

Abstract
Lymphomas with subtle patterns in the marrow can be a diagnostic challenge, unless a high index of suspicion is maintained. We present two patients with aggressive lymphomas who presented with cytopenias and the subsequent bone marrow examinations yielded surprising results. These cases highlight the potential usefulness of a bone marrow examination in the diagnosis of lymphomas in the absence of nodal or other tissue specimens.
Keywords: Intravascular large B cell lymphoma, IVLBCL, Hepatosplenic T-cell lymphoma, HSTL
Introduction
The diagnosis and classification of lymphomas has traditionally been made on excision biopsy specimens of lymph nodes. The role of bone marrow examination is to adequately stage the lymphoma and to evaluate response after therapy [1]. In some instances, however, the bone marrow can yield crucial primary diagnostic information, in the absence of nodal tissue. We present two patients, who presented with cytopenias. Their bone marrow examinations yielded the primary diagnosis. These cases are presented here to highlight the subtle findings that can be present in these lymphomas. Increased awareness of the morphologic findings can hasten the time to diagnosis and timely therapy.
Patient 1
A 68 year old male presented with intermittent fever, cough and profound weakness for 2 months. Past history was not significant for any major illness. Clinical examination showed pallor with splenomegaly (4 cm below costal margin) without lymphadenopathy. Laboratory investigation showed normocytic anemia with pancytopenia. The complete blood count (CBC) was as follows: WBC 3.19 × 109/L (normal range 4–11 × 109/L); Hb 67 g/L (normal range 140–180 g/L); platelets 80 × 109/L (normal range 150–400 × 109/L). The automated differential count was neutrophils 62 %, lymphocytes 26.5 %, monocytes 10 %, eosinophils 0 % and basophils 0.5 %. Enzyme lactate dehydrogenase was 376 IU/ml (normal range 100–250 IU/ml) and reticulocyte count was normal. Serum creatinine was normal. Liver function test revealed mild elevation of liver enzymes [Alanine transaminase 80 IU/ml (normal range 10–50 IU/ml); Aspartate transaminase 48 IU/ml (normal range 5–37 IU/ml0]. Infectious workup for tuberculosis was negative. Imaging by PET-CT showed metabolically-avid splenomegaly but no lymphadenopathy or any other FDG-avid lesion.
A bone marrow aspirate showed scattered large cells with bizarre morphology. The trephine biopsy revealed hypercellular marrow with an intravascular infiltrate of large lymphoid cells with moderately abundant cytoplasm, thick nuclear membranes, clumped chromatin and mitotic figures (Fig. 1). Immunostains showed that these cells were intensely positive for CD20 (Fig. 2) and CD45; and were negative for CD3, suggesting marrow involvement by Intravascular large B-cell lymphoma (IVLBCL).
Fig. 1.
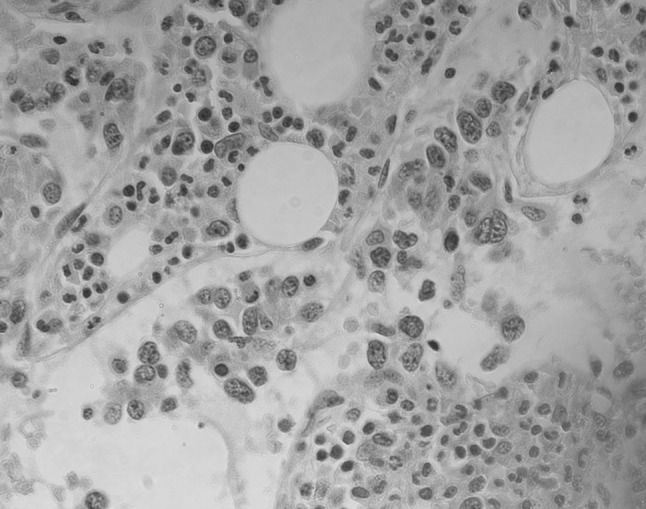
Hematoxylin and eosin-stained sections of trephine biopsy from patient 1 showing large lymphoid cells restricted to within blood vessel lumens. The interstitial marrow has trilineage hematopoiesis. (×400 magnification)
Fig. 2.
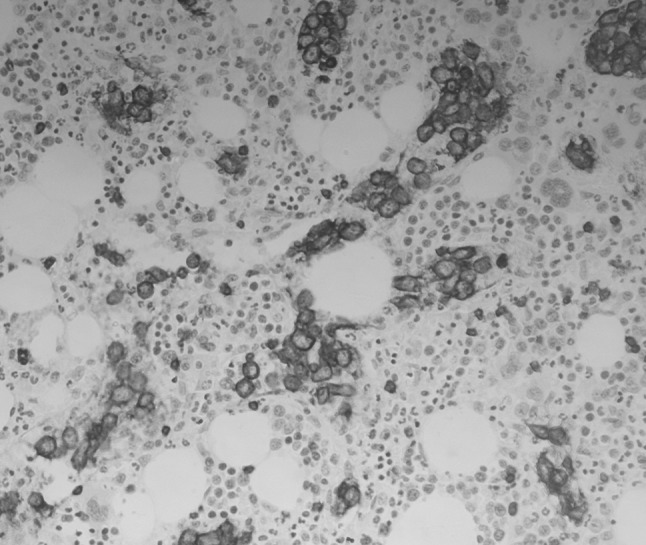
Trephine biopsy section from patient 1 showing intravascular CD20-positive lymphoma cells (×200 magnification)
Cerebrospinal fluid analysis was negative for lymphoma cells. He was started on steroid therapy along with other supportive measures. On the fifth day of admission, he developed sudden-onset perspiration and dizziness, followed by unconsciousness and hypotension. Blood gas analysis showed mild acidosis. He was started on intravenous fluids and ionotropic drugs. Electrocardiographic monitoring showed marked ST segment elevation. He developed asystole and could not be revived despite standard resuscitation protocol. The patient’s family did not consent to post mortem biopsies or sampling.
Patient 2
A 36 year old male patient presented with fever associated with chills, massive hepatosplenomegaly without lymphadenopathy and thrombocytopenia. Laboratory investigation showed normocytic anemia with severe thrombocytopenia. His CBC was as follows: WBC 11.46 × 109/L (normal range 4–11 × 109/L); Hb 89 g/L (normal range 140–180 g/L); platelets 3 × 109/L (normal range 150–400 × 109/L). The differential count was myelocyte 3 %, band forms 2 %, neutrophils 55 %, lymphocytes 25 %, monocytes 13 % and eosinophils 2 %. Nucleated RBCs were noted on the peripheral smear. Enzyme lactate dehydrogenase was 399 IU/ml (normal range 100–250 IU/ml). Serum triglycerides were noted to be 497 mg/dL (Normal range <150 mg/dL).
Approximately 2 months prior to this, he had been diagnosed with immune thrombocytopenia with a platelet count of 30 × 109/L. A bone marrow aspirate at that time showed unremarkable marrow. A biopsy was not done at that time. He was treated with high-dose dexamethasone and subsequently rituximab, and was found to be refractory to therapy.
A repeat bone marrow examination was performed. The aspirate smear showed florid hemophagocytosis (Fig. 3), and occasional clusters of large cells (Fig. 4). The bone marrow trephine showed hemophagocytosis as well as subtle infiltration by large cells (Fig. 5). Immunohistochemical staining showed that these cells were CD3-positive (Fig. 6), CD56-positive, and were in a sinusoidal as well as interstitial pattern. They were negative for CD20, and other T-cell markers. Abundant CD68-positive histiocytes were evident, reflecting concomitant hemophagocytosis. The unique pattern and CD3-positivity, along with the clinical history of massive hepatosplenomegaly made this entity consistent with Hepatosplenic T-cell lymphoma (HSTL). A liver biopsy could not be performed because of severe thrombocytopenia. The patient expired within 3 weeks of the second biopsy after receiving a single round of etoposide.
Fig. 3.
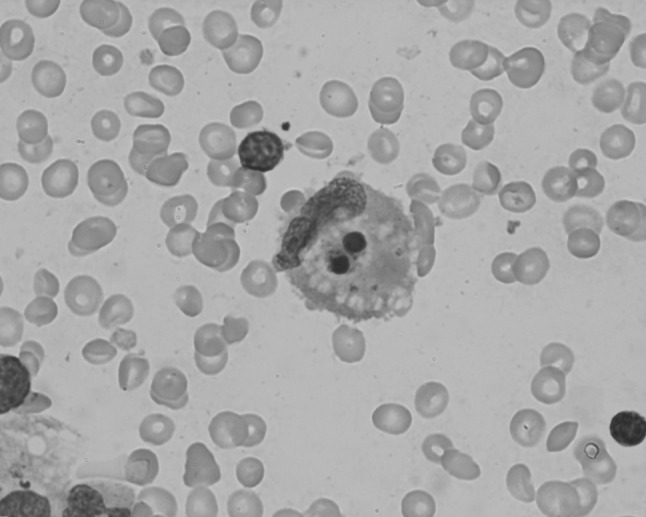
Leishman-stained bone marrow aspiration smear from patient 2 showing hemophagocytosis (Oil immersion lens, ×1000 magnification)
Fig. 4.

Leishman-stained bone marrow aspiration smear from patient 2 showing clusters of large lymphoid cells with fine chromatin (Oil immersion lens, ×1000 magnification)
Fig. 5.
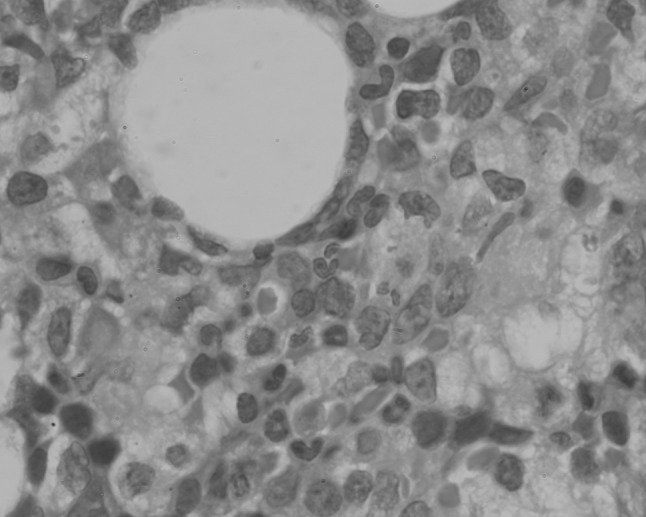
Hematoxylin and eosin-stained sections of trephine biopsy section from patient 2 showing sinusoidally arranged large cells (×400 magnification)
Fig. 6.
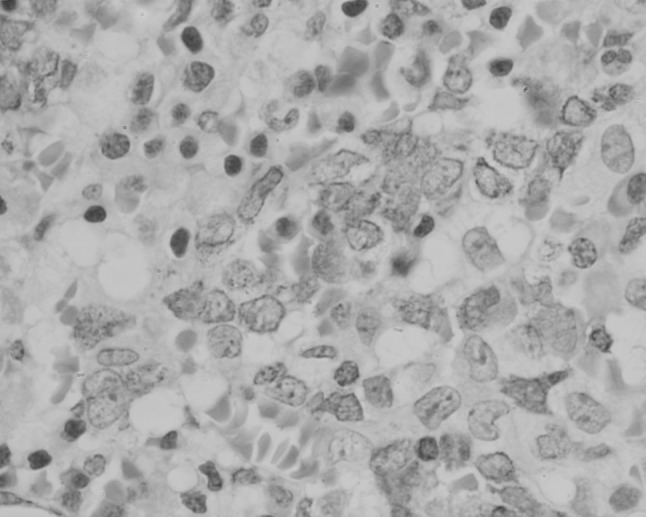
Trephine biopsy section from patient 2 showing distension of sinusoids by CD3-positive lymphoma cells (×400 magnification)
Discussion
IVLBCL is a rare subtype of non-Hodgkin lymphoma. When initially described, it was referred to as malignant angioendotheliomatosis and intravascular malignant lymphomatosis. However, in the 2008 World Health Organization classification; it has been classified as a subtype of diffuse large B-cell lymphoma (DLBCL) [2]. The hallmark of this type of DLBCL is the presence of lymphoma cells within blood vessel lumens, predominantly capillaries and post-capillary venules. The description of this entity is limited to case reports and series and estimated incidence is less than one person per one million individuals. It has been described in a wide range of ages (34–90 years) with equal male to female occurrence. Clinical presentation of IVBCL is quite variable and is dependent on organ compromise due to lymphoma cells causing microvasculopathy. Two clinical variants have been described—an Asian variant in which patients present with multi-organ failure, hepatosplenomegaly, pancytopenia and hemophagocytosis; and a Western variant in which patients present with central nervous system (CNS) involvement or cutaneous involvement [3].
Our patient presented with pancytopenia and splenomegaly. His bone marrow showed presence of B-cells restricted to intravascular spaces; and PET-CT showed no avid lesions except for splenomegaly. The other pathologic entity that can present with a similar morphology in the marrow is a diffuse large B-cell lymphoma with primary splenic and bone marrow involvement [4]. However, the pattern of marrow involvement in such lymphomas is a mix of interstitial and intravascular involvement, unlike that seen in our index case. The cell of origin for IVLBCL is still elusive. The presence of clonally rearranged immunoglobulin genes makes a transformed mature post-germinal B-cell likely [2, 5]. The neoplastic cells commonly express B-cell-associated antigens CD20 and CD79a. Aberrant expression of CD5 is seen in approximately one-third of cases [2]. A purported explanation for the restricted distribution of the malignant cells to the intravascular compartment is a defect of homing receptors on the lymphoma cells, with a lack of adhesion molecules CD29, CD54 and CD11a [6]. IVLBCL are aggressive lymphomas. As its occurrence is rare, there is lack of data comparing different treatment protocols. Majority of treatment strategies are extrapolated from DLBCL. Chemotherapy regimens in addition to anti-CD20 monoclonal antibodies have been reported with variable success [7]. Monotherapy with anti-CD20 antibodies leading to sustained remission has been described as well [8]. The treatment strategy for the Western variant of IVBCL with CNS involvement includes high dose methotrexate, as penetration to brain is poor for conventional chemotherapy used [9].
IVLBCL is frequently a late diagnosis in many patients with very short survival times. Our patient also had a rapid downhill course. We speculate that an acute coronary event due to lymphoma cells might be a plausible reason of demise. However, due to lack of post-mortem examination, this could not be confirmed.
HSTL as defined by the WHO 2008 classification, is a systemic extranodal cytotoxic T-cell neoplasm of γδ- type of T-cell receptor, with marked sinusoidal infiltration of spleen, liver and bone marrow [2]. It was first described in 1990 by Farcet and colleagues [10]. The postulated cell of origin is a mature peripheral γδ cytotoxic memory T-cell. It is common in adolescents and young adults with a male predominance [11]. An unusual association has been reported in patients with chronic immunosuppression, or after treatment with azathioprine and infliximab [12]. Patients present with marked splenomegaly and/or hepatomegaly without lymphadenopathy and systemic symptoms. Thrombocytopenia with anaemia and/or leucopenia is common. A characteristic cytogenetic abnormality seen isochromosome 7q [2].
Our patient presented with refractory thrombocytopenia, anaemia and marked hepatosplenomegaly. Morphologically, the marrow showed presence of CD3-positive T cells with an intra-sinusoidal distribution. The other lymphoproliferative neoplasm that can share this morphology is a T-large granular lymphocytic leukemia (T-LGL). However, T-LGL is a more indolent disease characterized by persistent (>6 months) elevation of peripheral blood large granular lymphocytes, neutropenia and concomitant autoimmune disorders in elderly patients [2]. Morphologically, the sinusoidal infiltrate in T-LGL has been described as occurring in linear arrays, versus an expanded sinusoidal infiltrate in HSTL [13].Immunophenotypically, T-LGL is a neoplasm of mature CD8-positive, TCR- αβ-positive cytotoxic T-cells. Mortality due to T-LGL is very uncommon.
HSTL is frequently associated with hemophagocytosis due to release of cytokines from the neoplastic T-cells, and the extent of this phenomenon may mask the presence of neoplastic cells [1]. In the absence of flow cytometry, immunophenotyping by immunohistochemistry is essential to classify this lymphoma. Classically, the neoplastic T-cells in HSTL are CD2+, CD3+, CD4−, CD5−, CD8±, T-cell receptor (TCR)γδ+ [2]. The median survival is less than 2 years, with a rapidly progressive clinical course [14]. This lymphoma is resistant to most chemotherapeutic agents, and complete clinical remission after conventional chemotherapy is rare [15]. The severity of thrombocytopenia appears to correlate with disease progression, and re-occurrence of thrombocytopenia after therapy heralds impending relapse [16]. As mentioned above, HSTL is frequently associated with hemophagocytosis, and its presence may also correlate with a rapidly downhill course [17]. Other factors that were associated with poor prognosis were male gender, absence of TCR gene rearrangement, failure to achieve complete response and HSTL arising in the post-transplant setting [18, 19].
Like the situation with IVLBCL, standardization of treatment in HSTL is hindered by the rarity of the lymphoma, and treatment regimens are frequently extrapolated from those of peripheral T-cell lymphoma (PTCL). Among the treatment regimens that have been used are corticosteroids, alkylating agents, purine analogues, anthracycline-containing regimens and cytarabine–cisplatinum combinations [15]. Response rates of 30–45 % complete remission with anthracycline-based chemotherapy have been seen, however with poor overall long term results [20]. Alemtuzumab, an anti-CD52 monoclonal antibody used in combination with purine analogs has shown no significant effect on survival [21, 22]. There have been case reports and small studies that have shown encouraging results with autologous or allogeneic transplant after first remission [15].
Summary
Both cases described above represent rare entities with aggressive and frequently fulminant course. They represent challenges in diagnosis as well as in treatment. In the absence of lymphadenopathy and/or accessible organs to biopsy, bone marrow examinations can frequently aid in diagnosis. A high index of suspicion and use of immunohistochemical stains (even limited panels) can help confirm these lymphomas. Better treatment modalities are needed for both of these distinct entities.
References
- 1.Foucar K, Reichard K, Czuchlewski D. Bone marrow pathology. 3. Chicago: ASCP Press; 2010. [Google Scholar]
- 2.Swerdlow SH, International Agency for Research on Cancer., World Health Organization . WHO classification of tumours of haematopoietic and lymphoid tissues. World Health Organization classification of tumours. 4. Lyon: International Agency for Research on Cancer; 2008. [Google Scholar]
- 3.Ferreri AJ, Dognini GP, Campo E, Willemze R, Seymour JF, Bairey O, Martelli M, De Renz AO, Doglioni C, Montalban C, Tedeschi A, Pavlovsky A, Morgan S, Uziel L, Ferracci M, Ascani S, Gianelli U, Patriarca C, Facchetti F, Dalla Libera A, Pertoldi B, Horvath B, Szomor A, Zucca E, Cavalli F, Ponzoni M. Variations in clinical presentation, frequency of hemophagocytosis and clinical behavior of intravascular lymphoma diagnosed in different geographical regions. Haematologica. 2007;92(4):486–492. doi: 10.3324/haematol.10829. [DOI] [PubMed] [Google Scholar]
- 4.Morice WG, Rodriguez FJ, Hoyer JD, Kurtin PJ. Diffuse large B-cell lymphoma with distinctive patterns of splenic and bone marrow involvement: clinicopathologic features of two cases. Mod Pathol. 2005;18(4):495–502. doi: 10.1038/modpathol.3800297. [DOI] [PubMed] [Google Scholar]
- 5.Yegappan S, Coupland R, Arber DA, Wang N, Miocinovic R, Tubbs RR, Hsi ED. Angiotropic lymphoma: an immunophenotypically and clinically heterogeneous lymphoma. Mod Pathol. 2001;14(11):1147–1156. doi: 10.1038/modpathol.3880450. [DOI] [PubMed] [Google Scholar]
- 6.Ponzoni M, Arrigoni G, Gould VE, Del Curto B, Maggioni M, Scapinello A, Paolino S, Cassisa A, Patriarca C. Lack of CD 29 (beta1 integrin) and CD 54 (ICAM-1) adhesion molecules in intravascular lymphomatosis. Hum Pathol. 2000;31(2):220–226. doi: 10.1016/S0046-8177(00)80223-3. [DOI] [PubMed] [Google Scholar]
- 7.Shimada K, Matsue K, Yamamoto K, Murase T, Ichikawa N, Okamoto M, Niitsu N, Kosugi H, Tsukamoto N, Miwa H, Asaoku H, Kikuchi A, Matsumoto M, Saburi Y, Masaki Y, Yamaguchi M, Nakamura S, Naoe T, Kinoshita T. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189–3195. doi: 10.1200/JCO.2007.15.4278. [DOI] [PubMed] [Google Scholar]
- 8.Bazhenova L, Higginbottom P, Mason J. Intravascular lymphoma: a role for single-agent rituximab. Leuk Lymphoma. 2006;47(2):337–341. doi: 10.1080/10428190500300837. [DOI] [PubMed] [Google Scholar]
- 9.Ponzoni M, Ferreri AJ, Campo E, Facchetti F, Mazzucchelli L, Yoshino T, Murase T, Pileri SA, Doglioni C, Zucca E, Cavalli F, Nakamura S. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168–3173. doi: 10.1200/JCO.2006.08.2313. [DOI] [PubMed] [Google Scholar]
- 10.Farcet JP, Gaulard P, Marolleau JP, Le Couedic JP, Henni T, Gourdin MF, Divine M, Haioun C, Zafrani S, Goossens M, et al. Hepatosplenic T-cell lymphoma: sinusal/sinusoidal localization of malignant cells expressing the T-cell receptor gamma delta. Blood. 1990;75(11):2213–2219. [PubMed] [Google Scholar]
- 11.Vose J, Armitage J, Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–4130. doi: 10.1200/JCO.2008.16.4558. [DOI] [PubMed] [Google Scholar]
- 12.Mackey AC, Green L, Liang LC, Dinndorf P, Avigan M. Hepatosplenic T cell lymphoma associated with infliximab use in young patients treated for inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 2007;44(2):265–267. doi: 10.1097/MPG.0b013e31802f6424. [DOI] [PubMed] [Google Scholar]
- 13.Chen YH, Chadburn A, Evens AM, Winter JN, Gordon LI, Chenn A, Goolsby C, Peterson L. Clinical, morphologic, immunophenotypic, and molecular cytogenetic assessment of CD4-/CD8-gammadelta T-cell large granular lymphocytic leukemia. Am J Clin Pathol. 2011;136(2):289–299. doi: 10.1309/AJCPTFFQ18JMYKDF. [DOI] [PubMed] [Google Scholar]
- 14.Tripodo C, Iannitto E, Florena AM, Pucillo CE, Piccaluga PP, Franco V, Pileri SA. Gamma-delta T-cell lymphomas. Nat Rev Clin Oncol. 2009;6(12):707–717. doi: 10.1038/nrclinonc.2009.169. [DOI] [PubMed] [Google Scholar]
- 15.Foppoli M, Ferreri AJ. Gamma-delta t-cell lymphomas. Eur J Haematol. 2014 doi: 10.1111/ejh.12439. [DOI] [PubMed] [Google Scholar]
- 16.Belhadj K, Reyes F, Farcet JP, Tilly H, Bastard C, Angonin R, Deconinck E, Charlotte F, Leblond V, Labouyrie E, Lederlin P, Emile JF, Delmas-Marsalet B, Arnulf B, Zafrani ES, Gaulard P. Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood. 2003;102(13):4261–4269. doi: 10.1182/blood-2003-05-1675. [DOI] [PubMed] [Google Scholar]
- 17.Nosari A, Oreste PL, Biondi A, Costantini MC, Santoleri L, Intropido L, Muti G, Pungolino E, Gargantini L, Morra E. Hepato-splenic gammadelta T-cell lymphoma: a rare entity mimicking the hemophagocytic syndrome. Am J Hematol. 1999;60(1):61–65. doi: 10.1002/(SICI)1096-8652(199901)60:1<61::AID-AJH10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 18.Gaulard P, Belhadj K, Reyes F. Gammadelta T-cell lymphomas. Semin Hematol. 2003;40(3):233–243. doi: 10.1016/S0037-1963(03)00137-9. [DOI] [PubMed] [Google Scholar]
- 19.Schafer E, Chen A, Arceci RJ. Sustained first remission in an adolescent with hepatosplenic T-cell lymphoma treated with T-cell leukemia induction, nucleoside analog-based consolidation, and early hematopoietic stem cell transplant. Pediatr Blood Cancer. 2009;53(6):1127–1129. doi: 10.1002/pbc.22129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weidmann E. Hepatosplenic T cell lymphoma. A review on 45 cases since the first report describing the disease as a distinct lymphoma entity in 1990. Leukemia. 2000;14(6):991–997. doi: 10.1038/sj.leu.2401784. [DOI] [PubMed] [Google Scholar]
- 21.Mittal S, Milner BJ, Johnston PW, Culligan DJ. A case of hepatosplenic gamma-delta T-cell lymphoma with a transient response to fludarabine and alemtuzumab. Eur J Haematol. 2006;76(6):531–534. doi: 10.1111/j.1600-0609.2006.00646.x. [DOI] [PubMed] [Google Scholar]
- 22.Jaeger G, Bauer F, Brezinschek R, Beham-Schmid C, Mannhalter C, Neumeister P. Hepatosplenic gammadelta T-cell lymphoma successfully treated with a combination of alemtuzumab and cladribine. Ann Oncol. 2008;19(5):1025–1026. doi: 10.1093/annonc/mdn119. [DOI] [PubMed] [Google Scholar]