

Abstract
We have developed a generalized in vitro compartmentalization based bead display selection strategy that allows for the identification of enzymes that can perform ligation reactions. While a number of methods have been developed to evolve such enzymes, many of them are limited in library size (106–107), do not select for enzymes using a scheme that allows for multiple turnover, or only work on enzymes specific to nucleic acids. This approach is amenable to screening libraries of up to 1012 protein variants by allowing beads to be overloaded with up to 104 unique mutants. Using this approach we isolated a variant of sortase A from Staphylococcus aureus that shows a 114-fold enhancement in kcat/m in the absence of calcium compared to the wild type and improved resistance to the inhibitory effects of cell lysates. Unlike the wildtype protein, the newly selected variant shows intracellular activity in the cytoplasm of eukaryotic cells where it may prove useful for intracellular labeling or synthetic biological applications.
Keywords: in vitro compartmentalization, sortase A, biotin protein ligase (BirA), directed evolution, microbead display, FACS
Introduction
Protein engineering holds great promise to develop the next generation of biocatalysts for both biomedical and industrial settings. Although much effort has been made to rationally design enzymes both from existing proteins and de novo, these enzymes often show greatly inferior characteristics when compared to naturally evolved proteins (Karanicolas et al. 2011; Khersonsky et al. 2012; Khersonsky et al. 2011). Directed evolution, a process which mimics natural evolution by selecting desirable variants from a library of mutants derived from a parental enzyme, has allowed researchers to enhance the activity of enzymes toward their natural substrate (Griffiths and Tawfik 2003), alter their substrate preference (Zhang et al. 1997), as well as alter physical properties such as fold stability (Miyazaki et al. 2000). The approach has also been very effective in helping de novo designed enzymes to achieve kinetic parameters similar to those of enzymes found in nature (Karanicolas et al. 2011; Khersonsky et al. 2011).
In vitro compartmentalization (IVC) is a powerful tool for directed evolution allowing for the use of extremely large libraries and has been employed for the development of a number of different enzymes including phosphotriesterases (Griffiths and Tawfik 2003), galactosidases (Mastrobattista et al. 2005) restriction enzymes (Doi et al. 2004) and polymerases (Ghadessy et al. 2001; Ong et al. 2006; Paul et al. 2013) among others. Indeed, we and other groups have previously extended the approach to the evolution of non-protein based enzymes, ribozymes (Levy et al. 2005; Zaher and Unrau 2007).
To engineer proteins using IVC, a water-in-oil emulsion is used to physically compartmentalize a DNA library with its resulting protein. Within each droplet, a mechanism to link the function of an enzyme to the gene is employed such that when the emulsion is broken, functional genes can be recovered. These functional genes are subsequently amplified by PCR and subjected to additional rounds of selection and mutagenesis until the desired characteristics have been obtained. Unlike techniques that utilize organisms (e.g. bacterial or yeast), the approach is not limited by transfection efficiencies (typically 106–109) and libraries of up to 1010 per mL of emulsion can be easily screened. In fact, emulsions as large as 50 mL have been employed to screen a library of RNA polymerase ribozyme variants (Zaher and Unrau 2007). Additionally, theoretical treatments have suggested that individual droplets can be overloaded with multiple genes, potentiating the screening of libraries even greater than 1010 mL in the early rounds of selection provided that stringency is increased in the later rounds (Levy et al. 2005).
Methods for performing the directed evolution of bond forming enzymes have previously and successfully been performed utilizing yeast surface display (Chen et al. 2011). However the use of yeast can significantly limit library sizes to ~106–7 variants (Chen et al. 2011). To the extent that larger libraries potentiate the identification of greater more diverse function (Dalby 2011), we sought to develop a general scheme to screen for bond forming enzyme using IVC. We recently demonstrated the selection of a bond forming enzyme, biotin ligase, BirA, with altered substrate specificity using an IVC approach in which functional enzymes encoded within dsDNA library attached desthiobiotin to the biotin acceptor peptide which had been covalently attached to dsDNA (Lu et al. 2014). However, while the selection scheme was successful, it only required that the selected enzymes perform a single catalytic event in order for it to pass through to the next round. Thus, the selected enzymes showed poor kinetic characteristics (Lu et al. 2014).
The use of microbeads coupled in combination IVC and fluorescence activated cell sorting (FACS) has previously been shown to allow for the selection of multiple turnover reactions for other enzymes such as phosphotriesterases (Griffiths and Tawfik 2003), T7 RNAP (Paul et al. 2013) and a ribozyme ligase (Levy et al. 2005) from libraries up to ~108 variants, a limitation imposed by sorting. Drawing from these works, here we report the development of a bead-based strategy for the directed evolution of bond forming enzymes by IVC. To achieve this, we set up an IVC based selection in which microbeads displayed both the library DNA and one half of the enzyme substrate (acceptor; Figure 1). Following compartmentalization with the components of an in vitro transcription translation reaction and the other half of the enzyme substrate (donor; Figure 1), the beads were emulsified. Compartments containing functional proteins resulted in beads tagged with the reaction product, which following dissolution of the emulsion can be labeled for detection. Flow cytometry was then to use the sort positive beads and the attached gene amplified by PCR.
Figure 1.
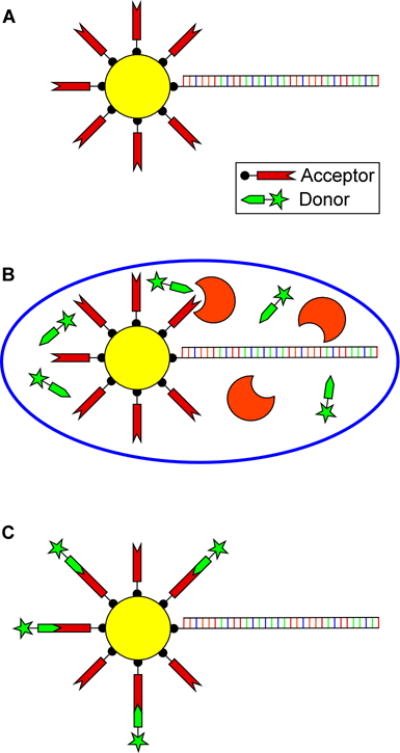
In vitro compartmentalization (IVC) based bead selection strategy. (A) Library DNA and the acceptor substrate are displayed on the surface of streptavidin coated microbeads. (B) These beads are then compartmentalized within a water in oil emulsion containing the components of an in vitro transcription and translation (TnT) reaction supplemented with the donor substrate. Beads with functional genes attached will be tagged with the reaction product on the bead’s surface. (C) Upon breaking the emulsion, the reaction product is labeled for detection and sorted based on fluorescence intensity. The recovered functional genes can then be reamplified using PCR for further screening.
To validate our approach, we preformed control experiments utilizing BirA, which allowed us to not only confirm the methodology, but to demonstrate that the approach can utilized to identify functional protein variants from extremely large libraries, up to 1012 variants by loading beads with as many as 104 unique protein sequences. Then, using this approach we isolated a variant of sortase A from Staphylococcus aureus that shows an improved resistance to the inhibitory effects of cell lysates and a 114-fold enhancement in kcat/Km in the absence of calcium compared to the wild type. More importantly unlike the wildtype protein, our SrtA variant is capable of functioning when expressed in the cytoplasm of mammalian cells.
Results
Modeling Selection with BirA
We have previously demonstrated the selection of the bond forming enzyme, biotin ligase, BirA, using an IVC approach in which functional enzymes encoded within dsDNA library attached desthiobiotin to the biotin acceptor peptide which had been covalently attached to dsDNA (Lu et al. 2014). However, due to the design of the selection only a single turnover was required for a variant to pass to the next round limiting the ability to select for enzyme variants with enhanced kinetic properties. Drawing from this, we initiated control experiments to assess the potential to adapt BirA function to our bead display system. In short, initial experiments were performed using beads bearing ~100,000 copies of the BirA acceptor peptide and a dsDNA encoding BirA immobilized on at a ratio of 0, 0.5, 1 and 2 genes per bead. Beads were subsequently combined with the components of an in vitro transcription and translation (TnT) reaction supplemented with 400 uM biotin and emulsified. In parallel, a similar set of experiments were performed but without emulsification. Following incubation overnight at 30°C, the emulsion was broken, beads were washed and the degree of biotinyation determined by labeling the beads with fluorescently labeled streptavidin followed by detection using flow cytometry.
When reactions were performed without emulsification (Figure 2A, non-emulsified) a significant level of BirA activity was observed in all samples including reactions that did not contain dsDNA encoding BirA (Figure 2A, non-emulsified; grey) suggesting that the TnT lysate contained significant levels of active, endogenous BirA. Nonetheless, BirA activity from the presence of BirA produced within the TnT reaction could still be observed (Figure 2A, non-emulsified, red, blue, green and orange traces). Interestingly, when reactions were performed with emulsification, the activity of the endogenous BirA, while still present, was significantly reduced (Figure 2A, emulsified; grey). This is likely due to inactivation of much of the endogenous BirA during the emulsification process. More importantly, activity from BirA produced from the translation of the attached gene could be clearly observed (Figure 2A, emulsified; red, blue, green and orange traces).
Figure 2.
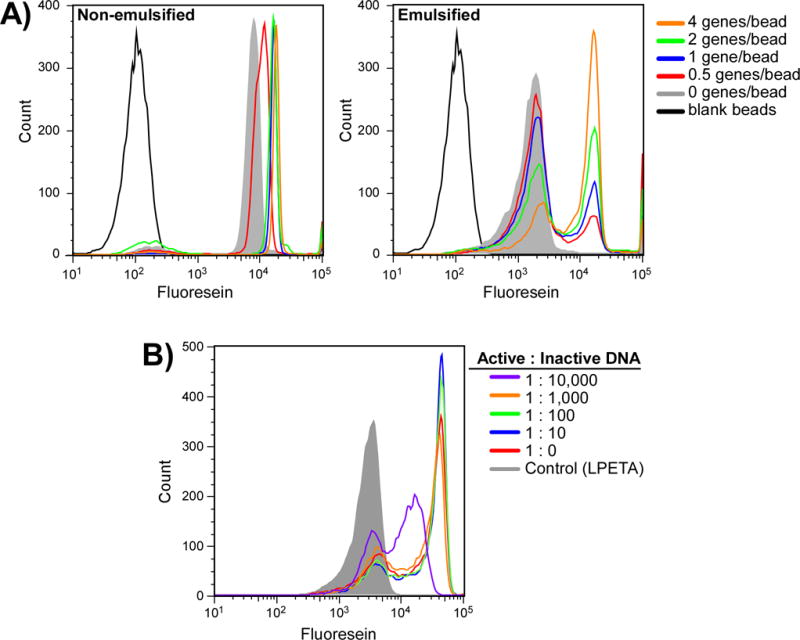
Modeling selection using BirA. (A) Beads displaying the BirA acceptor peptide along with 0, 0.5, 1, 2 or 4 BirA genes per bead were suspended in a TnT reaction supplemented with 200 uM biotin. Reactions were allowed to proceed both non-emulsified (left) and emulsified (right) at 30°C overnight after which the beads were labeled with fluorescein labeled streptavidin and analyzed by flow cytometry. (B) Titration to assess maximal library size. The BirA gene was displayed on the bead surface along with increasing ratios of an unrelated gene (Sortase A). Beads were suspended in a TnT reaction supplemented with 200 uM biotin and emulsified. Reactions were allowed to proceed overnight at 30°C after which the beads were labeled with a streptavidin fluorescein conjugate and analyzed by flow cytometry.
Having established that we could detect a single dsDNA encoding BirA on a bead, we next sought to assess the potential to load beads with multiple genes. Indeed, while IVC allows for facile screening of up to ~1010 compartments and thus 1010 protein variants per mL of emulsion, the use of bead display and FACS limits the library size to ~108 beads; a limitation imposed by sorting beads at ~20,000/s for 1.5 hrs. We thus sought to see if a single functional gene on a bead could still be detected, even if displayed along with multiple (10 – 10,000) non-functional genes.
We mixed different ratios of dsDNA encoding the BirA gene with a gene encoding a different protein, SrtA, which does not possess biotin ligase activity, at ratios ranging from 1:0 up to 1:10,000 BirA:SrtA genes. Mixtures were subsequently immobilized on beads at a ratio of 1 functional gene per bead along with ~ 100,000 copies of the biotin acceptor peptide. Following emulsification within a TnT reaction supplemented with 400uM biotin, reactions were incubated overnight after which the emulsion was broken, the beads labeled with fluorescent streptavidin, and subsequently analyzed by flow cytometry. As shown in Figure 2B, overloading bead with as many as 1000 non-functional genes has little effect on the observed activity of a single BirA gene in a compartment. Indeed, a clear difference between the positive and negative populations can even be seen when beads were loaded with 10,000 non-functional genes, suggesting that starting libraries as large as 1012 are amenable to selection.
Engineering Sortase A (SrtA) using IVC
Sortases are a family of transpeptidases involved in peptidoglycan biosynthesis that are ubiquitous in gram-positive bacteria (Paterson and Mitchell 2004). Sortase A from S. Aureus recognizes the C-terminal sorting signal LPXTG, where X can be any amino acid, and upon binding, cleaves the threonine-glycine peptide bond forming an acyl-enzyme intermediate. In nature, this intermediate is resolved by the N-terminal amine of a pentaglycine branched lipid II, a cell wall component, resulting in the formation of a new peptide bond and covalently linking lipid II to a protein substrate (Paterson and Mitchell 2004). In the lab, this provides an easy way to seamlessly ligate proteins together in a highly specific manner. Indeed, since the discovery of this enzyme, bioengineers have seen the utility of such an enzyme in the manipulation of proteins. To date, sortase A from S. aureus has been used for a multitude of applications from surface labeling of live cells (Tanaka et al. 2008) to immobilization of proteins to surfaces (Sinisi et al. 2012) to engineering unnatural protein fusions (Witte et al. 2013). As there is much interest in this protein, we sought to use our selection scheme to identify novel protein variants with altered function.
Initially, we performed control experiments to assess the ability of wild type sortase A (sa.SrtA) to function in the TnT lysate and label beads tagged with an LPETG acceptor peptide with the fluorescently labeled peptide donor, GGG-FAM. Interestingly, while purified recombinant protein produced in E. coli from plasmid or from dsDNA using our TnT system was capable of labeling beads with fluorescent peptide when performed in 50 mM Tris-HCl, 150 mM NaCl, 5 mM CaCl2, pH 7.5 (Ton-That et al. 2000) as determined by flow cytometry the protein proved non-functional when assays were performed within the TnT system or when recombinant purified protein was added to S30 lysate, suggesting that a component of the TnT mixture was inhibiting function (Figure 3A). When we assayed sa.SrtA function in the presence of a variety of additives including synthetic substrate-like peptides (LPETA), purified protein (BSA), and cell lysate based sources (yeast extract, S30 E. coli lysate, tryptone), the enzyme also showed greatly diminished activity (Supplemental Figure S1).
Figure 3.
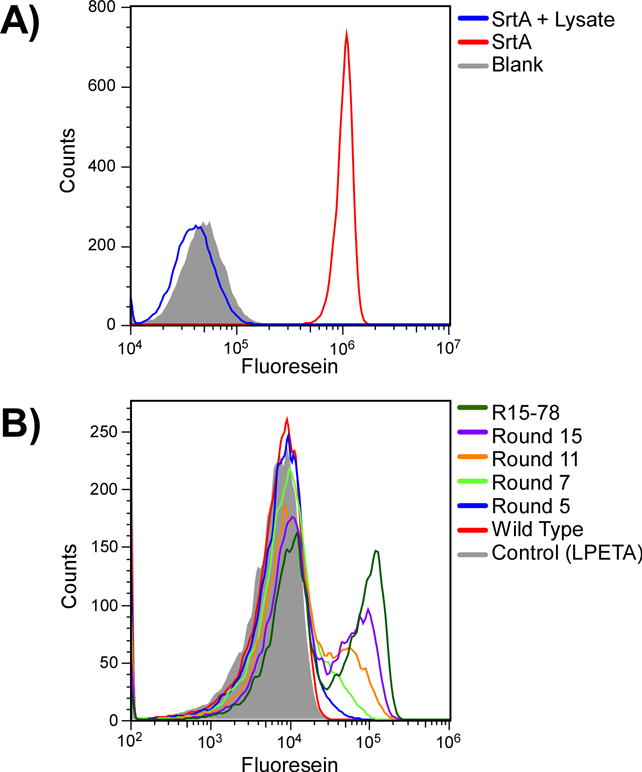
Evolving sortase variants with activity in cell lysates. (A) Wildtype sortase A is non-functional in S30 lysate. Purified wild type sortase A was incubated with beads displaying LPETG peptide and the donor peptide, GGG-FAM, in the presence (blue) or absence (red) of E. coli S30 lysate. Blank, unlabeled beads are shown in grey. (B) Progress of the selection for improved SrtA variants with IVC. Individual rounds of selection were assayed for activity under conditions which mimic the selection. dsDNA from Rounds 5, 7, 11 and 15 was immobilized on beads bearing the LPETG acceptor peptide at a ratio of 1 gene per bead. After emulsification, reactions were allowed to proceed overnight at 30°C after which the emulsion was broken and the beads analyzed by flow cytometry. For comparison, experiments were performed using the wild type protein and clone R15-78, a clone identified from Round 15.
We mutagenized sa.SrtA to a level of ~10% using error prone PCR (Fromant et al. 1995) (Supplemental Figure S2). For the initial round of selection, we chose to immobilize ~1000 dsDNA variants on from this library per bead on 108 beads along with the SrtA acceptor peptide, LPETG, for a starting library size of ~1011 variants. Beads were added to a TnT reaction containing 5 uM donor peptide (GGG-FAM) and incubated for 4 hrs at 30°C after which the emulsion was broken and the beads sorted. For the first five rounds of selection, we employed the use of an antibody based amplification kit to boost any fluorescent signal on the beads. The amplification system provides an increase in the signal by utilizing a rabbit anti-fluorescein specific primary antibody labeled with AlexaFluor 488 and a secondary goat anti-rabbit antibody labeled also with AlexaFluor 488. This allows for the potential to enrich for sub-optimal SrtA variants that can be improved upon additional rounds of mutagenesis. During each sort, beads corresponding to the top 1% of the fluorescent population were collected. Library molecules were subsequently amplified by PCR and the process repeated. For subsequent rounds, we decreased the number of genes per bead (Table I) such that by Round 5 beads were loaded at a ratio of ~2 beads per gene.
Table I.
Ratio of genes to beads used for each round of selection. Double bars indicate rounds which were preceeded by mutagenesis.
| round | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 |
| # genes per bead | 1000 | 100 | 1 | 1 | 0.5 | 100 | 1 | 100 | 1 | 1 | 0.5 | 100 | 1 | 1 | 0.5 |
By Round 5 the library showed a significant improvement in activity as determined by flow cytometry, but still required the use of antibody based FITC amplification to observe a signal (Figure 3 and Supplemental Figure S3). Therefore to further enhance the catalytic potential of the library, prior to Round 6 we re-mutagenized the library using a combination of DNA shuffling (Lorimer and Pastan 1995) and error-prone PCR. The selection was then reinitiated starting with 100 genes per bead, and reducing this down to 1 per bead in round 7. Following round 7, antibody-based amplification was no longer necessary for detecting a positive population. The mutagenesis process was repeated two additional times, before round 8 and round 12 (Table I). By round 15, a significant proportion of the library showed greatly enhanced activity (Figure 3).
Characterization of novel SrtA variants
Sequence analysis of the SrtA-Round 15 library revealed the presence of several families of sequences (Supplemental Figure S4). We initially assayed selected clones (R15-25, 35, 68 and 78) for function using a bead-based assay that mimicked the selection conditions. As shown in Figure 4A, when emulsified, all of the selected clones demonstrate bead labeling activity significantly greater than that observed with the wildtype protein under the same conditions (Figure 4A and B). Consistent with our previous findings, under these conditions the sa.SrtA displayed little to no activity in these assays, showing only trace signal following amplification (Figure 4B). When a similar assay was performed without emulsification (data not shown), only trace bead labeling could be observed consistent with previous observations that enzyme function with a compartment is more efficient that that observed in bulk solution (Tawfik and Griffiths 1998). However, even here, the enhanced activity of the mutants when compared to the wildtype protein becomes apparent upon amplification (Figure 4C). Perhaps more importantly, when purified proteins generated in E. coli were assayed in buffer, all proteins demonstrate activity on par with that observed for the wildtype (Figure 4D). Interestingly, the most abundant family was not the most active (Supplemental Figure S5).
Figure 4.

Analysis of selected clones from the Round 15 library. (A and B) Clones R15-25, 35 and 78 isolated from Round 15 all showed enhanced activity in assays when assayed in emulsion under selection-like conditions. To allow for better comparison, these assays were performed at a ratio of 100 genes per bead to eliminate the presence of any population of negative beads. Following emulsification, reactions were allowed to proceed overnight at 30°C after which the emulsion was broken and the beads were analyzed by flow cytometry without (A) or with additional signal amplification (B). (C) Analysis of selected clones without emulsification. Assays were performed as in (A) and (B) omitting the emulsification step. Analysis required amplification. (D) Flow cytometric analysis of purified clones. Beads bearing LPETG peptide were incubated in TBST with 5 uM GGG-FAM and 1.5 uM purified enzyme for 2 h at 37°C. Beads were then washed and analyzed by flow cytometry.
Among the clones we tested, one, R15-78, contained the mutations E105V and E108G. Since E105 and E018 are known to be involved in calcium binding (Naik et al. 2006), it appeared likely these clones would be Ca2+ independent. To assess this in more detail, we first performed assays using purified protein and a fluorescently quenched peptide substrate as previously described in buffer in both the presence and absence of added Ca2+ (Huang et al. 2003). Under these assay conditions, in the presence of Ca2+, all of the selected clones performed better than the wildtype protein and displayed activity more similar to that observed for recently reported SrtA variant (Chen et al. 2011) (Supplemental Figure S6). However, when assays were performed in the absence of Ca2+ as predicted, only the clone R15-78, which contains the E105V and E108G, displayed significant activity.
To more accurately determine the kinetic parameters of this new variant, we employed an HPLC based assay for determining enzyme kinetics (Kruger et al. 2004). For comparison, we measured kcat and Km for R15-78, sa.SrtA, and a SrtA variant recently reported by David Liu et al. (Liu-tase; (Chen et al. 2011)) in the presence and absence of Ca2+. Consistent with the literature, sa.SrtA and Liu-tase displayed kcat and Km values of 0.39/s and 2.22 mM and 5.03/s and 0.07 mM in the presence of Ca2+, with much lower activity observed in its absence (0.019/s and 7.28 mM, .47/s and 4.56 mM; Figure 5). Consistent with our previous results, R15-78 displayed Ca2+ independent kinetics, kcat+ = 0.69/s, Km+ = 2.20 mM and kcat− = 0.57/s, Km− = 1.94 mM.
Figure 5.
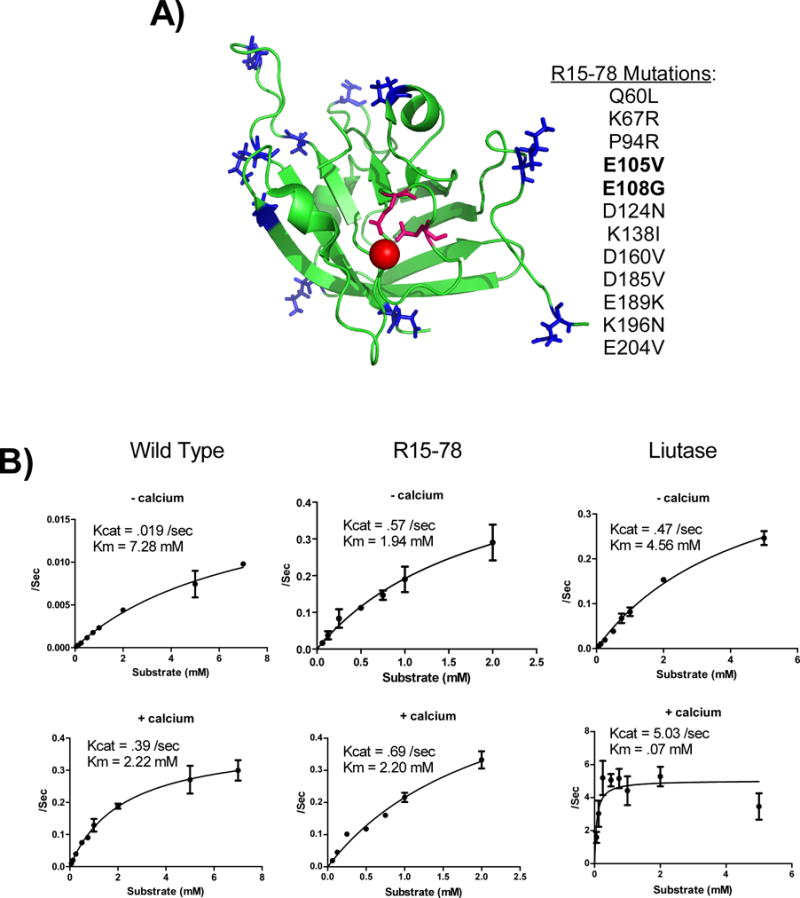
Characterization of R15-78. (A) List of mutations observed in R15-78. The positions of each mutation was mapped to the structure of the wildtype protein (PDB ID: 2KID) are indicated in blue with the two Asp mutations (E105V and E108G) which are likely to engender calcium independence indicated in pink. (B) Comparison of the kinetic properties of R15-78 with the wildtype protein and a variant isolated by Chen et al. (Liutase) in the absence (top) and presence of 2mM calcium. All data represents the average of at least 2 independent assays. Error bars show standard deviation. Please note the scale of the y-axis isn’t fixed.
Application of a Ca2+ independent SrtA
Recent work by the Ploegh lab has suggested that the ability to utilize SrtA from S. aureus as a labeling agent within live mammalian cells is inhibited by the protein’s dependence on Ca2+ which is present in the cytoplasm at vanishing low concentration (~100 nM; (Clapham 2007)). Indeed, using the calcium-independent SrtA from a different bacterial species, S. pyogenes, an enzyme that displays markedly inferior enzymatic properties (kcat = 0.0070 s−1 km = 0.53 mM in the presence of 5 mM Ca2+ (Race et al. 2009)), Strijbis et al. were able to demonstrate in vivo sortase activity in both yeast and mammalian cells (Strijbis et al. 2012).
We used a GFP circularization assay to assess the potential for our Ca2+-independent variant, R15-78, to function in mammalian cells. In this assay cells transfected with a GFP reporter bearing an N-terminal glycine residue and a C-terminal LPETG (Gly-GFP-LPETG) can be monitored for sortase active by monitoring the circularization product of GFP by gel electrophoresis (Antos et al. 2009) (Figure 6A). Using this assay we co-transfected Gly-GFP-LPETG into HEK293T cells along with a second vector encoding SrtA from S. pyogenes (sp.SrtA), S. aureus (sa.SrtA), or our Ca2+-independent variant, R15-78. As shown in Figure 6B, circularized GFP was detected in the lanes containing sp.SrtA and R15-78. Consistent with previous reports, no circularized product was detected in the lanes containing sa.SrtA. Perhaps, more importantly, consistent with the in vitro activities of these enzymes, it appears that at comparable expression levels R15-78 yields more circularized product than sp.SrtA.
Figure 6.
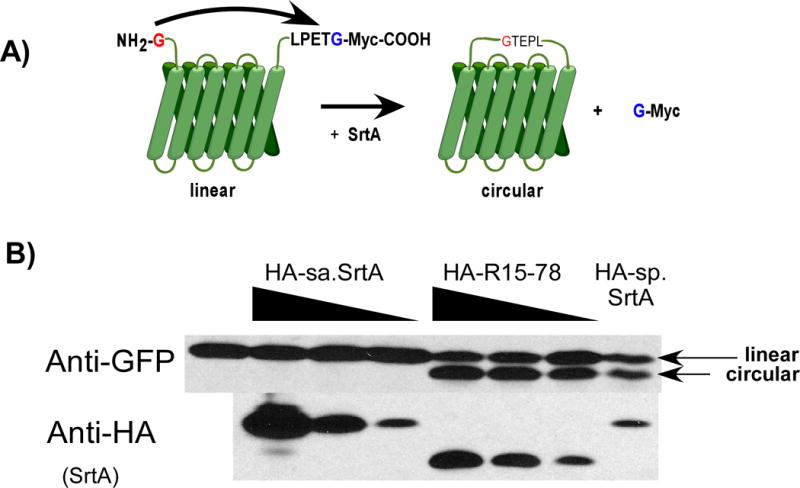
GFP circularization assay to detect intracellular sortase activity. (A) A GFP construct bearing an N-terminal glycine and C-terminal LPETG peptide is transfected into cells. In the presence of sortase activity, the LPETG peptide is cleaved and attached to the glycine at the N-terminus resulting in a circularized protein. This circularized product can be detected via Western blot as a band of lower apparent molecular weight. (B) HEK293T cells were transfected with the GFP construct along with HA tagged wild type sortase A from S. aureus (HA-sa.SrtA), mutant R15-78 (HA-R15-78) or wild type sortase A from S. pyogenes (HA-sp.SrtA). The production of circularized GFP was assessed by Western using an anti-GFP antibody. Production of HA-sa.SrtA, HA-R15-78 or HA-sp.SrtA was detected using anti-HA antibody. The linear and circular GFP products are indicated.
Materials and Methods
Primers and peptides
All oligonucleotides were obtained from integrated DNA technologies. The biotinylated peptides used for all bead assays were obtained from Genscript (Piscataway, NJ). The FRET peptide used in the progress curves was obtained from Anaspec, and the peptides used for the kinetic assays were synthesized in house on an ABI 433A peptide synthesizer. Streptavidin coated polystyrene microbeads were obtained from Bangs Laboratories. Transcription and translation reagents were made based on (Liu et al. 2005). A complete list of primer sequences can be found in the Supplemental Methods.
Model BirA selection
A linear BirA template for the transcription and translation reaction was made by amplification with the appropriate upstream and downstream regulatory regions (Supplemental Methods). The template was then amplified by PCR using a double biotinylated 5′ primer so it could be immobilized on streptavidin coated polystyrene microbeads. This dsDNA was then added to ~108 microbeads (Bang’s Labs CP01N) in TBST (50 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween 20) such that on average there were 0, 0.5, 1, 2 or 4 genes on each bead. Following a 30 min incubation, the beads were saturated by the addition of excess biotinylated BirA acceptor peptide (Biotin-GGGSGGGSGLNDIFEAQKIEWHE). Following a 15 min incubation the beads were washed 3X with 500uL TBST and resuspended in 50 ul RTS E.coli HY reaction mix (5 PRIME) supplemented with 200 uM Biotin and emulsified on ice by the addition to 500 ul of an oil mixture containing light mineral oil, 4.5% Span-80, 0.5% Tween-80 and 0.1% Triton X-100 as previously described (Ghadessy et al. 2001; Levy and Ellington 2008). The mixture was stirred on ice for 4 min then subsequently incubated at 30°C overnight. To break the emulsion, we added 200 ul TBST + 5% BSA each reaction followed by 800 ul diethyl ether. After mixing the sample was centrifuged at 21,000 × g for 10 min to separate the phases. The organic phase was removed and the aqueous phase was washed with 600 ul light mineral oil and centrifuged again. The pelleted beads were then removed and transferred into a new 1.7 ml eppendorf tube. The beads were then dispersed by sonication and washed in TBST, then 6 M guanidine HCl + 50 mM PO4 + 10 mM Tris-Cl, then once more in TBST. Beads were labeled by incubation with 0.1 uM streptavidin-AlexaFluor 488 for 30 min and washed 3x in TBST prior to analysis by flow cytometry. Analyses were performed using an Eclipse, EC800 flow cytometer (Sony biotechnologies, San Jose California).
Library generation
The library for the selection for SrtA variants was generated by mutagenic PCR using the mutagenic PCR protocol described by Fromant et al. (Fromant et al. 1995) in a manner previously described (Levy and Ellington 2008). The reaction buffer contained 10 mM Tris pH 8.7, 25 mM KCl, 50 ug/ml BSA, 0.12 mM dATP, 0.1 mM dCTP, 0.55 mM dGTP, 3.9 mM dTTP, 0.5 mM MnCl2, and 4.8 mM MgCl2. 10 pg of wild type S. aureus sortase A was amplified for 25 cycles. The resulting product was then gel purified and 10 pg of the purified product was amplified under the same conditions for another 25 cycles, yielding a final rate of mutagenesis of ~10% as determined by conventional sequencing (Supplemental Figure S2).
At rounds 5, 7, and 11, the library was re-mutagenized using a combination mutagenic PCR and gene shuffling, as originally described by Lorimer and Pastan (Lorimer and Pastan 1995), and previously employed by us (Levy and Ellington 2008). Prior to mutagenesis, the library was amplified with gene specific primers to remove the upstream and downstream regulatory regions. Mutagenic PCR was performed as described above. Gene shuffling was carried out by incubating ~2 ug library DNA in a 50 ul reaction containing 50 mM Tris pH 7.4, 10 mM MnCl2, and 0.3 U DNAseI. The digestion was carried out for 4 min at 15°C followed by a 10 min deactivation step at 90°C. The fragments were cleaned up using a centricep column (Princeton Separations) and amplified in the absence of additional primers using KOD DNA polymerase for 10 cycles. The resulting product was then gel purified and reamplified using RTS.ext forward and reverse primers.
To re-mutagenize the round 11 library, we used the nucleoside analog protocol described by Gherardi et al. (Zaccolo et al. 1996). Briefly, 10 fmol template was added to a 20 ul reaction containing 20 mM Tris pH 8.3, 10 mM NH4SO4, 10 mM KCl, 2 mM MgSO4, 0.1% Triton X-100, 2.5 U Taq polymerase (ThermoFisher), and 500 uM each of dATP, dTTP, dGTP, dCTP, dPTP, and 8-oxodGTP. This reaction was then cycled 20 times yielding approximately 10% mutagenesis. 1 ul of this reaction was then used in a second PCR reaction under the same conditions as above but excluding the nucleoside analogs. The resulting product was then gel purified and then re-amplified using adaptor primers for Gibson assembly (Supplemental Methods).
Gibson based library assembly
Gibson ligation was used to append upstream and downstream regulatory regions required for efficient in vitro transcription and translation to the initial library and subsequent rounds following each round of mutagenesis and/or shuffling. In short, the library was amplified using gene specific Gibson adaptor primers (Supplemental Methods) bearing an 18 base pair extension that overlaps with the RTS upstream and RTS downstream fragments. Assemblies were performed as follows, gel purified library (100ng) was incubated with a 2-fold molar excess of the upstream and downstream regulatory regions wre incubated a 20 ul reaction containing 100 mM Tris pH 7.5, 10 mM MgCl2, 0.4 mM each of dGTP, dCTP, dATP, dTTP, 10 mM DTT, 50 mg/ml PEG-8000, 1 mM NAD, 0.08 U T5 exonuclease (Epicenter), 0.5 U Phusion polymerase (NEB), and 80 U Taq ligase (NEB). Reactions were incubated for 1 hr at 50°C and the extent of assembly (typically greater than 90%) confirmed by gel electrophoresis. 5 ul of this reaction was then used as the template in a 100 ul PCR reaction using RTS F and RTS R primers which are specific for the RTS upstream and downstream fragments. The resulting product was then gel purified and re-amplified with a double biotinylated forward primer to be used in the selection.
IVC selection scheme
Biotinylated libraries were attached to streptavidin coated polystyrene microbeads at the indicated gene to bead ratios (Table I). Following a 30 min incubation the beads were saturated with a biotinylated SrtA acceptor peptide (biotin-GGGSGGLPETGG). Following an additional 15 min incubation the beads were washed 3 × 500 uL of TBST, re-suspended in 50 uL of TnT mix along with 5 uM acceptor peptide (GGG-FAM). To prevent premature translation of enzymes, following the additional of TnT mix all reactions were kept on ice. Emulsions formed as described above, on ice by adding the beads in TnT mix to 500 ul of light mineral oil containing 4.5% Span-80, 0.5% Tween-80, and 0.1% Triton X-100. Upon adding 50 ul of the selection reaction, the mixture was stirred for 4 min on ice and transferred to a 1.7 ml Eppendorf tube and incubated at 30°C for 4 hrs. To break the emulsion, we added 200 ul 1x TBST containing 5% BSA followed by 800 ul of water saturated diethyl ether. After mixing the sample was centrifuged at 21,000 × g for 10 min to separate the phases. The organic phase was removed and the aqueous phase was washed with 600 ul light mineral oil remove the resulting detergent layer at the interface and centrifuged again. After the oil wash, the pelleted beads were transferred to a new tube and washed 3 times in 1x TBST + 1% BSA.
For the first 5 rounds of selection, an antibody-based FITC amplification kit (Invitrogen) was used following the included instructions. Briefly, the beads were pelleted and resuspended in 1 ml 1x TBST + 1% BSA. Primary antibody was added to the beads at a 1:400 dilution and incubated 30 min on ice. The beads were washed and the secondary was added at the same dilution for 30 min. The beads were then washed in 3 × 500 ul 1x TBST + 1% BSA and diluted in FACS tubes to a concentration of ~ 2.5 × 107 beads per ml for sorting.
Bead sorting was performed using a FACSaria III flow cytometer using a 70 um nozzle at a rate of 22,000 events per second. The sort gate was set such that the 1% brightest beads were collected. BSA was added to the sorted beads to a final concentration of 5% and the beads are pelleted in a 1.7 ml tube by centrifuging at 21,000 × g for 15 min. The pelleted beads were re-suspended in water and amplified using Maxima hot start Taq in the presence of 10% DMSO using 5.RTS.F and RTS.hisR. The amplified DNA was gel purified on a 2% agarose gel and the received dsDNA re-amplified with a double biotinylated forward primer for use in the next round be used in the next round.
Cloning and recombinant expression of sortase variants
The sortase library was cloned into PCR4-TOPO and transformed into TOP10 cells as per the kit directions (Invitrogen). The plasmids were then isolated using a Qiagen mini prep kit and sequenced using an ABI 3730 at the Albert Einstein College of Medicine Genomics Core.
For protein expression, plasmids were transformed into KRX cells (Promega, Madison Wisconsin). A single colony was picked and used to inoculate a 5 ml starter culture containing LB + 50 ug/ml kanamycin containing 0.4% glucose. These cultures were incubated at 37°C and allowed to grow over night and used to inoculate 500 ml TB containing 50 ug/ml kanamycin. The large scale cultures were grown at 37°C to an OD600 of 0.8–1.2 at which point they were induced with a final concentration of 0.1% rhamnose for 4 hrs before being collected by centrifugation. The cells were then lysed by sonication and purified via FPLC using a Ni-NTA column and size exclusion column (Cells were lysed in 50 mM Tris pH 7.5, 300 mM NaCl, and 10 mM imidazole by sonication and the cleared lysate applied to resin. Elution was performed using 50 mM Tris pH 7.5, 300 mM NaCl, and 250 mM imidazole). The fractions containing sortase were then pooled and concentrated using an Amicon Ultra centrifugal concentrator with a 10 kDa cutoff.
Enzyme Kinetics
Enzyme kinetics were measured using the HPLC based assay originally published by McCafferty et al. (Kruger et al. 2004). Briefly, enzyme was diluted at a concentration ranging from 12.5 nM to 840 nM in 150 mM NaCl, 300 mM Tris pH 7.5, 5 mM EGTA, either 0 or 10 mM CaCl2, 2 mM triglycine, and Abz-LPETG-Dnp ranging in concentration from 0 to 5 mM. A 15 ul reaction was incubated at 37° C for 2–30 min and then quenched using 7.5 ul 1 N HCl. The reactions were then diluted to 100 uM in 0.1% TFA in H2O and 15 ul was injected into an Aligent 1200 series LC/MS with a Waters XBridge C18 column. The product was separated from the substrate using a linear gradient of 0 to 100% Acetonitrile + 0.1% TFA over 15 min. Percent completion was determined by relative peak area and used to determine umol of product produced. This information was then used to determine the turnovers per second and fit to the Michaelis-Menten equation to determine kcat and Km. In the case of R15-78, substrate inhibition prevented data points above 2 mM Abz-LPETG-Dnp from being used.
GFP Circularization
GFP circularization assays were performed in a manner similar to that previously described (Strijbis et al. 2012). In short, HEK293T cells were transfected with pcDNA3.1-Gly-GFP-LPETG-Myc along with empty pUC19 (Control), pcDNA3.1-HA-S. pyogenese SrtA, or pcDNA3.1-HA-R15-78 using Lipofectamine2000 according to manufacturer instructions. 24 hrs later the cells were lysed in 1X SDS-PAGE loading dye, boiled for 10 min, and run on a 12% polyacrylamide gel. The gel was then transferred onto nitrocellulose paper and probed with anti-GFP-HRP (Stefanovic and Hegde 2007) or an anti-HA (Abcam 18181) followed by anti-mouse or anti-rabbit horseradish peroxidase secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA). The blots were then incubated with SuperSignal West Pico (Pierce, Rockford Il) chemiluminescent substrate according to manufacturer instructions and exposed on Crystalgen blue sensitive film.
Discussion
Protein engineering methods are important tools for the development of novel proteins with optimized function. Here we have developed and implemented in vitro method for performing the directed evolution of proteins which make use of IVC, microbead display and FACS to engineer bond forming protein enzymes.
We initially assessed our approach using biotin ligase (BirA) a protein known to function in in vitro translation systems (Cox et al. 2002; Lu et al. 2014) and which we have previously engineered using IVC, albeit using a different strategy (Lu et al. 2014). These initial experiments with BirA not only allowed us to establish the feasibility of our method, but also provided us with a means to examine the limits of our approach. That is, because FACS based selection schemes are limited to the number of events that can be practically sorted we assessed the ability to overload each microbead with multiple genes per bead such that larger libraries could be screen. Theoretical treatments have previously suggested the ability to perform selections with IVC which employed larger amounts of DNA library molecules than the number of actual compartments (Levy et al. 2005). Our experiments here demonstrate that library size could be increased from ~108, a practical limitation enforced by sorting ~20,000 events per hr, to as much as ~1012 by loading as many as 104 genes on a single bead. Although, we note that at this dilution of functional genes, the signal observed on each bead is diminished ~5-fold, a likely consequence of the dilution of the total number of functional proteins per compartment.
Having established the approach with BirA, we then applied the method to a different enzyme, sortase A from S. aureus (sa.SrtA). Interestingly, while functional in vitro in buffered solutions, sa.SrtA has been shown to be nonfunctional in the cellular environment (e.g mammalian cells, yeast cells; (Strijbis et al. 2012)), a finding confirmed in our own experiments in vitro and in vivo, as the wildtype protein activity was readily inhibited by the addition of cellular extracts and homogenates such as tryptone or E. coli S30 lysate.
IVC based selection schemes have previously been used to evolve and/or identify protein variants that function in the presence of inhibitors. For example, Ghadessy et al. used an IVC based scheme to identify Taq polymerase variants capable of functioning it the presence of heparin sulfate, a known inhibitor of this enzyme (Ghadessy et al. 2001). Similarly, Paegel and Joyce were able to evolve RNA ligase ribozymes with increased resistance to inhibition by neomycin using microfluidic based IVC selection (Paegel and Joyce 2010). Thus, we sought to use our approach to identify SrtA variants that could function within our system.
Previous work by Drummond et al. showed that higher rates of mutagenesis yielded libraries with greater numbers of improved variants despite also containing more non-functional sequences (Drummond et al. 2005). While this may pose a limitation for screens or selections performed using small (~106) libraries, because of our ability to screen a large number of sequences, 1011–12, we reasoned that using a high rate of mutagenesis, ~10%, might be advantageous for our purposes.
We screened a starting library of ~1011 variants to identify protein variants which could function in our S30 E. coli in vitro translations system and performed 15 iterative rounds of selection employing multiple rounds of both random mutagenesis and DNA shuffling. We identified a number of novel protein variants that demonstrate significant activity both in our bead based assay, conducted in S30 lysate, as well as in assays performed using recombinant protein. Of particular note, we identified one sequence variant, R15-78, which demonstrates both increase activity in the presence of cell lysate and perhaps more importantly Ca2+ independent activity. Recent work by the Ploegh lab has suggested that the ability to utilize SrtA from S. aureus as a labeling agent within live mammalian cells is inhibited by the protein’s dependence on Ca2+ which is present in the cytoplasm at vanishing low concentration (~100 nM; (Clapham 2007)). Consistent with this finding, R15-78 proved capable functioning in vivo as determined using an GFP circularization assay (Strijbis et al. 2012).
Taken as a whole, our work provides a new approach which can be used to engineer bond-forming enzymes from very large libraries. However, more immediately we have identified a new variant of sortase A which may prove useful for labeling, tracking or modifying proteins in live cells.
Supplementary Material
References
- Antos JM, Popp MW, Ernst R, Chew GL, Spooner E, Ploegh HL. A straight path to circular proteins. J Biol Chem. 2009;284(23):16028–36. doi: 10.1074/jbc.M901752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Dorr BM, Liu DR. A general strategy for the evolution of bond-forming enzymes using yeast display. Proceedings of the National Academy of Sciences. 2011;108(28):11399–11404. doi: 10.1073/pnas.1101046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 2007;131(6):1047–58. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Cox JC, Hayhurst A, Hesselberth J, Bayer TS, Georgiou G, Ellington AD. Automated selection of aptamers against protein targets translated in vitro: from gene to aptamer. Nucleic Acids Res. 2002;30(20):e108. doi: 10.1093/nar/gnf107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby PA. Strategy and success for the directed evolution of enzymes. Curr Opin Struct Biol. 2011;21(4):473–80. doi: 10.1016/j.sbi.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Doi N, Kumadaki S, Oishi Y, Matsumura N, Yanagawa H. In vitro selection of restriction endonucleases by in vitro compartmentalization. Nucleic Acids Res. 2004;32(12):e95. doi: 10.1093/nar/gnh096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromant M, Blanquet S, Plateau P. Direct random mutagenesis of gene-sized DNA fragments using polymerase chain reaction. Anal Biochem. 1995;224(1):347–53. doi: 10.1006/abio.1995.1050. [DOI] [PubMed] [Google Scholar]
- Ghadessy FJ, Ong JL, Holliger P. Directed evolution of polymerase function by compartmentalized self-replication. Proc Natl Acad Sci U S A. 2001;98(8):4552–7. doi: 10.1073/pnas.071052198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths AD, Tawfik DS. Directed evolution of an extremely fast phosphotriesterase by in vitro compartmentalization. Embo J. 2003;22(1):24–35. doi: 10.1093/emboj/cdg014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Aulabaugh A, Ding W, Kapoor B, Alksne L, Tabei K, Ellestad G. Kinetic mechanism of Staphylococcus aureus sortase SrtA. Biochemistry. 2003;42(38):11307–15. doi: 10.1021/bi034391g. [DOI] [PubMed] [Google Scholar]
- Karanicolas J, Corn JE, Chen I, Joachimiak LA, Dym O, Peck SH, Albeck S, Unger T, Hu W, Liu G, et al. A de novo protein binding pair by computational design and directed evolution. Mol Cell. 2011;42(2):250–60. doi: 10.1016/j.molcel.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O, Kiss G, Rothlisberger D, Dym O, Albeck S, Houk KN, Baker D, Tawfik DS. Bridging the gaps in design methodologies by evolutionary optimization of the stability and proficiency of designed Kemp eliminase KE59. Proc Natl Acad Sci U S A. 2012;109(26):10358–63. doi: 10.1073/pnas.1121063109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O, Rothlisberger D, Wollacott AM, Murphy P, Dym O, Albeck S, Kiss G, Houk KN, Baker D, Tawfik DS. Optimization of the in-silico-designed kemp eliminase KE70 by computational design and directed evolution. J Mol Biol. 2011;407(3):391–412. doi: 10.1016/j.jmb.2011.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger RG, Dostal P, McCafferty DG. Development of a high-performance liquid chromatography assay and revision of kinetic parameters for the Staphylococcus aureus sortase transpeptidase SrtA. Anal Biochem. 2004;326(1):42–8. doi: 10.1016/j.ab.2003.10.023. [DOI] [PubMed] [Google Scholar]
- Levy M, Ellington AD. Directed evolution of streptavidin variants using in vitro compartmentalization. Chem Biol. 2008;15(9):979–89. doi: 10.1016/j.chembiol.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Griswold KE, Ellington AD. Direct selection of trans-acting ligase ribozymes by in vitro compartmentalization. Rna. 2005;11(10):1555–62. doi: 10.1261/rna.2121705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DV, Zawada JF, Swartz JR. Streamlining Escherichia coli S30 extract preparation for economical cell-free protein synthesis. Biotechnol Prog. 2005;21(2):460–5. doi: 10.1021/bp049789y. [DOI] [PubMed] [Google Scholar]
- Lorimer IA, Pastan I. Random recombination of antibody single chain Fv sequences after fragmentation with DNaseI in the presence of Mn2+ Nucleic Acids Res. 1995;23(15):3067–8. doi: 10.1093/nar/23.15.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu WC, Levy M, Kincaid R, Ellington AD. Directed evolution of the substrate specificity of biotin ligase. Biotechnol Bioeng. 2014;111(6):1071–81. doi: 10.1002/bit.25176. [DOI] [PubMed] [Google Scholar]
- Mastrobattista E, Taly V, Chanudet E, Treacy P, Kelly BT, Griffiths AD. High-throughput screening of enzyme libraries: in vitro evolution of a beta-galactosidase by fluorescence-activated sorting of double emulsions. Chem Biol. 2005;12(12):1291–300. doi: 10.1016/j.chembiol.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Miyazaki K, Wintrode PL, Grayling RA, Rubingh DN, Arnold FH. Directed evolution study of temperature adaptation in a psychrophilic enzyme. J Mol Biol. 2000;297(4):1015–26. doi: 10.1006/jmbi.2000.3612. [DOI] [PubMed] [Google Scholar]
- Naik MT, Suree N, Ilangovan U, Liew CK, Thieu W, Campbell DO, Clemens JJ, Jung ME, Clubb RT. Staphylococcus aureus Sortase A transpeptidase. Calcium promotes sorting signal binding by altering the mobility and structure of an active site loop. J Biol Chem. 2006;281(3):1817–26. doi: 10.1074/jbc.M506123200. [DOI] [PubMed] [Google Scholar]
- Ong JL, Loakes D, Jaroslawski S, Too K, Holliger P. Directed evolution of DNA polymerase, RNA polymerase and reverse transcriptase activity in a single polypeptide. J Mol Biol. 2006;361(3):537–50. doi: 10.1016/j.jmb.2006.06.050. [DOI] [PubMed] [Google Scholar]
- Paegel BM, Joyce GF. Microfluidic compartmentalized directed evolution. Chem Biol. 2010;17(7):717–24. doi: 10.1016/j.chembiol.2010.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson GK, Mitchell TJ. The biology of Gram-positive sortase enzymes. Trends Microbiol. 2004;12(2):89–95. doi: 10.1016/j.tim.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Paul S, Stang A, Lennartz K, Tenbusch M, Uberla K. Selection of a T7 promoter mutant with enhanced in vitro activity by a novel multi-copy bead display approach for in vitro evolution. Nucleic Acids Res. 2013;41(1):e29. doi: 10.1093/nar/gks940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race PR, Bentley ML, Melvin JA, Crow A, Hughes RK, Smith WD, Sessions RB, Kehoe MA, McCafferty DG, Banfield MJ. Crystal structure of Streptococcus pyogenes sortase A: implications for sortase mechanism. J Biol Chem. 2009;284(11):6924–33. doi: 10.1074/jbc.M805406200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinisi A, Popp MW, Antos JM, Pansegrau W, Savino S, Nissum M, Rappuoli R, Ploegh HL, Buti L. Development of an influenza virus protein array using Sortagging technology. Bioconjug Chem. 2012;23(6):1119–26. doi: 10.1021/bc200577u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic S, Hegde RS. Identification of a targeting factor for posttranslational membrane protein insertion into the ER. Cell. 2007;128(6):1147–59. doi: 10.1016/j.cell.2007.01.036. [DOI] [PubMed] [Google Scholar]
- Strijbis K, Spooner E, Ploegh HL. Protein ligation in living cells using sortase. Traffic. 2012;13(6):780–9. doi: 10.1111/j.1600-0854.2012.01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Yamamoto T, Tsukiji S, Nagamune T. Site-specific protein modification on living cells catalyzed by Sortase. Chembiochem. 2008;9(5):802–7. doi: 10.1002/cbic.200700614. [DOI] [PubMed] [Google Scholar]
- Tawfik DS, Griffiths AD. Man-made cell-like compartments for molecular evolution. Nat Biotechnol. 1998;16(7):652–6. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- Ton-That H, Mazmanian SK, Faull KF, Schneewind O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. Sortase catalyzed in vitro transpeptidation reaction using LPXTG peptide and NH(2)-Gly(3) substrates. J Biol Chem. 2000;275(13):9876–81. doi: 10.1074/jbc.275.13.9876. [DOI] [PubMed] [Google Scholar]
- Witte MD, Theile CS, Wu T, Guimaraes CP, Blom AEM, Ploegh HL. Production of unnaturally linked chimeric proteins using a combination of sortase-catalyzed transpeptidation and click chemistry. Nat Protocols. 2013;8(9):1808–1819. doi: 10.1038/nprot.2013.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M, Williams DM, Brown DM, Gherardi E. An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J Mol Biol. 1996;255(4):589–603. doi: 10.1006/jmbi.1996.0049. [DOI] [PubMed] [Google Scholar]
- Zaher HS, Unrau PJ. Selection of an improved RNA polymerase ribozyme with superior extension and fidelity. RNA. 2007;13(7):1017–26. doi: 10.1261/rna.548807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Dawes G, Stemmer WP. Directed evolution of a fucosidase from a galactosidase by DNA shuffling and screening. Proc Natl Acad Sci U S A. 1997;94(9):4504–9. doi: 10.1073/pnas.94.9.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.