

Abstract
Background:
Stroke is one of the most common causes of death and the leading cause of disability in adults. Cerebral ischemia/reperfusion injury causes cerebral edema, hemorrhage, and neuronal death.
Aims:
In post-ischemic reperfusion, free radical production causes brain tissue damage by oxidative stress. Pregabalin, an antiepileptic agent was shown to have antioxidant effects. The aim of this study was to evaluate the neuroprotective and antioxidant effects of pregabalin on ischemia and reperfusion in rat brain injury.
Study Design:
Animal experimentation.
Methods:
Male Wistar rats weighing (250–300 g) were randomly divided into six groups, each consisting of 6 rats: control (C), pregabalin (P), ischemia (I), pregabalin + ischemia (PI), ischemia + reperfusion (IR) and ischemia + reperfusion + pregabalin (PIR). Rats were initially pre-treated with 50 mg/kg/d pregabalin orally for two days. Then, animals that applied ischemia in I, PI, IR and PIR groups were exposed to carotid clamping for 30 minutes and 20 minutes reperfusion was performed in the relevant reperfusion groups.
Results:
NR2B receptor levels were significantly lower in the PIR group in comparison to the IR group. In the PIR group, Thiobarbituric acid reactive substance (TBARS) level had statistically significant decrease compared with IR group. Glutathione peroxidase (GSH-PX) levels were also significantly increased in the PIR group compared with I, IR and control groups. In the PI and PIR groups, catalase (CAT) levels were also significantly increased compared with I and IR groups (p=0.03 and p=0.07, respectively).
Conclusion:
Pregabalin may protect the damage of oxidative stress after ischemia + reperfusion. This result would illuminate clinical studies in the future.
Keywords: Cerebral ischemia, ischemia-reperfusion, neuroprotective effects, pregabalin
Stroke is one of the most common causes of death and the leading cause of disability in adults. The incidence of stroke increases with age. Early neurological deterioration occurs in up to one-third of patients (1). Efficacious stroke treatment requires recanalization of the occluded cerebral blood vessels. However, reperfusion after cerebral ischemia may also contribute to brain injury. Cerebral ischemia/reperfusion injury causes cerebral edema, hemorrhage, and neuronal death.
Increased reactive oxygen species (ROS) production overwhelms endogenous antioxidant systems and leads to oxidative stress. Free radicals induce peroxidation, and low levels of endogenous antioxidants cause brain tissue to become highly vulnerable to the effects of oxidative stress by high oxygen consumption during reperfusion (2).
Oxidative stress occurs when the antioxidant defense system is overwhelmed by the production of ROS. Antioxidants and other cell redox state modulating enzyme systems act as the first-line of defense against ROS in all cellular and extracellular compartments (3). Superoxide dismutase (SOD), CAT, and GSH-PX are the most important antioxidant enzymes against ROS. On the other hand, markers of lipid peroxidation, such as TBARS, which react with malondialdehyde (MDA), and nitric oxide (NO) are increased in oxidative stress (4).
Experimental and clinical studies have shown that antioxidants can protect the development of ischemia/reperfusion injury by using endogenous antioxidant enzymes and the suppression of free radical generation (5).
It was shown that NMDA receptors are important mediators in ischemia (6). These receptors have three subunits, namely NR1 (a–h), NR2 (A–D), and NR3 (A–B). NR1 and NR3 subunits do not have a binding site for excitatory amino acids. Only NR2 subunits have excitatory amino acid binding sites (7).
In acute ischemia, sodium (Na++), chloride (Cl−) and calcium (Ca++) ions increase in cells secondary to interactions with glutamate receptors. NOS enzyme activity decreases in ischemic phase of ischemia, and then increases during reperfusion (8–10).
Some antiepileptic drugs were shown to have neuroprotective effects by reducing cell death after cerebral ischemia (11). The antiepileptic drug pregabalin, which has a gamma-aminobutyric acid-like (GABA) structure, potently binds the CaVα2δ-1 subunit of voltage-gated calcium channels and reduces Ca++ influx at the presynaptic nerve endings and reduces the release of several neurotransmitters such as glutamate and noradrenaline (12–15). In cerebral ischemia, the main reason for cell death is the increase of excitatory amino acids (5). Anti-inflammatory and anti-apoptotic effects of pregabalin were previously shown by the histopathological and biochemical methods, indicating that pregabalin might be a neuroprotective agent for the treatment of ischemia-reperfusion injury (16). This study aims to investigate whether pregabalin exerts neuroprotective effects on ischemia and reperfusion injury of brain tissue.
MATERIALS AND METHODS
Animals
Adult male Wistar rats weighing 250–300 g were purchased from the animal research laboratory of our university. Animals were allowed to acclimatize for at least 7 days prior to experimentation. Rats were group housed (six rats in a cage) under 12:12 h light/dark cycles at room temperature (24±1°C) and a relative humidity of 50±10% with access to food and water ad libitum. All experimental procedures were approved by the local animal care committee (Ethic number: 16/06/2009-15/3) and carried out in accordance with the guidelines of the European Communities Council Directive (86/609/EEC).
Experimental procedures
Rats were randomly assigned into one of the following six groups: control, pregabalin, ischemia, pregabalin + ischemia, ischemia and reperfusion, and pregabalin + ischemia and reperfusion. Before any interventions, rats in the P, PI and PIR groups were pre-treated with 50 mg/kg/d pregabalin (17) (Pfizer Pharmaceuticals, New York, NY, USA) orally for two days to reach maximum plasma concentration (18,19). We followed the clinical dosage that is commonly used for neuropathic pain (40–80 mg/kg/d). The other groups were only administered distilled water. After this pretreatment phase, animals in the I, PI, IR and PIR groups were exposed to carotid clamping.
For carotid clamping, the rats were anaesthetized using intraperitoneal injection of 10% ketamine plus 2% xylazine mixture. Both common carotid arteries were exposed over a midline incision and dissection on the ventral side of the neck between the sternocleidomastoid and sternohyoid muscles parallel to the trachea. The trachea was exposed followed by the left and right common carotid artery. The right common carotid artery was freed from all muscles, ligaments, and its adventitial sheath, and the vagus nerve was carefully separated and maintained. The induction of ischemia was performed by occluding the right internal carotid artery with clamps. Ischemia was reversed after 30 minutes by removing the internal artery clamps (20). During all procedures, animals were observed for the maintenance of dilated pupils, the absence of a cornea reflex on exposure to light and the maintenance of rectal temperature at 37±0.5°C. The development of hypothermia was prevented using a heating lamp on the surgery table. Animals that did not meet these criteria and or had seizures were excluded. Sham control animals underwent surgery without carotid clamping. Unless otherwise stated, for all of the parameters, rats were decapitated and the brains were isolated after 20 minutes of reperfusion (21).
Tissue preparation
The hippocampus and frontal lobe of each rat was dissected on ice pickets which were wetted with cold phosphate buffer (50 mM), and the tissue was placed in eppendorf tubes that were filled with phosphate buffer (50 mM) and frozen at −80°C until assays.
Each hippocampus and frontal lobe was weighed and homogenized in ice-cold buffer [50 mm Tris-HCl (pH 7.5), 0.15 M NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 25 μg/mL leupeptin, 25 μg/mL aprotinin, 1 mM sodium orthovanadate, 10 μM benzamidine and 4 mM p-nitrophenyl phosphate]. After obtaining the supernatant fractions, upon centrifugation of the homogenates at 10000xg for 10 min, an aliquot was taken for protein determination (22). The oxidative stress parameters TBARS, NO and anti-oxidative enzymes SOD, GSH-PX and CAT were determined from the frontal lobes of rats. The receptor expressions NR2A and NR2B were determined from the hippocampi of rats.
Western blotting
Anti-NR2A, anti-NR2B were purchased from Millipore (Billerica, MA, USA), mouse monoclonal antibody to β-actin was purchased from Abcam Company (Cambridge, USA) and prestained molecular weight marker was purchased from Sigma-Aldrich Co. (Steinheim, Germany). All other reagents were analytical grade or the highest grade available. Equal amounts of protein for each sample (50 μg of protein per lane) were separated by SDS/PAGE on 7.5% minigels, blotted electrophoretically to immobilen membrane, and incubated in tris-buffered saline for 30 min with Tween 20 (TBST) [50 mm Tris-HCl (pH 7.5–8.0), 150 mM NaCl, and 0.1% Tween 20] containing 3% bovine serum albumin (BSA). Blots were incubated overnight with anti-NR2A (1/500), anti-NR2B (1/500) and finally with anti-β-actin (1/5000) in 1% BSA. Blots were subjected to three additional 10-min washings in TBST and were incubated with alkaline phosphatase conjugated monoclonal anti-rabbit IgG (1/10000) in 1% BSA for 1 h at room temperature, and 3 additional washings were performed with TBST for 10 min. The membrane was incubated in 20 mL of fresh reagent solution (BCIP/NBT) until color development. Images of immunoblots were analyzed with a computerized image analysis system (Kodak MM 2000 Image Station, USA). Immunoblotting for β-actin was used as an internal standard to confirm equal protein loading and sample transferring. The ratios of such subunit/β-actin of all the individual samples (control group and simultaneously other experiment groups) were calculated for each blot. The mean control group values for each antibody was assumed as 100 and, each value of experimental groups was proportioned with the control group in order to simplify the values so that the data were expressed as a percentage of the control group (23).
Determination of oxidative products and antioxidant enzymes
Measurement of SOD was based on the principle that xanthine reacts with xanthine oxidase to generate superoxide radicals, which react with 2-(4-iodophenyl)-3-(4-nitrophenol)-5-phenyltetrazolium chloride (INT) to form a red formazan dye. SOD activity was measured by the degree of inhibition of this reaction (24). A commercial kit (RANDOX) applied to an autoanalyzer (Olympus AU 2700, USA) was used to determine the activities of SOD.
The determination of GSH-PX activity was based on the method of Paglia and Valentine (25). Briefly, the method was as follows: GSH-PX catalyzed the oxidation of glutathione by cumene hydroperoxide. In the presence of glutathione reductase and NADPH, the oxidized glutathione was immediately converted to the reduced form with the concomitant oxidation of NADPH to NADP+. The decrease in absorbance of NADPH was measured at a wavelength of 340 nm. A commercial kit (RANSEL) which was applied to an autoanalyzer (Olympus AU 2700, USA) was used to determine the activities of GSH-PX.
Catalase activity was measured according to the method of Aebi (26). The principle of the assay is based on the determination of the rate constant (k, s−1) of decomposition of hydrogen peroxide by the enzyme catalase. The rate constant was calculated from the formula: k=(2.3/_t)(a/b) log(A1/A2), where A1 and A2 are the absorbance values of hydrogen peroxide at t1 (0 second) and t2 (15 seconds), a is the dilution factor, and b is the protein level of tissue homogenate. A Shimadzu UV-1601 spectrophotometer (Shimadzu, Kyoto, Japan) was used to estimate the activities of the enzyme CAT.
Nitric oxide activity was measured by using a commercial CAYMAN Chemical Nitrate/Nitrite Colorimetric Assay Kit (USA). This kit uses a simple two-step process for the measurement of total nitrate/nitrite concentration. The first step involved the conversion of nitrate to nitrite utilizing nitrate reductase. The second step was the addition of the Griess Reagents, which converted nitrite into a deep purple azo compound. Photometric measurement of the absorbance at 540 nm due to this azo chromophore accurately determined the NO2 concentration.
TBARS activity was measured by using a commercial CAYMAN Chemical TBARS Assay Kit (USA). The MDA-TBA adduct formed by the reaction of MDA and TBA under high temperature (1000 °C) and acidic conditions was measured colorimetrically at 530–540 nm. For the colorimetric evaluation of NO and TBARS, the Organon Teknika Reader 530 was used as the plate reader.
Statistical analyses
SPSS 18.0 (SPSS Inc., Chicago, Illinois, USA) for Windows was used for statistical analyses. Data were given as mean±standard deviation (SD). Kruskal-Wallis test was used to compare the values between groups. A 5% margin of error considering the 95% confidence interval p<0.05 was taken as statistically significant. Significance was set at p<0.01 for Bonferroni corrections.
RESULTS
For evaluation of the protein expression of the receptors, data were normalized for β-actin compared to the mean of the control group. Optical densities of other groups were calculated in comparison with the control value, and results were given as % concentration values (Table 1).
TABLE 1.
NR2A and NR2B levels of rat’s frontal lobe
| Groups | NR2A | NR2B |
|---|---|---|
| Control | 100.1±22 | 100.2±12 |
| Ischemia | 134.4±25.7 | 144.8±10.4a |
| Ischemia + Reperfusion | 145.9±6.9 | 229.3±35.6a |
| Pregabalin + Ischemia | 166.8±28.4 | 117.3±9.3c |
| Pregabalin + Ischemia + Reperfusion | 148.1±22.8 | 140±9.7b |
| Pregabalin | 129.9±28.9 | 95±3.4 |
p<0.05 vs control;
p<0.05 vs to IR(cerebral ischemia reperfusion-injured rats);
p<0.05 vs to I (cerebral ischemia rats without reperfusion)
NR2A expressions showed no significant differences between groups (p>0.05) (Figure 1, Table 1). However, NR2B receptor expression was significantly higher in the ischemia group compared to controls (p=0.02). The IR group also showed significantly increased NR2B receptor expression when compared to the control group (p=0.02). NR2B receptor levels in the PI group were lower than levels in the I group (borderline significant) (p=0.05). NR2B receptor expression was significantly lower in the PIR group in comparison to the IR group (p=0.02) (Figure 1, Table 1).
FIG. 1.
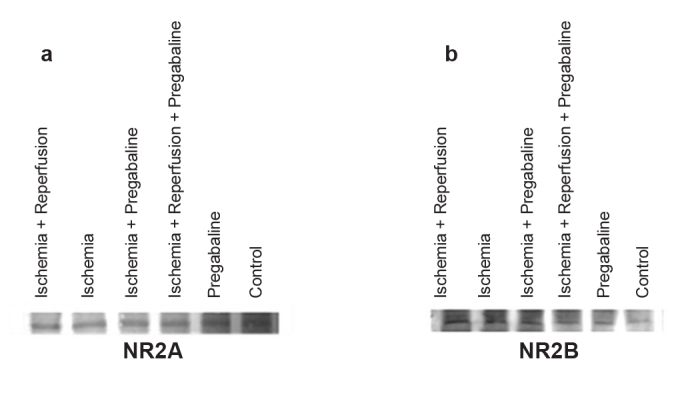
a, b. Bands of NR2A receptors (a), bands of NR2B receptors (b)
Oxidative stress parameters are shown in Table 2. TBARS levels were similar in the C and P groups; however, there were significant increases in I and IR groups as expected (p=0.004 and p=0.004, respectively). Mean TBARS levels in the PI and I groups were comparable, with no significant differences (p=0.81). However TBARS levels were decreased in the PIR group when compared to the IR group (borderline significant) (p=0.05). Mean NO levels only showed a significant increase in the ischemia group compared to controls (p=0.04).
TABLE 2.
Oxidative stress parameters of rat’s frontal lobe
| Groups | TBARS (µmolar/grprotein) | NO (µmolar/grprotein) | GSH-PX (nmol/grprotein) | SOD (U/grprotein) | CAT (kU/grprotein) |
|---|---|---|---|---|---|
| Pregabalin + Ischemia (n=6) | 23.16±2.2 | 1.88±0.2 | 34.26±2.4 | 2.47±0.3 | 4.66±0.3c |
| Ischemia (n=6) | 25.12±2.9a | 6.70±1.4a | 33.38±3.7a | 1.90±0.1a | 3.88±0.3a |
| Ischemia+ Reperfusion (n=6) | 23.84±1.2a | 4.49±0.7 | 35.05±3.0a | 2.25±0.3a | 5.57±0.5a |
| Pregabalin + Ischemia + Reperfusion (n=6) | 17.75±0.9b | 2.06±0.4 | 48.51±1.1b,c | 2.54±0.2 | 6.77±0.7b |
| Pregabalin (n=6) | 11.39±1.1 | 0.55±0.2 | 49.23±1.8 | 2.03±0.2 | 7.46±0.4 |
| Control (n=6) | 11.09±0.5 | 0.74±0.3 | 46.89±1.8 | 2.53±0.1 | 7.40±0.4 |
p<0.05 vs control;
p<0.05 vs to IR (cerebral ischemia reperfusion-injured rats);
p<0.05 vs to I (cerebral ischemia rats without reperfusion)
There was a statistically significant decrease in GSH-PX values in the I group when compared with the control and P groups (p=0.02). No statistically significant difference was detected among the I, PI, and IR groups. However, the mean GSH-PX level in the PIR group was significantly higher than in the I and IR groups (p=0.02 and p=0.004, respectively). In a similar way, a significant SOD reduction was observed in the I group when compared with the C group (p=0.02). In the IR group, the results were similar to controls. Reperfusion after ischemia reduced the SOD levels up to the values of the control group. Although SOD levels were higher in the PIR group than in the IR group, this did not reach statistical significance (p=0.26).
The most significant CAT level decrease was in the I group in comparison to the control and pregabalin groups (p=0.004 and p=0.004). The mean CAT level was also higher in PI group when compared with the I group (p=0.03) and in the PIR group compared with the IR group (p=0.07).
DISCUSSION
Recently, various approaches for the neuroprotection of cerebral ischemia were supported. Ischemia treatment requires recanalization of occluded cerebral blood vessels. However, cerebral ischemia/reperfusion may also contribute to brain injury. After cerebral ischemia/reperfusion, the production of reactive oxygen species dramatically increases and leads to oxidative stress (27).
Oxidative stress reactions and membrane lipid peroxidation cause oxidative damage of the brain as a result of the formation of free oxygen radicals (28). In rats, following brain ischemia and reperfusion damage, endogenous antioxidant enzymes such as SOD, CAT, and GSH-PX were found to be reduced (29) and levels of the lipid peroxidation markers nitric oxide and TBARS were increased (4).
Several antioxidant agents including CAPE, theanine and caffeine, selenium and melatonin have been reported to prevent brain ischemia and reperfusion damage (30–32). Neuroprotective drugs affect areas of ischemic tissue that are not irreversibly damaged. The synergistic effect of ischemic tissue reperfusion therapies with the use of neuroprotective agents may increase the protective effects of this damage.
Pregabalin, a GABA analogue, is a potent new generation antiepileptic drug that is used for the treatment of partial onset seizures, painful diabetic neuropathy, and post-herpetic neuralgia (33). Besides these clinical indications, it has some new properties that we need to focus on.
Our data support the potential use of pregabalin as a neuroprotective factor following cerebral ischemia and reperfusion secondary to its antioxidant, anti-inflammatory and anti-apoptotic effects (18).
The GABA analogue pregabalin is effective in the prevention of injury by various factors. For example, the anti-apoptotic and neuroprotective effects of pregabalin have been proved. Pregabalin decreased the production of caspase-3 and phosphorylated p38 MAPK, as well as decreasing the proliferation of astrocytes. This influence on spinal cord injury might be a possible approach for achieving neuroprotection following central nervous system trauma, including spinal cord injury (18). These data support the neuroprotective effect of pregabalin on spinal ischemia.
The protective effect of gabapentin (another GABA analogue) against oxidative damage in mice by using GABAergic modulation was also shown (34). Hence, pregabalin may protect tissues from oxidative damage and neuronal damage by using GABAergic modulation.
First, we analyzed the antioxidant effects by measuring the expression of NR2A and NR2B receptors. The increased production of these hippocampal receptors on ischemic brain damage was previously shown (35). We observed the biomarkers of oxidative stress (TBARS and NO), and antioxidant activities of SOD, GSH-PX and CAT. In groups treated with an antioxidant drug, the antioxidant enzymes activity must be increased to compensate for this activity (36–38).
Memantin, which is an NMDA receptor antagonist, was shown to be effective for the prophylaxis of ischemic injury. The combination of memantin with a calcium channel blocker nimodipin increased this effect (39). This shows the importance of blocking NMDA receptors for ischemia prophylaxis. We used pregabalin in our study to reduce NMDA receptors and the damage secondary to calcium increase in cells with impending ischemia.
In our study, there were no significant changes in NR2A receptor expression, but NR2B receptor expression was significantly higher in the ischemia and ischemia reperfusion groups in comparison to controls. In the I and IR groups, all of the antioxidant enzymes were decreased and biomarkers of oxidative damage (TBARS and NO) were increased as expected. According to these findings oxidative damage was present in brain tissues.
In a study of 24 hour reperfusion after middle cerebral artery occlusion, the enzyme CAT was shown to be significantly decreased (40). Our study demonstrated that CAT activity in the I and IR groups was significantly decreased as well. Pregabalin treatment significantly increased that activity in the PI and PIR groups.
Another study investigated the antioxidant effect of calycosin at 24 h of reperfusion after 2 h of middle cerebral artery occlusion, and demonstrated that treatment with calycosin led to a significant increase in the activities of GSH-PX when compared with the model group (41). In our study, pregabalin administration increased the GSH-PX and CAT activity in the PIR group. TBARS, which is an indicator of damage, decreased in the PIR group with borderline significance. In our study, damage groups with pregabalin treatment had fewer NR2B receptors. Therefore, pregabalin may have antioxidant activity.
In conclusion, pregabalin treatment after cerebral ischemia and reperfusion decreased lipid peroxidation (TBARS) and the activity of antioxidant enzymes such as GSH-PX and CAT with no effects on NO levels and SOD activity. According to these findings, Pregabalin may be useful for the prevention of ischemia reperfusion injury in cerebral infarction cases which limit the life of patients. Thereby, the disability and mortality of cerebral infarction can be reduced using the neuroprotective agent pregabalin. However, longer treatment with pregabalin administration is needed for further studies.
Footnotes
Ethics Committee Approval: Ethics committee approval was received for this study from the ethics committee of the Local Animal Care Committee and carried out in accordance with the guidelines of the European Communities Council Directive (86/609/EEC).
Informed consent: N/A.
Peer-review: Externally peer-reviewed.
Author contributions: Concept - S.A.; Design - S.D., S.A.; Supervision - S.D.; Resource - S.A., H.A.; Materials - S.A., H.A., İ.O.; Data Collection &/or Processing - S.A., H.A.; Analysis &/or Interpretation D.K.D., İ.O.; Literature Search - S.A., H.A., S.D.; Writing - S.A., H.A.; Critical Reviews - H.A., S.D.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: This research has been supported by the Süleyman Demirel University Unit of Scientific Research (Project Number: 17U3-YL-08).
REFERENCES
- 1.Siegler JE, Martin-Schild S. Early Neurological Deterioration (END) after stroke: the END depends on the definition. Int J Stroke. 2011;6:211–2. doi: 10.1111/j.1747-4949.2011.00596.x. http://dx.doi.org/10.1111/j.1747-4949.2011.00596.x. [DOI] [PubMed] [Google Scholar]
- 2.Mohagheghi F, Khalaj L, Ahmadiani A, Rahmani B. Gemfibrozil pretreatment affecting antioxidant defense system and inflammatory, but not Nrf-2 signaling pathways resulted in female neuroprotection and male neurotoxicity in the rat models of global cerebral ischemia-reperfusion. Neurotox Res. 2013;23:225–37. doi: 10.1007/s12640-012-9338-3. http://dx.doi.org/10.1007/s12640-012-9338-3. [DOI] [PubMed] [Google Scholar]
- 3.Volkova IuV, Sukhova LL, Davydov VV, Goloborod’ko AV. The activity of the first line enzymes of the antioxidant defence in the liver of pubertal rats during stress. Biomed Khim. 2012;58:573–8. doi: 10.18097/pbmc20125805573. http://dx.doi.org/10.18097/pbmc20125805573. [DOI] [PubMed] [Google Scholar]
- 4.Wu JQ, Kosten TR, Zhang XY. Free radicals, antioxidant defense systems, and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;46:200–6. doi: 10.1016/j.pnpbp.2013.02.015. http://dx.doi.org/10.1016/j.pnpbp.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 5.Pérez-Asensio FJ, de la Rosa X, Jiménez-Altayó F, Gorina R, Martínez E, Messeguer A, et al. Antioxidant CR-6 protects against reperfusion injury after a transient episode of focal brain ischemia in rats. J Cereb Blood Flow Metab. 2010;30:638–52. doi: 10.1038/jcbfm.2009.237. http://dx.doi.org/10.1038/jcbfm.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee G, MacLean DM, Ulrich H, Zhao X, Aronowski J, Jayaraman V. RNA based antagonist of NMDA receptors. ACS Chem Neurosci. 2014;5:559–67. doi: 10.1021/cn500041k. http://dx.doi.org/10.1021/cn500041k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guard DB, Swartz TD, Ritter RC, Burns GA, Covasa M. Blockade of hindbrain NMDA receptors containing NR2 subunits increases sucrose intake. Am J Physiol Regul Integr Comp Physiol. 2009;296:921–8. doi: 10.1152/ajpregu.90456.2008. http://dx.doi.org/10.1152/ajpregu.90456.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brouns R, De Deyn PP. The complexity of neurobiological processes in acute ischemic stroke. Clin Neurol Neurosurg. 2009;111:483–95. doi: 10.1016/j.clineuro.2009.04.001. http://dx.doi.org/10.1016/j.clineuro.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Lasoń W, Chlebicka M, Rejdak K. Research advances in basic mechanisms of seizures and antiepileptic drug action. Pharmacol Rep. 2013;65:787–801. doi: 10.1016/s1734-1140(13)71060-0. http://dx.doi.org/10.1016/S1734-1140(13)71060-0. [DOI] [PubMed] [Google Scholar]
- 10.Maher A, El-Sayed NS, Breitinger HG, Gad MZ. Overexpression of NMDAR2B in an inflammatory model of Alzheimer’s disease: Modulation by NOS inhibitors. Brain Res Bull. 2014;109:109–16. doi: 10.1016/j.brainresbull.2014.10.007. http://dx.doi.org/10.1016/j.brainresbull.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 11.Schwer CI, Lehane C, Guelzow T, Zenker S, Strosing KM, Spassov S, et al. Thiopental inhibits global protein synthesis by repression of eukaryotic elongation factor 2 and protects from hypoxic neuronal cell death. PLoS One. 2013;22(8):e77258. doi: 10.1371/journal.pone.0077258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez JA, Kasamatsu M, Rosales-Hernandez A, Hanson LR, Frey WH, Toth CC. Comparison of central versus peripheral delivery of pregabalin in neuropathic pain states. Mol Pain. 2012;11(8):3. doi: 10.1186/1744-8069-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Cunningham MO, Woodhall GL, Thompson SE, Dooley DJ, Jones RS. Dual effects of gabapentin and pregabalin on glutamate release at rat entorhinal synapses in vitro. Eur J Neurosci. 2004;20:1566–76. doi: 10.1111/j.1460-9568.2004.03625.x. http://dx.doi.org/10.1111/j.1460-9568.2004.03625.x. [DOI] [PubMed] [Google Scholar]
- 14.Fink K, Dooley DJ, Meder WP, Suman-Chauhan N, Duffy S, Clusmann H, et al. Inhibition of neuronal Ca(2+) influx by gabapentin and pregabalin in the human neocortex. Neuropharmacology. 2002;42:229–36. doi: 10.1016/s0028-3908(01)00172-1. http://dx.doi.org/10.1016/S0028-3908(01)00172-1. [DOI] [PubMed] [Google Scholar]
- 15.Kammerer M1, Brawek B, Freiman TM, Jackisch R, Feuerstein TJ. Effects of antiepileptic drugs on glutamate release from rat and human neocortical synaptosomes. Naunyn Schmiedebergs Arch Pharmacol. 2011;383:531–42. doi: 10.1007/s00210-011-0620-3. http://dx.doi.org/10.1007/s00210-011-0620-3. [DOI] [PubMed] [Google Scholar]
- 16.Ha KY, Carragee E, Cheng I, Kwon SE, Kim YH. Pregabalin as a neuroprotector after spinal cord injury in rats: biochemical analysis and effect on glial cells. J Korean Med Sci. 2011;26:404–11. doi: 10.3346/jkms.2011.26.3.404. http://dx.doi.org/10.3346/jkms.2011.26.3.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.André V, Rigoulot MA, Koning E, Ferrandon A, Nehlig A. Long-term pregabalin treatment protects basal cortices and delays the occurrence of spontaneous seizures in the lithium-pilocarpine model in the rat. Epilepsia. 2003;44:893–903. doi: 10.1046/j.1528-1157.2003.61802.x. http://dx.doi.org/10.1046/j.1528-1157.2003.61802.x. [DOI] [PubMed] [Google Scholar]
- 18.Ha KY, Kim YH, Rhyu KW, Kwon SE. Pregabalin as a neuroprotector after spinal cord injury in rats. Eur Spine J. 2008;17:864–72. doi: 10.1007/s00586-008-0653-6. http://dx.doi.org/10.1007/s00586-008-0653-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ben-Menachem E. Pregabalin pharmacology and its relevance to clinical practice. Epilepsia. 2004;45:13–8. doi: 10.1111/j.0013-9580.2004.455003.x. http://dx.doi.org/10.1111/j.0013-9580.2004.455003.x. [DOI] [PubMed] [Google Scholar]
- 20.Safwen K, Selima S, Mohamed E, Ferid L, Pascal C, Mohamed A, et al. Protective effect of grape seed and skin extract on cerebral ischemia in rat: implication of transition metals. Int J Stroke. 2015;10:415–24. doi: 10.1111/ijs.12391. http://dx.doi.org/10.1111/ijs.12391. [DOI] [PubMed] [Google Scholar]
- 21.Rahman A, Ustundag B, Ozercan IH, Burma O, Cekirdekci A, Ozveren MF, et al. The effect of flavonoids on reduction of reperfusion injury after global cerebral ischemia. TGKDCD. 1999;7:430–4. [Google Scholar]
- 22.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folinphenol reagents. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 23.Liu Y, Belayev L, Zhao W, Busto R, Belayev A, Ginsberg MD. Neuroprotective effect of treatment with human albumin in permanent focal cerebral ischemia: histopathology and cortical perfusion studies. Eur J Pharmacol. 2001;428:193–201. doi: 10.1016/s0014-2999(01)01255-9. http://dx.doi.org/10.1016/S0014-2999(01)01255-9. [DOI] [PubMed] [Google Scholar]
- 24.Woolliams JA, Wiener G, Anderson PH, McMurray CH. Variation in the activities of glutathione peroxidase and superoxide dismutase and in the concentration of copper in the blood in various breed crosses of sheep. Res Vet Sci. 1983;34:253–6. [PubMed] [Google Scholar]
- 25.Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 1967;70:158–69. [PubMed] [Google Scholar]
- 26.Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–6. doi: 10.1016/s0076-6879(84)05016-3. http://dx.doi.org/10.1016/S0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- 27.Wang Z, Liu T, Gan L, Wang T, Yuan X, Zhang B, et al. Shikonin protects mouse brain against cerebral ischemia/reperfusion injury through its antioxidant activity. Eur J Pharmacol. 2010;643:211–7. doi: 10.1016/j.ejphar.2010.06.027. http://dx.doi.org/10.1016/j.ejphar.2010.06.027. [DOI] [PubMed] [Google Scholar]
- 28.Reddy VD, Padmavathi P, Kavitha G, Saradamma B, Varadacharyulu N. Alcohol-induced oxidative/nitrosative stress alters brain mitochondrial membrane properties. Mol Cell Biochem. 2013;375:39–47. doi: 10.1007/s11010-012-1526-1. [DOI] [PubMed] [Google Scholar]
- 29.Ozerol E, Bilgic S, Iraz M, Cigli A, Ilhan A, Akyol O. The protective effect of erdosteine on short-term global brain ischemia/reperfusion injury in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:20–4. doi: 10.1016/j.pnpbp.2008.09.024. http://dx.doi.org/10.1016/j.pnpbp.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 30.Ahmad A, Khan MM, Ishrat T, Khan MB, Khuwaja G, Raza SS, et al. Synergistic effect of selenium and melatonin on neuroprotection in cerebral ischemia in rats. Biol Trace Elem Res. 2011;139:81–96. doi: 10.1007/s12011-010-8643-z. http://dx.doi.org/10.1007/s12011-010-8643-z. [DOI] [PubMed] [Google Scholar]
- 31.Cengiz N, Colakoglu N, Kavakli A, Sahna E, Parlakpinar H, Acet A. Effects of caffeic acid phenethyl ester on cerebral cortex: structural changes resulting from middle cerebral artery ischemia reperfusion. Clin Neuropathol. 2007;26:80–4. doi: 10.5414/npp26080. http://dx.doi.org/10.5414/NPP26080. [DOI] [PubMed] [Google Scholar]
- 32.Sun L, Tian X, Gou L, Ling X, Wang L, Feng Y, et al. Beneficial synergistic effects of concurrent treatment with theanine and caffeine against cerebral ischemia-reperfusion injury in rats. Can J Physiol Pharmacol. 2013;91:562–9. doi: 10.1139/cjpp-2012-0309. http://dx.doi.org/10.1139/cjpp-2012-0309. [DOI] [PubMed] [Google Scholar]
- 33.Blommel ML, Blommel AL. Pregabalin: An antiepileptic agent useful for neuropathic pain. Am J Health Syst Pharm. 2007;64:1475–82. doi: 10.2146/ajhp060371. http://dx.doi.org/10.2146/ajhp060371. [DOI] [PubMed] [Google Scholar]
- 34.Kumar A, Goyal R. Possible involvement of GABAergic modulation in the protective effect of gabapentin against immobilization stress-induced behavior alterations and oxidative damage in mice. Fundam Clin Pharmacol. 2007;21:575–81. doi: 10.1111/j.1472-8206.2007.00524.x. http://dx.doi.org/10.1111/j.1472-8206.2007.00524.x. [DOI] [PubMed] [Google Scholar]
- 35.Liu Z, Zhao W, Xu T, Pei D, Peng Y. Alterations of NMDA receptor subunits NR1, NR2A and NR2B mRNA expression and their relationship to apoptosis following transient forebrain ischemia. Brain Res. 2010;18:1361, 133–9. doi: 10.1016/j.brainres.2010.09.035. http://dx.doi.org/10.1016/j.brainres.2010.09.035. [DOI] [PubMed] [Google Scholar]
- 36.Ogunro PS, Bolarinde AA, Owa OO, Salawu AA, Oshodi AA. Antioxidant status and reproductive hormones in women during reproductive, perimenopausal and postmenopausal phase of life. Afr J Med Med Sci. 2014;43:49–57. [PubMed] [Google Scholar]
- 37.Qian H, Liu L. Protective effect of hydrogen sulfide on mice with experimental viral myocarditis and its mechanism. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2014;30:708–12. [PubMed] [Google Scholar]
- 38.Gozzi GJ, Pires AD, Martinez GR, Rocha ME, Noleto GR, Echevarria A, et al. The antioxidant effect of the mesoionic compound SYD-1 in mitochondria. Chem Biol Interact. 2013;205:181–7. doi: 10.1016/j.cbi.2013.07.004. http://dx.doi.org/10.1016/j.cbi.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 39.Babu CS, Ramanathan M. Post-ischemic administration of nimodipine following focal cerebral ischemic-reperfusion injury in rats alleviated excitotoxicity, neurobehavioural alterations and partially the bioenergetics. Int J Dev Neurosci. 2011;29:93–105. doi: 10.1016/j.ijdevneu.2010.08.001. http://dx.doi.org/10.1016/j.ijdevneu.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 40.Maheshwari A, Badgujar L, Phukan B, Bodhankar SL, Thakurdesai P. The protective effect of Etoricoxib against middle cerebral artery occlusion induced transient focal cerebral ischemia in rats. Eur J Pharmacol. 2011;667:230–7. doi: 10.1016/j.ejphar.2011.05.030. http://dx.doi.org/10.1016/j.ejphar.2011.05.030. [DOI] [PubMed] [Google Scholar]
- 41.Guo C, Tong L, Xi M, Yang H, Dong H, Wen A. Neuroprotective effect of calycosin on cerebral ischemia and reperfusion injury in rats. J Ethnopharmacol. 2012;144:768–74. doi: 10.1016/j.jep.2012.09.056. http://dx.doi.org/10.1016/j.jep.2012.09.056. [DOI] [PubMed] [Google Scholar]