

Abstract
Visceral leishmaniasis (VL) is endemic in many parts of India. Rarely, it may be complicated by hemophagocytic lymphohistiocytosis (HLH) that has varied presentation and course. We describe two cases of VL complicated by HLH that were markedly different in clinical presentation, course and management. First case presented with Fever of unknown origin whereas second case had fever with severe bleeding manifestations. VL was diagnosed by bone marrow aspiration and serum rk39 immunodiagnostic test respectively in these cases. HLH was diagnosed by HLH 2004 diagnostic criteria. VL was treated by intravenous amphotericin B in both cases. HLH was managed by treating primary disease in the first case whereas steroid was given for management in the second case. High index of suspicion is crucial for early diagnosis of HLH to reduce morbidity and mortality.
Keywords: Hemophagocytic lymphohistiocytosis (HLH), Visceral leishmaniasis, Fever of unknown origin, Kala-azar
Introduction
Fever may occasionally pose great challenge in diagnosis and management. Visceral leishmaniasis (VL), a common cause of fever in many endemic areas, is increasing at alarming rate in many parts of the world [1]. Infrequently, it is complicated with HLH which is a rare and potentially fatal condition. Diagnosis of HLH requires high index of suspicion and pose real challenge when associated with diseases like VL because of overlapping clinical features leading to significant under-diagnosis.
Here, we discuss two cases of VL associated HLH with markedly different presentation, diagnostic features and therapeutic course despite having the same diagnosis. This highlights the need of having clinical awareness and high index of suspicion for early diagnosis of this challenging and highly fatal disease, if not treated on time [2].
Case Report
Case 1
Fifty three year old female presented with the complaint of fever with loss of appetite and weight for the last 3 months. The fever was intermittent high grade and associated with chills, rigor and profuse sweating. There were no other systemic complaints. Clinical examination revealed mild pallor, enlarged right axillary lymph node and hepatosplenomegaly. Other systemic examinations were non-contributory.
Initial hematological examination showed moderate anemia (hemoglobin 7.2 g/dl), leucopenia (2300 cells/µl) and normal platelets (176,000/µl). Peripheral smear showed normochromic normocytic anemia with reactive lymphocytes and toxic granules. Liver function tests showed hypoalbuminemia with reversal of A:G ratio (1:2.4) while kidney function tests were normal. Serological markers for HIV, HCV, HBV, brucellosis as well as widal test were negative. The blood culture reports were sterile. CECT abdomen showed hepatosplenomegaly. Further investigations revealed high serum ferritin (1427 ng/ml), hypofibrinogenemia (124 mg/l) and raised CRP levels (5 mg/l). On the basis of HLH 2004 criteria, a diagnosis of secondary HLH was made. Bone marrow aspirate examination revealed LD bodies, which confirmed VL as the primary cause (Fig. 1). Workup for other infective causes like EBV serology, CMV pp65 antigen turned out to be negative.
Fig. 1.
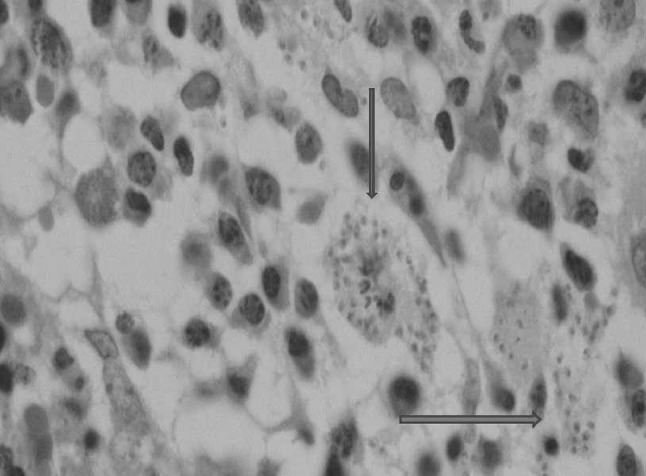
Photomicrograph of cellular bone marrow biospy showing extra as well as intracellular LD bodies
She was started i.v. amphotericin B deoxycholate (total dose 1 mg/kg), which was discontinued after 6 days due to deranged kidney function tests (urea 37 mg/dl, creatinine 1.8 mg/dl). Her kidney function tests normalized after 1 week and she was again started with amphotericin B under close supervision. Her symptoms and biochemical derangements improved in 2 weeks.
Patient improved clinically with resolution of fever and hepatosplenomegaly and was discharged after 3 weeks.
Case 2
A 30 year old truck driver, resident from endemic area of kala-azar, presented to our emergency department with severe epistaxis, severe thrombocytopenia and prolonged aPTT. In emergency department, he was managed by nasal packing and transfused three units of packed RBC and six units of platelet-rich plasma. He gave the history of fever with bleeding manifestations for the last 1 month. Fever was high-grade, associated with chills, rigors and with bleeding from multiple sites and generalized purpuric rashes. On examination, he had tachycardia (120/min), normal blood pressure (100/70 mm of Hg) with fever (103.5 °F), pallor and pedal edema. There were multiple ecchymotic and purpuric rashes all over the body. Abdominal examination revealed hepatosplenomegaly with ascites. Other systemic examinations were unremarkable.
Initial hemogram showed pancytopenia (hemoglobin 5.9 mg/dl, leukocyte count 2800/µl and platelet count 10,000/µl). Urine and blood cultures were negative. Liver enzymes were elevated (SGOT 216 IU/l, SGPT 142 IU/l) with hypoalbuminemia (2.5 g/dl) with reversal of A:G ratio (globulin 4.1 g/dl). Other biochemical investigations revealed raised serum LDH (1376 U/l), markedly raised serum ferritin (34,661.8 µg/l) and serum triglycerides (346 mg/dl). These biochemical reports raised the possibility of HLH syndrome. Hence, bone marrow aspiration and biopsy was done which revealed active hemophagocytosis and histiocytic predominance (Fig. 2). NK cell activity was also found to be low as compared to healthy family control. Thus, 7 out 8 HLH 2004 diagnostic criteria were met which confirmed our diagnosis of sHLH. Screening for secondary causes for HLH including common infective, autoimmune and neoplastic conditions tuned out to negative except for serum rk39 immunodiagnostic test which was positive. In view of clinical presentation along with positive rk39, in a patient residing in endemic area, a diagnosis of VL with secondary HLH was made.
Fig. 2.
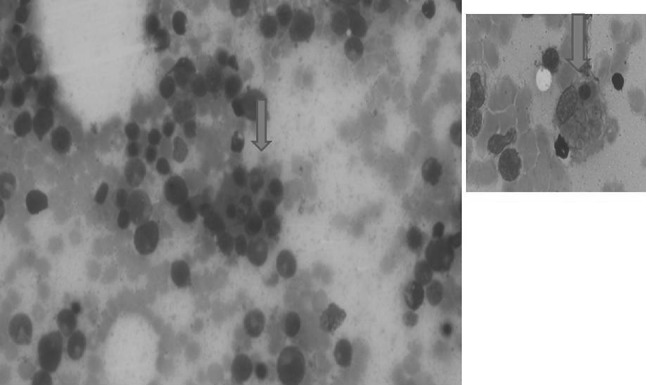
Photomicrograph with arrow showing a macrophage with hemophagocytosis
The patient was treated with liposomal amphotericin B in a dose of 3 mg/kg (210 mg) for 5 days with repeat dosing at day 8, day 15 and day 28. In view of sHLH, he was started on injection dexamethasone 18 mg/day in divided doses.
The patient improved clinically (resolution of fever and hepatosplenomegaly) and hematologically (hemoglobin increased to 9.6 g/dl, TLC increased to 9600/µl and platelet count to 90,000/µl) in a week. There was no further bleeding manifestations. The patient was discharged after 4 weeks with normal hematological and biochemical parameters.
Discussion
HLH is a rare clinico-pathological entity caused by dysregulation in natural killer T cell function and cytokines production. It results in inappropriate activation and proliferation of monocyte-macrophage-histiocytic lineage with uncontrolled hemophagocytosis. The primary form of the disease occurs due to genetic mutations and found in young children [3]. Another form of the disease occurs secondary to conditions like infections, malignancies, autoimmune diseases or immunosuppression [4]. Most common infectious etiologies include EBV among virus and VL among parasites [5]. The HLH syndrome, which is similar in both forms, is diagnosed as per HLH-2004 [6] guidelines that includes eight criteria—fever, splenomegaly, cytopenia of at least two cell lines, hypofibrinogenemia or hypertriglyceridemia, demonstrable hemophagocytosis, ferritin level >500 µg/l, low/absent NK cell activity and soluble CD25 >2400 U/ml. Fulfillment of any five criteria is required for diagnosis. Both of our patients satisfied the above criteria.
Being a rare entity with non-specific signs and symptoms, diagnosing HLH is challenging as this may be easily confused with any systemic infection. High index of clinical suspicion along with high serum ferritin levels is highly valuable in guiding the diagnosis. Serum ferritin is an important inflammatory biomarker which is routinely measured in most centers. It is highly elevated in four rare conditions like HLH, AOSD, catastrophic antiphospholipid syndrome and septic shock-together postulated as hyperferritiemic syndrome [7]. Serum ferritin level >10,000 µg/l has a 90 % sensitivity and 96 % specificity for diagnosis of HLH [8] and markedly high level of ferritin (34,661.8 µg/l) in our second case practically confirmed the diagnosis. Even for levels >500 µg/l, it has a sensitivity and specificity of 82 and 42 % respectively.
Both our patients were residents of Bihar, a state in India which is a high endemic zone for VL [9]. Still there have been very few case reports of HLH, making us think that HLH may be significantly under-recognized entity in VL as it may either cause early mortality [10] or just be in-apparent [11] resolving with only specific therapy, as in our first case. Thus, reinforcing need for having high clinical awareness about HLH-2004 diagnostic criteria.
The microscopic diagnosis of Leishmaniasis is difficult and repeated bone marrow studies may be required as first bone marrow is negative in 36.3 % of cases for LD bodies due to pauci-microbial nature of disease in its early course [2]. Thus, serological diagnosis might be helpful in making earlier diagnosis of VL. The rapid diagnostic test (rk39) has 97 % sensitivity and 98.9 % specificity, good prognostic value as well as earlier sero-conversion [12, 13]. It can be a valuable serologic tool complimenting parasitological diagnosis.
Management of HLH is challenging for obvious reasons. The entity, being rare, is not considered at onset. Moreover, its recognition is not easy and the clinical picture is often misleading. For familial HLH, specific immunosuppression with etoposide, dexamethasone and cyclosporine A is important [6]. However in sHLH, associated with infections, intense immunosuppression may have fatal consequences especially in countries like India with high prevalence of tuberculosis.
The cornerstone of the treatment of HLH associated with VL is treatment of primary disease. In a systematic review, liposomal amphotericin B was found successful in all reported cases whereas antimonials were successful in only 65.4 % of cases [2]. If the HLH syndrome is recognized early and specific antimicrobial therapy is started, majority of the patients show prompt resolution of signs and symptoms without need of any adjunctive therapy [2], as in first case. In others with more severe presentation, like second case, additive steroid therapy along with antimicrobial treatment might be required. Few studies have reported beneficial role of IVIG in severe HLH [14] or coexistent sepsis, in which it buys time for evaluation in severely ill patients [2]. However, because of the inconsistent results and high cost of IVIG, adjunctive therapy with IVIG should be initiated carefully.
Acknowledgments
Conflict of interest
All participated authors in this study declare no financial, professional, or personal conflict of interests.
Consent
Informed written consent of the patient was taken.
References
- 1.Shaw J. The leishmaniases–survival and expansion in a changing world. Mem Inst Oswaldo Cruz. 2007;102:541–547. doi: 10.1590/S0074-02762007000500001. [DOI] [PubMed] [Google Scholar]
- 2.Rajagopala S, Dutta U, Chandra KSP, Bhatia P, Varma N, Kochhar R. Visceral leishmaniasis associated hemophagocytic lymphohistiocytosis—case report and systematic review. J Infect. 2008;56:381–388. doi: 10.1016/j.jinf.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Henter J-I, Arico M, Elinder G. Familial hemophagocyticlymphohistiocytosis (primary HLH) Hematol Oncol Clin North Am. 1998;12:417–433. doi: 10.1016/S0889-8588(05)70520-7. [DOI] [PubMed] [Google Scholar]
- 4.Janka G, Elinder G, Imashuku S. Infection and malignancy associated hemophagocytic syndromes—secondary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 1998;12:435–444. doi: 10.1016/S0889-8588(05)70521-9. [DOI] [PubMed] [Google Scholar]
- 5.Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814–822. doi: 10.1016/S1473-3099(07)70290-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 7.Rosário C, Zandman-Goddard G, Meyron-Holtz EG, D’Cruz DP, Shoenfeld Y. The hyperferritinemic syndrome—macrophage activation syndrome, Still’s disease, septic shock and catastrophic anti-phospholipid syndrome. BMC Med. 2013;11:185. doi: 10.1186/1741-7015-11-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dierickx D, Vandenberghe P, Verhoef G. Hemofagocytaire lymfohistiocytose. Ned Tijdschr Hematol. 2006;3:214–220. [Google Scholar]
- 9.Leishmaniasis: disease and burden [Internet] (2014 July 06), Geneva: WHO 2014. http://www.who.int/leishmaniasis/burden/en/
- 10.Sipahi T, Tavil B, Oksal A. Visceral leishmaniasis and pseudomo-nas septicemia associated with hemophagocytic syndrome and myelodysplasia in a Turkish child. Turk J Pediatr. 2005;47:191–194. [PubMed] [Google Scholar]
- 11.Gagnaire MH, Galambrun C, Stephan JL. Hemophagocytic syndrome: a misleading complication of visceral leishmaniasis in children-a series of 12 cases. Pediatrics. 2000;106:E58. doi: 10.1542/peds.106.4.e58. [DOI] [PubMed] [Google Scholar]
- 12.Mathur P, Samantaray J, Chauhan NK. Evaluation of a rapid im-munochromatographic test for diagnosis of kala-azar and post kala-azar dermal leishmaniasis at a tertiary care centre of north India. Indian J Med Res. 2005;122:485–490. [PubMed] [Google Scholar]
- 13.Chappuis F, Rijal S, Soto A, Menten J, Boelaert M. A meta-analysis of the diagnostic performance of the direct agglutination test and rK39 dipstick for visceral leishmaniasis. BMJ. 2006;333:723. doi: 10.1136/bmj.38917.503056.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emmenegger U, Frey U, Reimers A, Fux C, Semela D, Cottagnoud P, et al. Hyperferritinemia as indicator for intravenous immunoglobulin treatment in reactive macrophage activation syndromes. Am J Hematol. 2001;68:4–10. doi: 10.1002/ajh.1141. [DOI] [PubMed] [Google Scholar]