribozyme: enhanced cleavage of certain oligonucleotide substrates that form mismatched ribozyme-substrate complexes. Biochemistry. 1988;27:8924–8931. doi: 10.1021/bi00425a008. [DOI] [PubMed] [Google Scholar]
- 61.Bergeron LJ, Ouellet J, Perreault JP. Ribozyme-based gene-inactivation systems require a fine comprehension of their substrate specificities; the case of delta ribozyme. Curr Med Chem. 2003;10:2589–2597. doi: 10.2174/0929867033456486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Champoux JJ, Schultz SJ, Ribonuclease H. properties, substrate specificity and roles in retroviral reverse transcription. FEBS J. 2009;276:1506–1516. doi: 10.1111/j.1742-4658.2009.06909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 64.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 65.Malone JH, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011;9:34. doi: 10.1186/1741-7007-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cooper DA, Jha BK, Silverman RH, Hesselberth JR, Barton DJ. Ribonuclease L and metal-ion–independent endoribonuclease cleavage sites in host and viral RNAs. Nucleic Acids Res. 2014;42:5202–5216. doi: 10.1093/nar/gku118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cooper DA, Banerjee S, Chakrabarti A, García-Sastre A, Hesselberth JR, Silverman RH, Barton DJ. Ribonuclease L targets distinct sites in Influenza A virus RNAs. J Virol. 2014;89:2764–2776. doi: 10.1128/JVI.02953-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Han JQ, Wroblewski G, Xu Z, Silverman RH, Barton DJ. Sensitivity of hepatitis C virus RNA to the antiviral enzyme ribonuclease L is determined by a subset of efficient cleavage sites. J Interferon Cytokine Res. 2004;24:664–676. doi: 10.1089/jir.2004.24.664. [DOI] [PubMed] [Google Scholar]
- 69.Fang H, Taylor JS. Serial analysis of mutation spectra (SAMS): a new approach for the determination of mutation spectra of site-specific DNA damage and their sequence dependence. Nucleic Acids Res. 2008;36:6004–6012. doi: 10.1093/nar/gkn595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Köster H, Tang K, Fu DJ, Braun A, van den Boom D, Smith CL, Cotter RJ, Cantor CR. A strategy for rapid and efficient DNA sequencing by mass spectrometry. Nat Biotechnol. 1996;14:1123–1128. doi: 10.1038/nbt0996-1123. [DOI] [PubMed] [Google Scholar]
- 71.Monforte JA, Becker CH. High-throughput DNA analysis by time-of-flight mass spectrometry. Nat Med. 1997;3:360–362. doi: 10.1038/nm0397-360. [DOI] [PubMed] [Google Scholar]
- 72.Edwards JR, Ruparel H, Ju J. Mass-spectrometry DNA sequencing. Mutat Res - Fund Mol M. 2005;573:3–12. doi: 10.1016/j.mrfmmm.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 73.Quinlivan EP, Gregory JF. DNA methylation determination by liquid chromatography–tandem mass spectrometry using novel biosynthetic [U-15N] deoxycytidine and [U-15N] methyldeoxycytidine internal standards. Nucleic Acids Res. 2008;36:e119. doi: 10.1093/nar/gkn534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chan CT, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010;6:e1001247. doi: 10.1371/journal.pgen.1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Su D, Chan CT, Gu C, Lim KS, Chionh YH, McBee ME, Russell BS, Babu IR, Begley TJ, Dedon PC. Quantitative analysis of ribonucleoside modifications in tRNA by HPLC-coupled mass spectrometry. Nat Protoc. 2014;9:828–841. doi: 10.1038/nprot.2014.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Murtola M, Wenska M, Strömberg R. PNAzymes that are artificial RNA restriction enzymes. J Am Chem Soc. 2010;132:8984–8990. doi: 10.1021/ja1008739. [DOI] [PubMed] [Google Scholar]
- 77.Zimmermann K, Mannhalter JW. Technical aspects of quantitative competitive PCR. BioTechniques. 1996;21:268–279. doi: 10.2144/96212rv01. [DOI] [PubMed] [Google Scholar]
- 78.Rottman J, Tompkins W, Tompkins M. A reverse transcription-quantitative competitive polymerase chain reaction (RT-qcPCR) technique to measure cytokine gene expression in domestic mammals. Vet Pathol. 1996;33:242–248. doi: 10.1177/030098589603300217. [DOI] [PubMed] [Google Scholar]
- 79.Nguyen UT, Bittova L, Müller MM, Fierz B, David Y, Houck-Loomis B, Feng V, Dann GP, Muir TW. Accelerated chromatin biochemistry using DNA-barcoded nucleosome libraries. Nat Methods. 2014;11:834–840. doi: 10.1038/nmeth.3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Haldane JBS. Enzymes. The MIT Press; 1965. [Google Scholar]
- 81.Bender ML, Kezdy FJ. Mechanism of action of proteolytic enzymes. Annu Rev Biochem. 1965;34:49–76. doi: 10.1146/annurev.bi.34.070165.000405. [DOI] [PubMed] [Google Scholar]
- 82.Brot FE, Bender ML. Use of the specificity constant of alpha-chymotrypsin. J Am Chem Soc. 1969;91:7187–7191. [Google Scholar]
- 83.Fersht A. Catalysis, binding and enzyme-substrate complementarity. Proc R Soc Lond B Biol Sci. 1974;187:397–407. doi: 10.1098/rspb.1974.0084. [DOI] [PubMed] [Google Scholar]
- 84.Monod J, Changeux JP, Jacob F. Allosteric proteins and cellular control systems. J Mol Biol. 1963;6:306–329. doi: 10.1016/s0022-2836(63)80091-1. [DOI] [PubMed] [Google Scholar]
- 85.Conway A, Koshland DE. Negative cooperativity in enzyme action. Binding of diphosphopyridine nucleotide to glyceraldehyde-3-phosphate dehydrogenase. Biochemistry. 1968;7:4011–4023. doi: 10.1021/bi00851a031. [DOI] [PubMed] [Google Scholar]
- 86.Levitzki A, Koshland D. Negative cooperativity in regulatory enzymes. Proc Natl Acad Sci USA. 1969;62:1121–1128. doi: 10.1073/pnas.62.4.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hill AV. The combinations of haemoglobin with oxygen and with carbon monoxide. I. Biochem J. 1913;7:471–480. doi: 10.1042/bj0070471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ainslie GR, Shill JP, Neet KE. Transients and cooperativity: a slow transition model for relating transients and cooperative kinetics of enzymes. J Biol Chem. 1972;247:7088–7096. [PubMed] [Google Scholar]
- 89.Ferdinand W. The interpretation of non-hyperbolic rate curves for two-substrate enzymes. A possible mechanism for phosphofructokinase. Biochem J. 1966;98:278–283. doi: 10.1042/bj0980278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cornish-Bowden A, Cardenas ML. Specificity of non-Michaelis-Menten enzymes: necessary information for analyzing metabolic pathways. J Phys Chem B. 2010;114:16209–16213. doi: 10.1021/jp106968p. [DOI] [PubMed] [Google Scholar]
- 91.Henry RA, Kuo YM, Bhattacharjee V, Yen TJ, Andrews AJ. Changing the selectivity of p300 by acetyl-CoA modulation of histone acetylation. ACS Chem Biol. 2015;10:146–156. doi: 10.1021/cb500726b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cook PF, Cleland WW. Enzyme kinetics and mechanisms. Taylor & Francis Group; New York, NY, USA: 2007. [Google Scholar]
- 93.Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, Larocca C, Saldanha SA, Abagyan R, Sun Y, Meyers DJ, Marmorstein R, Mahadevan LC, Alani RM, Cole PA. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cárdenas M, Rabajille E, Niemeyer H. Kinetic cooperativity of glucokinase with glucose. Arch Biol Med Exp (Santiago) 1979;12:571–580. [PubMed] [Google Scholar]
- 95.Cardenas ML, Rabajille E, Niemeyer H. Fructose is a good substrate for rat liver’glucokinase’(hexokinase D) Biochem J. 1984;222:363–370. doi: 10.1042/bj2220363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cardenas ML, Rabajille E, Niemeyer H. Suppression of kinetic cooperativity of hexokinase D (glucokinase) by competitive inhibitors. Eur J Biochem. 1984;145:163–171. doi: 10.1111/j.1432-1033.1984.tb08536.x. [DOI] [PubMed] [Google Scholar]
- 97.Larion M, Miller BG. Homotropic allosteric regulation in monomeric mammalian glucokinase. Arch Biochem Biophys. 2012;519:103–111. doi: 10.1016/j.abb.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lin SX, Neet KE. Demonstration of a slow conformational change in liver glucokinase by fluorescence spectroscopy. J Biol Chem. 1990;265:9670–9675. [PubMed] [Google Scholar]