

Introduction
Caudal regression syndrome is a rare congenital abnormality in which a segment of the spine and spinal cord fails to develop. The severity of the morphologic derangement inversely correlates with residual spinal cord function [1].
Cases of lower spine agenesis with additional anomalies of the axial skeleton and internal organs are now considered as defects of blastogenesis, originating in the primary developmental field and/or the progenitor fields, representing polytopic field defects. Sacral agenesis or caudal dysplasia is an anomaly found more often in offspring of diabetic women. Severe forms are commonly associated with cardiac, renal and respiratory problems, which are responsible for early neonatal death.
Case Report
A four year old male child born to a diabetic mother was referred for magnetic resonance imaging (MRI) of the spine to exclude spinal dysraphysm. The child was born with imperforate anus for which a corrective surgery in multiple stages was carried out, following which the patient had impaired control over the bladder and bowel movements. The child had numbness over both the lower limbs. The legs showed wasting of both proximal and distal muscles with impairment in walking. The buttocks were flattened and the gluteal cleft shortened. There was dextrocardia associated with tricuspid regurgitation.
Clinical and ultrasound examinations revealed bilateral undescended testes. Radiographic examination revealed partial absence of distal sacrum and complete absence of coccyx (Fig. 1). MRI of the spine was performed with sagittal and axial T1WI, T2WI (Fig. 2) along with MR myelography. The multiplanar study demonstrated abrupt wedge-shaped truncation of the tip of the conus, at the level of LV1- “cord terminus“. Partial absence of distal sacrum and complete absence of coccyx was also demonstrated.
Fig. 1.
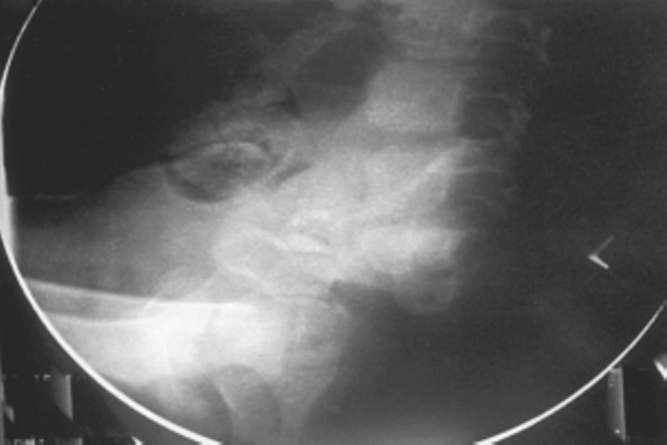
Lateral pelvic radiograph showing complete coccygeal and partial sacral agenesis.
Fig. 2.
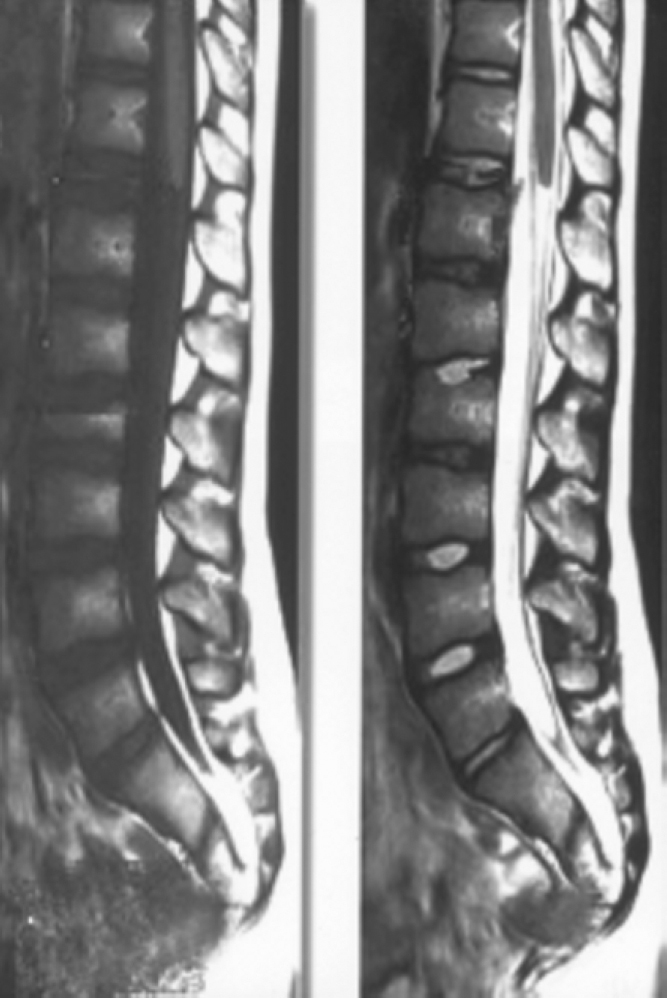
T1 and T2 weighted sagittal images of lumbosacral spine showing partial agenesis of sacrum, truncated cord and filum terminale.
Discussion
Caudal regression syndrome is an uncommon malformation seen in 0.1-0.25:10,000 of normal pregnancies. However it occurs in about one in 350 infants of diabetic mothers, representing an increase of about 200 times. The orthopedic, gastrointestinal, genitourinary and cardiac anomalies are commonly seen with this condition [1]. The clinical picture features a shortened trunk due to the absence of the spine, flexion-rotation-abduction contracture of the hip joints, flexion contracture of the knee joints and clubfeet.
Caudal regression syndromes with lumbo sacracral agenesis may range from absent coccyx as an isolated finding without neurologic sequelae, to sacral or lumbosacral agenesis. Other anomalies include imperforate anus, malformed genitalia, renal dysplasia or aplasia and sirenomyelia (fused lower extremities). In extreme cases, the last intact vertebra is DV11 or DV12 [2]. Caudal regression syndrome may have spinal anomalies involving the caudal segments of spine resulting from disturbed development of the caudal cell mass. The caudal cell mass gives rise to conus of the spinal cord, filum and distal nerve roots via canalization and retrogressive differentiation. It gives rise to lower spine and associated neural structures at the same time as primitive cloaca develops into ano-rectal and genitourinary structures. The embryologic insult occurs at the mid posterior axis mesoderm. The aetiology of caudal regression syndrome remains unclear. If the cord is tethered in addition to the lumbosacral dysgenesis, it will have a stretched, elongated shape similar to what is seen in a tethered cord without lumbosacral hypogenesis. When tethering is not present, the MR will reveal a characteristic wedge-shaped appearance of cord terminus. The sacrum should then be scrutinized to assess the degree of hypogenesis [3].
The associated orthopaedic anomalies may range from deformities of feet, flexion contractures of hips and knees, dislocation of hips, pelvic deformity, kyphoscoliosis and absence of ribs. Anorectal atresia, inguinal hernia, abdominal wall defects, malrotation of gut, imperforate anus are the gastrointestinal anomalies. Tracheo-esophageal, rectovaginal, rectourethral fistulas are encountered along with the condition. Vesicoureteral reflux, hydronephrosis, fused kidneys, renal agenesis, ectopic ureters, transposition of external genitalia due to mullerian duct agenesis besides the neural tube defects, congenital heart defects, strabismus, midline facial cleft have also been recorded.
Diagnosis can be made in the first trimester by noting the short crown – rump length [4]. Sonography in second or third trimester can also demonstrate the absence of sacrum and shortened femurs. The legs may be flexed and abducted at the hips, and there may be clubfeet. Sonography may detect associated urinary anomalies, such as renal agenesis, cystic dysplasias, caliectasis and gastro-intestinal anomalies, such as duodenal atresia [5].
Renshaw [6], had classified the spectrum of caudal regression syndrome into Type I with total or partial unilateral sacral agenesis, Type II with variable lumbar and total sacral agenesis and the ilia articulating with sides of the lowest vertebra, Type III with variable lumbar and a total sacral agenesis, the caudal end plate of the lowest vertebra resting above either fused ilia or an iliac amphiarthrosis, Type IV with fusion of soft tissues of both the lower limbs, Type V also known as “sirenomielia” or “mermaid syndrome” wherein there is a single femur and tibia. Pinter and Reece [7], have demonstrated that hyperglycemia induced alterations in neural tube closure include disordered cells, decreased mitoses, and changes indicating premature maturation. Altered oxidative metabolism from maternal diabetes may cause increased production of free oxygen radicals in the developing embryo, which may be teratogenic. If caudal regression is associated with bilateral renal agenesis, the defect is lethal. Severe sacral agenesis is associated with lower extremity neurologic compromise, and severe bowel and bladder dysfunction [8].
Conflicts of Interest
None identified
References
- 1.Tortori-Donati P, Fondelli MP, Rossi A, Charles AR, Armando C, Valeria C. Segmental spinal dysgenesis: neuro-radiologic findings with clinical and embryologic correlation. Am J Neuroradiol. 1999;20:445–456. [PMC free article] [PubMed] [Google Scholar]
- 2.Osborn Anne G. 1sted. Vol. 19. Mosby Inc; Missouri: 1994. pp. 809–810. (Diagnostic Neuroradiology. Normal anatomy and congenital anomalies of the spine and spinal cord). [Google Scholar]
- 3.Stark David D, Bradley Willam G., Jr. Magnetic Resonance Imaging. In: Armstrong Derek C., editor. 3rded. Vol. 89. Mosby Inc; Missouri: 1999. pp. 1924–1925. (Pediatric spine). [Google Scholar]
- 4.Baxi L, Warren W, Collins MH. Early detection of caudal regression syndrome with transvaginal scanning. Obstet Gynecol. 1990;75:486. [PubMed] [Google Scholar]
- 5.Carol MR, Stephanie R Wilson, Charboneau William, Johnson Jo-Ann M. Diagnostic ultrasound. In: Sauerberi Eric E., editor. 3rded. Vol. 38. Mosby Inc; Missouri: 2005. p. 1296. (The Fetal Spine). [Google Scholar]
- 6.Renshaw TS. Vol. 1. Raven Press; New York: 1994. p. 2214. (Sacral Agenesis. The Pediatric Spine — Principles and Practice). [Google Scholar]
- 7.Pinter E, Reece EA. Arachidonic acid prevents hypreglycemia-associated yolk sac damage and embryopathy. Am J Obstet Gynecol. 1986;166:691. doi: 10.1016/s0002-9378(86)80001-1. [DOI] [PubMed] [Google Scholar]
- 8.Mcgahan John P, Gianluigi P, Nyberg David A. 1sted. Vol. 7. Williams & Wilkins; Lippincott: 2003. pp. 321–323. (Neural tube defects and the spine: Diagnostic imaging of fetal anomalies). [Google Scholar]