

Abstract
Introduction
Polycythemia vera (PV) is one of the most common forms of myeloproliferative neoplasms. Acute myeloid leukemia secondary to PV is well reported, and the mechanism has been clarified to some extent. Only a limited number of cases have been reported about the development of acute lymphoblastic leukemia (ALL) in the course of PV, and the possible underlying mechanism has not been explored well.
Case presentation
A 75-year-old patient who developed ALL 3 years after he was diagnosed with PV. The presence of remarkable splenomegaly, typical immunophenotyping of the peripheral blood and increased expression of serum fibrosis markers indicated the existence of extramedullary hematopoiesis which may ascribe to myelofibrosis. After the treatment of dosage-modulated chemotherapy, the patient got complete remission.
Conclusion
The JAK2 mutation may the underlying factor that contributes to the development of ALL, and the existence of MF may indicate the progression to post- polycythemic MF, which may be a risk factor for the accelerated transformation.
Keywords: Polycythemia vera, Acute lymphocytic leukemia, JAK2 V617F mutation, Post-polycythemic MF
Background
Polycythemia vera (PV) is one of the most common myeloproliferative neoplasms; it is typically characterized by excessive proliferation of erythroid cells, and sometimes, increase in the granulocyte and platelet counts. Pruritus, hypertension, hemorrhage and thrombosis are common complications. The JAK2 V617F mutation is found in 95 % of PV cases, and has been the main indication for the diagnosis of PV [1]. Acute myeloid leukemia (AML) secondary to PV is observed in 10 % of PV patients who undergo hydroxyurea treatment, usually within 13 years of the treatment [2]. The risk of AML transformation and the molecular pathogenesis have been explored to some extent [3]. On the other hand, development of acute lymphoblastic leukemia (ALL) after the diagnosis of PV is rare. As far as we know, only eight cases of ALL accompanying PV have been reported [4], and there is no consensus on the underlying cause or mechanism. Here, we report a case of PV wherein the patient developed ALL 3 years later.
Case Presentation
A 75-year-old man was admitted to the hospital in April 2012 for edema in both lower limbs for 15 days. No splenomegaly was observed, and the routine blood test showed that the white blood cell (WBC) count was 10.3 × 109/L; red blood cell (RBC) count, 9.04 × 1012/L; hemoglobin (Hb), 182 g/L; mean cell volume (MCV), 69.1 fl; hematocrit (HCT), 62.5 %; and platelets (PLT), 11 × 109/L. The serum iron concentration was 5.1 μmol/L (normal range 9.0–32 μmol/L) and ferritin concentration was 50.8 ng/mL (normal range 7.0–323 ng/mL). Finally, examination of the bone marrow smear and detection of the JAK2 V617F mutation led to the diagnosis of PV. Hydroxyurea was administered at a dose of 500–1000 mg/day. Hydroxyurea treatment at this dosage was continued for 3 years, and peripheral RBC separation was performed twice, in 2013 and in 2014.
On 6 February 2015, the patient presented with distention in the left lower abdomen, accompanied by fatigue and night sweats but no pain. A routine blood test showed the WBC count to be 15.91 × 109/L; RBC count, 7.63 × 1012/L; Hb, 191 g/L; MCV, 84.7 fl; HCT, 64.6 %; and PLT, 50 × 109/L. However, the patient was not admitted, and he was not administered any treatment. One month later, he presented to the hospital again with aggravate symptoms mentioned above. The routine blood test showed that the WBC count was 23.2 × 109/L; Hb, 134 g/L; and PLT, 11 × 109/L. Examination of the bone marrow smear showed the presence of 78.5 % primary cells, with suppressed proliferation of granulocyte and RBCs. Myeloperoxidase (MPO), cholesterol esterase (CE) and naphthol butyrate esterase (NBE) were negative expression (≤5 %), and extracellular iron was absent. Immunophenotyping of the marrow using flow cytometry (FCM) showed that 38 % of the cells were abnormal, and mainly expressed HLA-DR, CD10, CD19, CD20, CD22, CD34, CD38, cCD79α and TdT (see Fig. 1). ALL was considered as a possible diagnosis, based on which 10 mg dexamethasone (DXM) per day was administered for 5 days.
Fig. 1.

Immunophenotyping of the marrow by flow cytometry when onset of acute lymphoblastic leukemia. Showed that 38 % of the cells were abnormal, and mainly expressed HLA-DR, CD10, CD19, CD20, CD22, CD34, CD38, cCD79α and TdT
Ten days later, the patient visited our hospital with remarkable splenomegaly, and the routine blood test showed the WBC count to be 5.9 × 109/L; RBC, 4.56 × 1012/L; Hb, 115 g/L; MCV, 76 fl; HCT, 34.5 %; and PLT, 9 × 109/L. Peripheral blood analysis showed the presence of 5 % irregularly shaped lymphocytes, 7 % neutrophilic myelocytes, 9 % neutral rod-shaped nuclear cells, 56 % segmented neutrocyte, 17 % lymphocytes, 6 % mononuclear leucocytes and 7 % immature RBCs. The serum lactic dehydrogenase level was 466 U/L (normal range 109–245 U/L). Abdominal ultrasound examination showed that the spleen was expanded, with uniform echogenicity and a diameter of about 7.0 cm. Abdominal CT showed a low-density shadow-shaped wedge that appeared patchy in the periphery of the spleen, which was assumed to be a splenic infarction (see Fig. 2). Reexamination of the bone marrow showed hypoplasia with 11 % lymphoblasts, characterized by a circular nucleus, loose meticulous chromatin, one to three nucleoli and a small amount of light blue cytoplasm (see Fig. 3). Further, the expression of peroxidase staining (POX) was negative (expression rate was 5 %). The karyotype of the patient was normal (46, XY [12]), and the presence of the JAK2 V617F mutation was confirmed. Further, SIL-TAL1, E2A-HLF, TEL-AML1, MLL-AF4, E2A-PBX1, HOX11, HOX11L2, CAML-AF10 and BCR-ABL were negative expression. Immunophenotyping of the peripheral blood using FCM revealed the presence of two groups of abnormal cells: P1 cells, 4.2 %; P2 cells, 2.5 %. The P1 cells mainly expressed CD33 (37.2 %), HLA-DR (70.9 %), CD117 (82.3 %), CD34 (73.6 %), CD38 (93.8 %), and CD11b (39.7 %), while the P2 cells mainly expressed CD19 (47.8 %), HLA-DR (50.8 %), CD34 (36.1 %), CD20 (47.3 %), CD22 (69.7 %), CD10 (71.2 %) and CD38 (61.6 %) (see Fig. 4). cCD79α and TdT expression was not detected. The patient had a history of hypertension for 2 years, with the diastolic pressure being 180/130 mmHg. Moreover, the patient had chronic hepatitis B viral hepatitis for 40 years and was positive for the surface antigen, core antibody and e antibody.
Fig. 2.
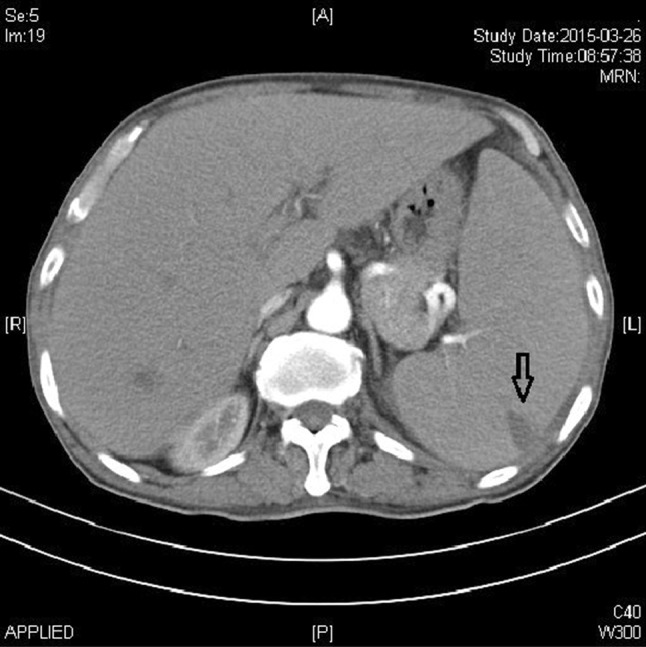
Abdominal computed tomography scan. Showed a low-density shadow-shaped wedge that appeared patchy in the periphery of the spleen (marked with black arrow), which was assumed to be a splenic infarction
Fig. 3.
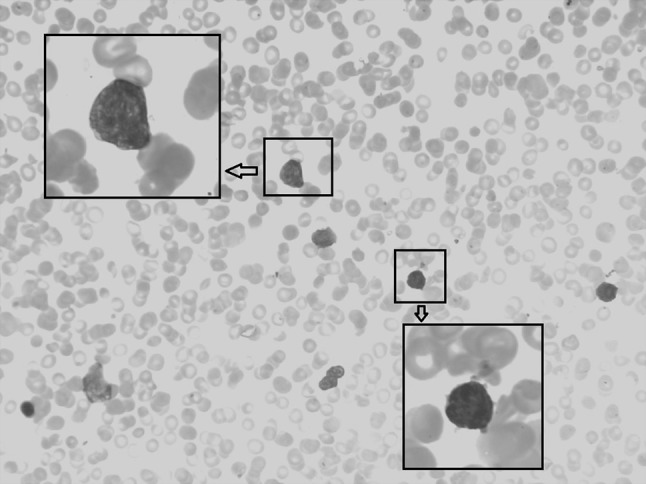
Morphological feature of bone marrow when onset of ALL. After Wright-Giemsa staining, slides were observed under a light microscope and photographed under 10 × 40 resolution (10 × 100 resolution under oil microscopy). Showed hypoplasia with 11 % lymphoblasts, characterized by a circular nucleus, loose meticulous chromatin, one to three nucleoli and a small amount of light blue cytoplasm (color figure online)
Fig. 4.

Immunophenotyping of the peripheral blood by flow cytometry. Revealed the presence of two groups of abnormal cells: P1 cells, 4.2 %; P2 cells, 2.5 %. The P1 cells mainly expressed CD33, HLA-DR, CD117, CD34, CD38 and CD11b. While the P2 cells mainly expressed CD19, HLA-DR, CD34, CD20, CD22, CD10 and CD38
On 24 March 2015, the patient was administered dosage-modulated VDCP chemotherapy, which included vindesine 4 mg qW × 3 weeks and DXM 10 mg qd × 14 days at gradually decreasing doses, pharmorubicin 30 mg and cyclophosphamide 400 mg on day +14. Lumber puncture and intrathecal chemotherapy were done before and after the chemotherapy, and the examination of cerebrospinal fluid was normal. One and a half months later, the routine blood test showed a WBC count of 16.8 × 109/L; Hb, 112 g/L; and PLT, 359 × 109/L. Peripheral blood analysis showed that it was composed of 1 % neutrophilic myelocytes, 6 % neutral rod-shaped nuclear cells, 63 % segmented neutrocyte, 15 % lymphocytes, 13 % mononuclear leucocytes, 2 % acidophilic cells and 7 % immature RBCs. The presence of the JAK2V617F mutation was confirmed again, and the serum LDH level was determined to be 484 U/L (normal range 109–245 U/L). Analysis of the bone marrow smear revealed excessive proliferation of erythroid cells (48.5 %), primitive and immature lymphocyte percentage of 3 %, and 37 megakaryocytes in a single smear (see Fig. 5). Bone marrow biopsy did not indicate myelofibrosis (MF), while the serum fibrosis marker levels were as follows: laminin, 108 μg/L (normal value <135 μg/L); collagen IV, 97.8 μg/L (normal range 54.77–84.77 μg/L); hyaluronic acid, 158 ng/mL (normal range 20–110 ng/L); procollagen III, 128 μg/L (normal range 20–120 μg/L). The patient is now undergoing the second round of chemotherapy with dosage-modulated VDCLP (VDCP and l-asaraginase).
Fig. 5.
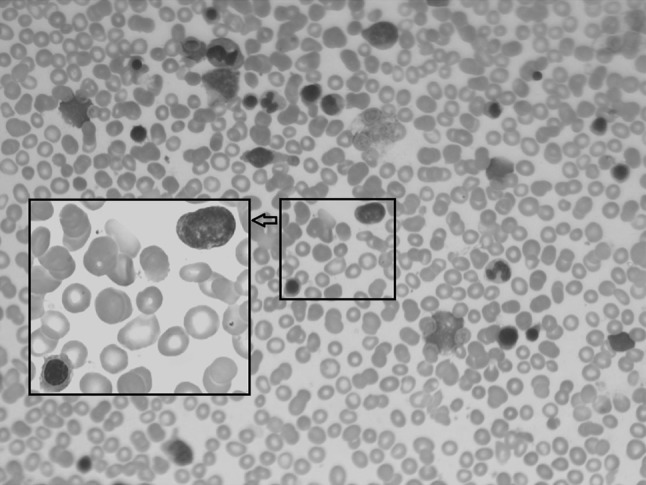
Morphological feature of bone marrow after first round of chemotherapy. After Wright-Giemsa staining, slides were observed under a light microscope and photographed under 10 × 40 resolution (10 × 100 resolution under oil microscopy). Revealed excessive proliferation of erythroid cells and existence of primitive and immature lymphocyte
Discussion and Conclusion
The mechanism of ALL development after the development of PV is not clear. Previous reports indicate that during the progression of PV, 38–56 % of patients may develop karyotype abnormalities [5]. It is believed that genetic alterations may be one of the initial factors causing leukemic transformation. The typical course of transformation was reported in a case in which the patient progressed from PV to chronic myelocytic leukemia (CML) and then ALL within a period of 11 years [6]. Another view is that the patient may have a group of primitive pluripotent stem cells, which may be the co-precursor cells of PV and ALL, and different clonal dominance may be manifested in different stages [7]. One study also suggested that PV and ALL originate from two completely different clones, based on cytogenetic analysis [8].
Although the JAK2 V617F mutation was determined to be the underlying cause of myeloproliferative neoplasms (MPNs) in 2008 [9], it was not clear whether the mutation had been previously present in all the eight MPN-ALL cases. It is clear that the JAK2 V617F mutation presents not only in most MPN cases, but also some cases of AML, CML and myelodysplastic syndrome [10–12]; moreover, it has also been detected in patients with chronic lymphocytic leukemia (CLL). So far, 30 CLL patients have been reported to have the JAK2 V617F mutation, two of whom show no evidence of MPN [13]. Additional animal experimentation has also confirmed the crucial effect of the JAK2 gene in the development of B cell lymphoma/leukemia [14]. Moreover, JAK2 V617F-positive MPN can also be accompanied by B cell lymphoma and plasmacytic malignancy [15, 16]. Besides, PV can also occur after the treatment of primary lymphatic diseases. It has been reported that a patient developed JAK2 V617F-positive PV after the treatment of primary diffuse large B cell lymphoma [17]. Further, two patients were diagnosed with primary ALL at the age of 2 and 4 years, and developed PV after they were treated for ALL for 7 and 6 years respectively [18, 19]. The median age of PV onset is about 60 years, and it is <20 years in only <0.1 % of the cases [20]. In the case of children, we cannot exclude the possibility that ALL and PV may develop from the same clone. All the studies so far have confirmed the relationship between PV and lymphatic diseases, and the role of the JAK2 V617F mutation, which leads to numerical and structural abnormalities of the JAK2 gene [21]. However, the mechanism underlying the effect of this mutation still needs to be studied in detail.
In our case, the patient developed ALL 3 years after he was diagnosed with PV, which is much shorter than the period reported previously (5–25 years) [4]. In the case of our patient, the detection of the JAK2 V617F mutation before and after the development of ALL, and the marrow presentation after the first round of chemotherapy proves the co-existence of PV and ALL. The possibility of disappearance of the JAK2 V617F mutation in AML secondary to MPN has been reported [22], but it is not clear whether the previously reported eight MPN-ALL cases were accompanied with PV after the development of ALL. In our case of ALL secondary to PV, the patient had normal B cells, and progressive enlargement of the spleen was observed before ALL was confirmed. Based on the peripheral cell compositions and the FCM results, the existence of extramedullary hematopoiesis can be confirmed in the present case. MF is the main cause of extramedullary hematopoiesis; it may also be the natural course of PV, and is also called post-polycythemic MF (post-PV PMF). So far, two cases of ALL after the development of post-PV PMF have been reported [23, 24]. In one case, the patient had PV for 17 years before MF was diagnosed, and developed ALL a year after MF. In the other case, PV was accompanied by MF, and the patient developed ALL 3 years later. The risk of leukemia transformation is the highest in cases of MPN accompanied by PMF, and the incidence may reach 21 % in high-risk PMF patients [25], with a median progression time of 31 months [26]. MF in the course of PV may be a risk factor for the development of ALL. In our case, the biopsy was not performed at the time of onset of ALL, and pathological examination after the chemotherapy showed the absence of MF. In general, the extent of splenomegaly is in accordance with the extensive degree of MF, and microscopic observation did not reveal any positive relationship of the splenomegaly with MF. MF may be focal or unevenly distributed [27]. We further detected the level of serum markers that are indicative of MF: elevated expression of collagen IV, hyaluronic acid and procollagen III were revealed. Based on these findings, post-PV PMF cannot be excluded in our case.
In conclusion, we report a rare case of ALL secondary to PV. The JAK2 V617F mutation may the underlying factor that contributes to the development of ALL, but the mechanism is still unclear. The existence of extramedullary hematopoiesis and elevated expression of markers of serum MF may indicate the progression to post-PV PMF, which may be a risk factor for the accelerated transformation of PV to ALL.
Compliance with Ethical Standards
Conflict of interest
None.
References
- 1.Anastasi J. The myeloproliferative neoplasms: insights into molecular pathogenesis and changes in WHO classification and criteria for diagnosis. Hematol Oncol Clin N Am. 2009;23(4):693–708. doi: 10.1016/j.hoc.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Najean Y, Rain JD. Treatment of polycythemia vera: the use of hydroxyurea and pipobroman in 292 patients under the age of 65 years. Blood. 1997;90(9):3370–3377. [PubMed] [Google Scholar]
- 3.Rampal R, Mascarenhas J. Pathogenesis and management of acute myeloid leukemia that has evolved from a myeloproliferative neoplasm. Curr Opin Hematol. 2014;21(2):65–71. doi: 10.1097/MOH.0000000000000017. [DOI] [PubMed] [Google Scholar]
- 4.Camós M, Cervantes F, Montoto S, Hernández-Boluda JC, Villamor N, Montserrat E. Acute lymphoid leukemia following polycythemia vera. Leuk Lymphoma. 1999;32(3–4):395–398. doi: 10.3109/10428199909167404. [DOI] [PubMed] [Google Scholar]
- 5.Rege-Cambrin G, Mecucci C, Tricot G, Michaux JL, Louwagie A, Van Hove W, Francart H, Van den Berghe H. A chromosomal profile of polycythemia vera. Cancer Genet Cytogenet. 1987;25(2):233–245. doi: 10.1016/0165-4608(87)90183-X. [DOI] [PubMed] [Google Scholar]
- 6.Roth AD, Oral A, Przepiorka D, Gollin SM, Chervenick PA. Chronic myelogenous leukemia and acute lymphoblastic leukemia occurring in the course of polycythemia vera. Am J Hematol. 1993;43(2):123–128. doi: 10.1002/ajh.2830430210. [DOI] [PubMed] [Google Scholar]
- 7.Neilson JR, Patton WN, Williams MD, Mayne EE, Boughton BJ. Polycythaemia rubra vera transforming to acute lymphoblastic leukaemia with a common immunophenotype. J Clin Pathol. 1994;47(5):471–472. doi: 10.1136/jcp.47.5.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anastasi J, Pettenati MJ, Le Beau MM, Kwaan HC, Weil SC. Acute lymphoblastic leukemia in a patient with longstanding polycythemia vera: cytogenetic analysis reveals two distinct abnormal clones. Am J Hematol. 1988;29(1):33–37. doi: 10.1002/ajh.2830290108. [DOI] [PubMed] [Google Scholar]
- 9.Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22(1):14–22. doi: 10.1038/sj.leu.2404955. [DOI] [PubMed] [Google Scholar]
- 10.Lee HJ, Daver N, Kantarjian HM, Verstovsek S, Ravandi F. The role of JAK pathway dysregulation in the pathogenesis and treatment of acute myeloid leukemia. Clin Cancer Res. 2013;19(2):327–335. doi: 10.1158/1078-0432.CCR-12-2087. [DOI] [PubMed] [Google Scholar]
- 11.Xu W, Chen B, Tong X. Chronic myeloid leukemia patient with co-occurrence of BCR-ABL junction and JAK2 V617F mutation. Int J Hematol. 2014;99(1):87–90. doi: 10.1007/s12185-013-1480-z. [DOI] [PubMed] [Google Scholar]
- 12.Ohyashiki K, Aota Y, Akahane D, Gotoh A, Miyazawa K, Kimura Y, Ohyashiki JH. The JAK2 V617F tyrosine kinase mutation in myelodysplastic syndromes (MDS) developing myelofibrosis indicates the myeloproliferative nature in a subset of MDS patients. Leukemia. 2005;19(12):2359–2360. doi: 10.1038/sj.leu.2403989. [DOI] [PubMed] [Google Scholar]
- 13.Yang YN, Qin YW, Wang C. JAK2 V617F detected in two B-cell chronic lymphocytic leukemia patients without coexisting Philadelphia chromosome-negative myeloproliferative neoplasms: a report of two cases. Oncol Lett. 2014;8(2):841–844. doi: 10.3892/ol.2014.2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.dos Santos NR, Ghysdael J. A transgenic mouse model for TEL-JAK2-induced B-cell lymphoma/leukemia. Leukemia. 2006;20(1):182–185. doi: 10.1038/sj.leu.2404026. [DOI] [PubMed] [Google Scholar]
- 15.Papageorgiou MV, Alexopoulou A, Kontopidou F, Filiotou A, Koskinas J, Pectasides D. Concomitant diagnosis of myeloproliferative neoplasm and non-Hodgkin’s lymphoma in a patient with portal vein thrombosis. Anticancer Res. 2011;31(4):1467–1469. [PubMed] [Google Scholar]
- 16.Malhotra J, Kremyanskaya M, Schorr E, Hoffman R, Mascarenhas J. Coexistence of myeloproliferative neoplasm and plasma-cell dyscrasia. Clin Lymphoma Myeloma Leuk. 2014;14(1):31–36. doi: 10.1016/j.clml.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 17.Elli EM, Belotti A, Cecchetti C, Realini S, Fedele M, Parma M, Pogliani EM. Development of JAK2V617F-positive polycythemia vera after chemotherapy-induced remission of primary central nervous system diffuse large B cell non-Hodgkin’s lymphoma: a case report and review of the literature. Acta Haematol. 2013;130(3):142–145. doi: 10.1159/000347159. [DOI] [PubMed] [Google Scholar]
- 18.Sutherland ND, Gonzalez-Peralta R, Douglas-Nikitin V, Hunger SP. Polycythemia vera in a child following treatment for acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2004;26(5):315–319. doi: 10.1097/00043426-200405000-00012. [DOI] [PubMed] [Google Scholar]
- 19.Hann HW, Festa RS, Rosenstock JG, Cifuentes E. Polycythemia vera in a child with acute lymphocytic leukemia. Cancer. 1979;43(5):1862–1865. doi: 10.1002/1097-0142(197905)43:5<1862::AID-CNCR2820430540>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 20.Danish EH, Rasch CA, Harris JW. Polycythemia vera in childhood: case report and review of the literature. Am J Hematol. 1980;9(4):421–428. doi: 10.1002/ajh.2830090409. [DOI] [PubMed] [Google Scholar]
- 21.Najfeld V, Cozza A, Berkofsy-Fessler W, Prchal J, Scalise A. Numerical gain and structural rearrangements of JAK2, identified by FISH, characterize both JAK2617V > F-positive and -negative patients with Ph-negative MPD, myelodysplasia, and B-lymphoid neoplasms. Exp Hematol. 2007;35(11):1668–1676. doi: 10.1016/j.exphem.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 22.Yu W, Chen L, Li J, Zhu H. Essential thrombocythemia with JAK2 V617F mutation transformed into acute myeloid leukemia without JAK2 V617F mutation: case report and literature review. Zhonghua Xue Ye Xue Za Zhi. 2014;35(12):1122–1123. doi: 10.3760/cma.j.issn.0253-2727.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 23.Ohanian M, Leventaki V, Verstovsek S, Estrov Z, Lin P, Yin C, Kantarjian H, Huh Y, Ravandi F. Acute lymphoblastic leukemia arising in post-polycythemic myelofibrosis: a rare entity. Leuk Lymphoma. 2012;53(9):1839–1841. doi: 10.3109/10428194.2012.663916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polliack A, Prokocimer M, Matzner Y. Lymphoblastic leukemic transformation (lymphoblastic crisis) in myelofibrosis and myeloid metaplasia. Am J Hematol. 1980;9(2):211–220. doi: 10.1002/ajh.2830090209. [DOI] [PubMed] [Google Scholar]
- 25.Tefferi A, Lasho TL, Jimma T, Finke CM, Gangat N, Vaidya R, Begna KH, Al-Kali A, Ketterling RP, Hanson CA, Pardanani A. One thousand patients with primary myelofibrosis: the mayo clinic experience. Mayo Clin Proc. 2012;87(1):25–33. doi: 10.1016/j.mayocp.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005;105(3):973–977. doi: 10.1182/blood-2004-07-2864. [DOI] [PubMed] [Google Scholar]
- 27.Arrago JP, Poirier O, Chomienne C, D’Agay MF, Najean Y. Type III aminoterminal propeptide of procollagen in some haematological malignancies. Scand J Haematol. 1986;36(3):288–294. doi: 10.1111/j.1600-0609.1986.tb01736.x. [DOI] [PubMed] [Google Scholar]