

Abstract
Congenital dyserythropoietic anemias are a rare group of inherited anemias characterized by ineffective erythropoiesis and distinct morphological abnormalities in the erythroblasts. Interferon alpha has been shown to be effective in type 1 congenital dyserythropoietic anemia but the optimal duration of therapy is undefined. We present here a 32-years-old female patient diagnosed with type 1 congenital dyserythropoietic anemia precipitated by pregnancy and treated successfully with a short course of interferon alpha resulting in a durable response. A literature search including PubMed database on previously published articles regarding congenital dyserythropoietic anemia type 1 patients treated with interferon is conducted.
Introduction
Congenital dyserythropoietic anemias (CDAs) are a highly heterogeneous group of rare, hereditary anemias characterized by abnormalities during late erythropoiesis leading to refractory anemia, reticulocytopenia, hypercellular bone marrow with ineffective erythropoiesis, distinctive dysplastic changes in the erythroblasts and development of secondary hemochromatosis. Table 1 summarizes the characteristic features of the three major types of congenital dyserythropoietic anemias. The diagnosis of CDA relies on hematologic findings and can be confirmed by molecular testing in an increasing number of patients [1, 2]. The correct diagnosis is often delayed leading to ineffective treatments and tissue damage related to iron overload. Interferon alpha (IFN-α) is known to be effective in CDA type 1 but data are sparse and the optimal duration of therapy is a pending issue. We report here a further patient with CDA-1 and a good response to IFN and analyze previously reported CDA-1 patients treated with IFN.
Table 1.
Summary of characteristic features of the three major types of congenital dyserythropoietic anemias
| CDA type | CDA type 1 | CDA type 2 | CDA type 3 |
|---|---|---|---|
| Inheritance | Autosomal recessive | Autosomal recessive | Autosomal dominant and sporadic |
| Frequency | Three times less frequent than CDA type II | Most frequent type | Very rare |
| Anemia | Macrocytic, mild to moderate | Normocytic or macrocytic, moderate | Macrocytic, mild to moderate |
| Erythroblast morphology in the bone marrow | |||
| Light microscopy | Megaloblastic, chromatin bridges | Normoblastic, binuclear erythroblasts (10–35 %) | Giant, multinuclear erythroblasts |
| Electron microscopy | ‘Swiss-cheese’ appearance of the heterochromatin | Peripheral double membranes | Nonspecific |
| Acid serum hemolysis test | Negative | Positive | Negative |
| Iron overload | Yes | Yes | No |
| Hemosiderinuria | No | No | Yes |
| Mutated gene | Codanin-1 | SEC23B | KIF23/MKLP1 |
| Chromosome | 15q15.1.3 | 20p11.23 | 15q21-25 |
| Described abnormalities | Skeletal or other dysmorphic features | Rare | Ocular abnormalities and tendency to develop plasma cell dyscrasias |
| Specific therapy | IFN-α | Splenectomy | None |
Case Report
In April 2013 32-years-old female patient was referred to the hospital with severe anemia, jaundice and splenomegaly. Severe anemia was first recognized in January 2013 at the 8th month of pregnancy (hemoglobin: 4.5 g/dl). She was diagnosed with hemolytic anemia and treated with erythrocyte transfusions, methylprednisolone and vitamin B12 at another hospital. In February 2013, postpartum hemoglobin level was 2.5 g/dl before admission to the emergency department of this hospital in April 2013. On examination she had fever (38.6 °C) and looked distressed. The conjunctivae were pale and icteric. A massively enlarged spleen was palpated. The remainder of the examination was unremarkable. She didn’t have any skeletal dysmorphic features.
The patient had a history of bipolar disorder, first diagnosed in 2006. Medications included olanzapine and sertraline. She did not use alcohol and tobacco. She had two healthy children and previous pregnancies were unremarkable. Her father had lung cancer and her mother had endometrial cancer. Her uncle was diagnosed with Wilson’s disease. The family history revealed no consanguinity.
The complete blood count revealed a hemoglobin: 2.93 g/dl, hematocrit: 8.2 %, MCV: 91 fl, RDW: 16 %. Leukocyte and platelet levels were normal, as was the differential count. The corrected reticulocyte was 1.47 %. Biochemical tests revealed total bilirubin: 5.4 mg/dl, conjugated bilirubin: 0.94 mg/dl, lactate dehydrogenase: 1049 U/liter (240–480 U/liter), iron: 232 μg/dl, total iron-binding capacity: 240 μg/dl, transferin saturation: 96 % and ferritin: 4598 ng/ml. Direct and indirect antiglobulin tests were negative. Haptoglobin was measured <28 (normal range 41–165 mg/dl). Vitamin B12 level was 476 pg/ml and folate 8.8 ng/ml. Copper level was normal.
During follow-up the fever resolved. Infectious causes such as brucellosis, malaria, leishmaniasis, human immunodeficiency virus, cytomegalovirus, parvovirus and Epstearr virus infections were excluded with specific laboratory tests.
Wilson’s disease was also excluded (no Kaiser-Fleischer ring, normal serum copper and seruloplasmin levels and normal liver biopsy results).
Peripheral blood film revealed macrocytic red blood cells with aniso-poikilocytosis and basophilic stippling (Fig. 1).
Fig. 1.
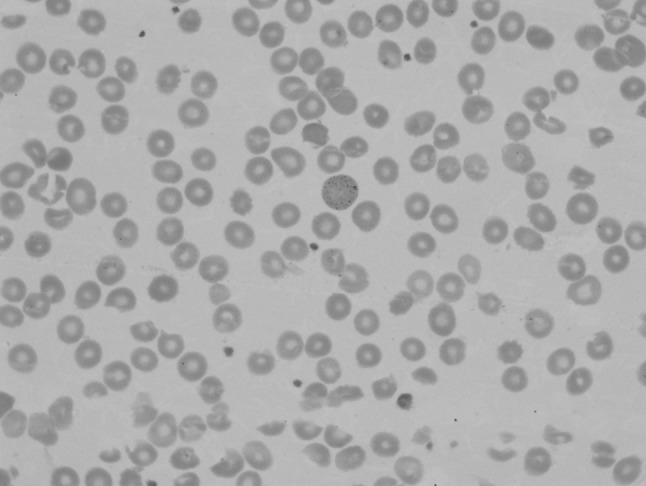
Peripheral blood smear showing basophilic stippling
Bone marrow was hypercellular with erythroid hyperplasia (myeloid/erythroid ratio of 0.13). There was notable megaloblastic development of erythroid precursors and dyserythropoiesis. Inter-nuclear chromatin bridges involving polychromatic erythroblasts were prominent (Fig. 2). At least 500 nucleated erythroblasts were examined and binucleated erythroblasts were less than 5 %. An iron stain of the bone marrow aspirate identified the presence of abundant storage iron and ringed sideroblasts (<15 %). Myeloid and megakaryocytic lineages were unremarkable with a lack of atypical bone marrow cells. Grade 1 reticulin fibrosis was seen. Flow cytometric analysis of the bone marrow aspirate was nondiagnostic. Conventional cytogenetic studies on the marrow aspirate showed a normal female karyotype.
Fig. 2.
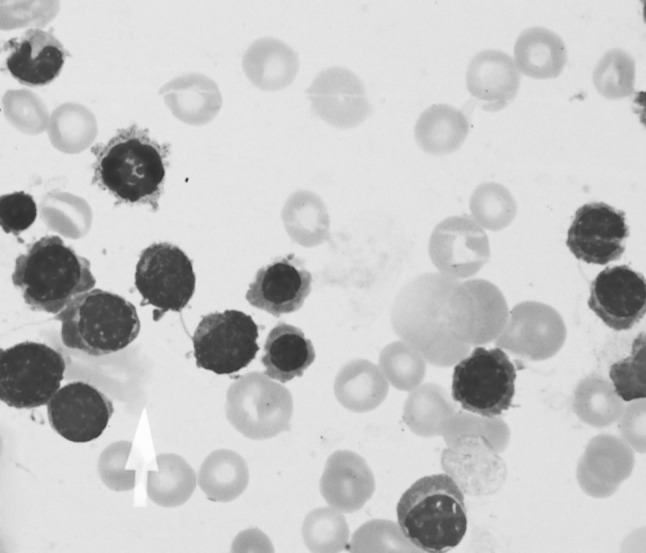
Light microscopy of bone marrow erythroblasts obtained by aspiration (×1000) demonstrating internuclear chromatin bridging
Ultrastructural erythroid features included spongy (Swiss-cheese) heterochromatin (Fig. 3).
Fig. 3.
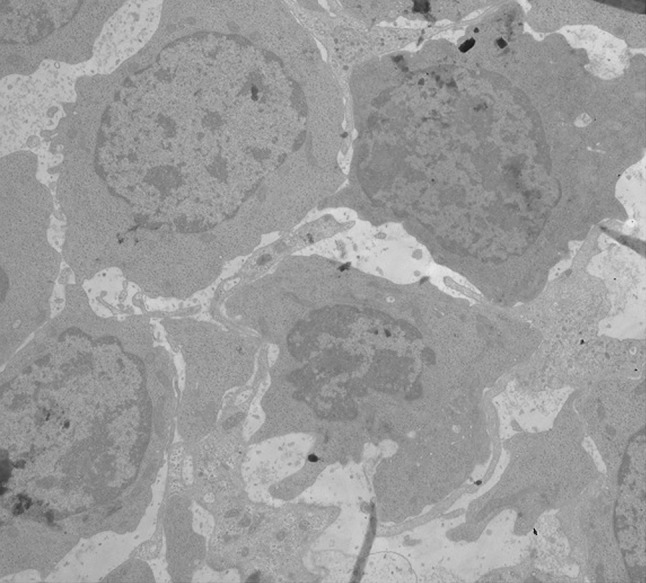
Electron micrographs of bone marrow late erythroblasts with spongy heterochromatin
Other causes of dyserythropoiesis were ruled out. Red cell enzyme, membrane disorders and thalassemias were excluded. G6PD and pyruvate kinase screening were normal. Hemoglobin electrophoresis showed HbA at 97.5 %, HbA2 at 2.4 % and HbF at 0.1 %. Osmotic fragility, sucrose lysis, acidified serum lysis (Ham’s test) tests and FLAER-based assays were unremarkable. Paroxysmal nocturnal hemoglobinuria, myelodysplastic syndrome and erythroleukemia were excluded. Hereditary or acquired sideroblastic anemias were excluded because sideroblasts were not prominent and acquired causes such as myelodysplastic syndromes, pyridoxine deficiency, lead poisoning and copper deficiency were ruled out. Olanzapine and sertraline are not known to produce sideroblastic anemia. A trial of pyridoxine was ineffective. Table 2 summarizes common causes of ineffective erythropoiesis and tests used to exclude them.
Table 2.
Common causes of ineffective erythropoiesis and tests used to exclude them
| Megaloblastic anemia | Normal vitamin B12 and folic acid level |
| Myelodysplastic syndrome | No dysplastic features in the bone marrow |
| Acute myeloid leukemia, FAB-type M6 | No blasts |
| Myelofibrosis | No bone marrow fibrosis |
| Sideroblastic anemia (inherited/acquired) | No microcytosis, no response to pyridoxine; causes of acquired sideroblastic anemia ruled out |
| Thalassemia syndromes | Normal hemoglobin electrophoresis |
| Hereditary spherocytosis | Normal osmotic fragility |
| Pyruvate kinase deficiency | Normal pyruvate kinase level |
Codanin-1 sequencing results did not show any mutation. Liver biopsy performed to exclude Wilson’s disease was consistent with extramedullary hematopoiesis. Findings of ineffective erythropoiesis, morphologically abnormal typical bone marrow erythroid precursors and exclusion of other causes of dyserythropoiesis although genetically not confirmed suggested the diagnosis of congenital dyserythropoietic anemia type 1. IFN-α 3 million units thrice weekly and deferasirox for iron chelation were initiated. After 2 weeks hemoglobin level raised to 8.2 g/dl. In September, after 3 months of therapy she had a hemoglobin of 11.5 g/dl. She did not experience any serious side effect. Hospitalization in the psychiatry clinic was necessary because of the previously diagnosed bipolar disease attack and IFN therapy was stopped after 4 months. The patient has mild anemia without any transfusion more than 1 year after cessation of IFN.
Discussion
Congenital dyserythropoietic anemia is a diagnosis of exclusion [3, 4]. Symptoms of CDA may first appear at any time between the neonatal period and late adulthood. The diagnosis can be made in episodes of erythropoietic stress such as pregnancy [4, 5]. The patient described here presented with severe anemia during pregnancy without having an episode of anemia before.
The patient had an uncle with Wilson’s disease, a diagnosis which must be considered in patients presenting with a Coombs-negative hemolytic episode and a history of neuropsychiatric disease [6]. Normal ceruloplasmin level, urinary copper excretion and absence of Kayser-Fleischer rings excluded diagnosis of Wilson’s disease. The liver biopsy showed findings of extramedullary hematopoiesis within the sinusoids, probably accompanying CDA. Neurological and psychiatric evaluation indicated a psychotic depression.
Presence of ringed sideroblasts in the bone marrow in CDA is a rare finding [7–9]. Absence of hypochromia and microcytosis, unresponsiveness to pyridoxine treatment, normal serum zinc level, exclusion of myelodysplastic syndromes, lead toxicity and copper deficiency made inherited or acquired sideroblastic anemia unlikely in our patient.
Light and electron microscopy findings of the bone marrow erythroblasts, the gold standard for CDA type 1 determination [4] were consistent with the diagnosis. The causative gene mutation for CDA type 1 (Codanin-1 mutation) was described first in 2002 [10]. So far, approximately 30 CDAN1 mutations have been reported in CDA type 1 patients. However no pathogenic CDAN1 mutations have been detected in 20 % of patients [11]. Although the case described above had the classic features of CDA-1, we failed to identify any mutation. This can be explained by the genetic heterogeneity of the disease.
CDA-1 patients are at high risk for complications regarding the pregnancy and delivery [5]. The patient underwent an elective cesarean section at term and the baby was born healthy.
Treatment of CDAs is mostly supportive and includes erythrocyte transfusions and iron chelation. Splenectomy can increase hemoglobin levels and eliminate the need for transfusions in CDA type 2 patients without having a beneficial effect on iron overload [4, 12]. Hematopoieitic stem cell transplantation is the only available curative treatment modality and can be considered in transfusion dependent, severe patients with human leukocyte antigen-identical donors [13].
CDA-1 patients treated with IFN have been reported so far [14–24] but data remained sparse. IFN treatment was started with the intention to increase hemoglobin level in our patient. Although most of the CDA-1 patients responded well to IFN therapy, patients having no favorable effect had also been reported [22]. Stopping IFN therapy in responding patients resulted in a drop in hemoglobin levels and optimal dose and duration of IFN treatment (life-long continuous or intermittent) is undefined. Table 3 summarizes information on previously published CDA-1 patients treated with IFN and the present patient. Our patient is unique in that response to IFN therapy emerged immediately and was durable after cessation of a short course of therapy. After a period of regular transfusion for 6 months, following a short course of IFN therapy (4 months) the patient is followed up without any treatment with Hb levels of 11–12 g/dl for 22 months. The apparent response to IFN treatment further supported the diagnosis of CDA type 1.
Table 3.
CDA-1 patients treated with interferon alpha previously reported in the literature
| Patient no. | Type of IFN | Age (at the time of therapy)/sex | Dose | Response | Time to response | Duration of therapy | Side effects | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 | IFN-alpha-2a, peginterferon-alpha-2b | 28/f | 3–4.5 MIU 2–3/weeks, 30 µg/weeks | Yes | 1 month | 9 years | No | 14, 15 |
| 2 | IFN-alpha-2a | 44/f | 3 MIU 3/weeks | Yes | Within 1 week | 14 weeks | N/A | 16 |
| 3 | IFN-alpha-2a | 30/f | 3 MIU 1–3/weeks | Yes | Within 5 weeks | 13.5 weeks | N/A | 17 |
| 4 | IFN-alpha-2a | 14 mo/f | 106 U 2–3/weeks | Yes | 6 days | 13 months | No | 18 |
| 5 | IFN-alpha | 64/f | 3 MIU 3/weeks | Yes | 1 month | 3 months | Myalgia, mild nausea | 19 |
| 6 | IFN-alpha | 31/m | 3 MIU 3/weeks | Yes | 1 month | 4 months | No | 19 |
| 7 | IFN-alpha-2b | 14/f | 3 MIU 2–3/weeks | Yes | N/A | 17 months | No | 20 |
| 8 | IFN-alpha | 3 mo/m | 1 MIU 3/weeks | Yes | 15 days | 57 months | No | 21 |
| 9, 10, 11 | IFN-alpha-2b | 2.5/f, 7/m, 11.5/m | 4.2 MIU 3/weeks, 3.9 and 6.5 MIU 3/week, 2.6 MIU 3–4/weeks | No | 13, 30 and 19 weeks | Fever, malaise, alopecia | 22 | |
| 12–14 | IFN-alpha-2a | 54/f, 54/f, 51/f | 3 MIU 3/weeks | Yes | Within 1 month | N/A | N/A | 23 |
| 15 | IFN-alpha-2b | 48/f | 2 MIU 1/week | Yes | Within 1 month | N/A | Neutropenia | 24 |
| 16 | IFN-alpha-2b, peginterferon-alpha-2b | 42/m | 3 MIU 3/weeks, 50 µg/weeks | Yes | Within 1 month | N/A | Neutropenia | 24 |
| 17 | IFN-alpha-2a | 32/f | 3 MIU 3/weeks | Yes | Within 2 weeks | 4 months | No | Present case |
References
- 1.Tamary H, Dgany O (1993) Congenital dyserythropoietic anemia type I. doi:NBK5313 [bookaccession] [PubMed]
- 2.Iolascon A, Heimpel H, Wahlin A, Tamary H. Congenital dyserythropoietic anemias: molecular insights and diagnostic approach. Blood. 2013;122(13):2162–2166. doi: 10.1182/blood-2013-05-468223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Renella R. Progress in the congenital dyserythropoietic anemias: Juicy but high-hanging fruits? Am J Hematol. 2010;85(12):913–914. doi: 10.1002/ajh.21900. [DOI] [PubMed] [Google Scholar]
- 4.Renella R, Wood WG. The congenital dyserythropoietic anemias. Hematol Oncol Clin North Am. 2009;23(2):283–306. doi: 10.1016/j.hoc.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 5.Shalev H, Avraham GP, Hershkovitz R, Levy A, Sheiner E, Levi I, Tamary H. Pregnancy outcome in congenital dyserythropoietic anemia type I. Eur J Haematol. 2008;81(4):317–321. doi: 10.1111/j.1600-0609.2008.01109.x. [DOI] [PubMed] [Google Scholar]
- 6.Walshe JM. The acute haemolytic syndrome in Wilson’s disease—a review of 22 patients. QJM. 2013;106(11):1003–1008. doi: 10.1093/qjmed/hct137. [DOI] [PubMed] [Google Scholar]
- 7.Mohamed KK, Alfaraidy A. Congenital dyserythropoietic anemia with sideroblasts and ringed forms. Ann Saudi Med. 2002;22(5–6):408. doi: 10.5144/0256-4947.2002.408. [DOI] [PubMed] [Google Scholar]
- 8.Soysal T, Akun E, Ozaras R, Aki H, Ozturk M, Tuzuner N. Congenital dyserythropoietic anemia type I with ringed sideroblasts. Haematologia (Budap) 2000;30(1):45–49. doi: 10.1163/15685590051129887. [DOI] [PubMed] [Google Scholar]
- 9.Brien WF, Mant MJ, Etches WS. Variant congenital dyserythropoietic anaemia with ringed sideroblasts. Clin Lab Haematol. 1985;7(3):231–237. doi: 10.1111/j.1365-2257.1985.tb00030.x. [DOI] [PubMed] [Google Scholar]
- 10.Dgany O, Avidan N, Delaunay J, Krasnov T, Shalmon L, Shalev H, Eidelitz-Markus T, Kapelushnik J, Cattan D, Pariente A, Tulliez M, Cretien A, Schischmanoff PO, Iolascon A, Fibach E, Koren A, Rossler J, Le Merrer M, Yaniv I, Zaizov R, Ben-Asher E, Olender T, Lancet D, Beckmann JS, Tamary H. Congenital dyserythropoietic anemia type I is caused by mutations in codanin-1. Am J Hum Genet. 2002;71(6):1467–1474. doi: 10.1086/344781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babbs C, Roberts NA, Sanchez-Pulido L, McGowan SJ, Ahmed MR, Brown JM, Sabry MA, Bentley DR, McVean GA, Donnelly P, Gileadi O, Ponting CP, Higgs DR, Buckle VJ. Homozygous mutations in a predicted endonuclease are a novel cause of congenital dyserythropoietic anemia type I. Haematologica. 2013;98(9):1383–1387. doi: 10.3324/haematol.2013.089490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heimpel H, Anselstetter V, Chrobak L, Denecke J, Einsiedler B, Gallmeier K, Griesshammer A, Marquardt T, Janka-Schaub G, Kron M, Kohne E. Congenital dyserythropoietic anemia type II: epidemiology, clinical appearance, and prognosis based on long-term observation. Blood. 2003;102(13):4576–4581. doi: 10.1182/blood-2003-02-0613. [DOI] [PubMed] [Google Scholar]
- 13.Modi G, Shah S, Madabhavi I, Panchal H, Patel A, Uparkar U, Anand A, Parikh S, Patel K, Shah K, Revannasiddaiah S (2015) Successful allogeneic hematopoietic stem cell transplantation of a patient suffering from type II congenital dyserythropoietic anemia a rare case report from Western India. Case Rep Hematol 792485. doi: 10.1155/2015/792485 [DOI] [PMC free article] [PubMed]
- 14.Lavabre-Bertrand T, Blanc P, Navarro R, Saghroun M, Vannereau H, Braun M, Wagner A, Taib J, Lavabre-Bertrand C, Navarro M. Alpha-interferon therapy for congenital dyserythropoiesis type I. Br J Haematol. 1995;89(4):929–932. doi: 10.1111/j.1365-2141.1995.tb08442.x. [DOI] [PubMed] [Google Scholar]
- 15.Lavabre-Bertrand T, Ramos J, Delfour C, Henry L, Guiraud I, Carillo S, Wagner A, Bureau JP, Blanc P. Long-term alpha interferon treatment is effective on anaemia and significantly reduces iron overload in congenital dyserythropoiesis type I. Eur J Haematol. 2004;73(5):380–383. doi: 10.1111/j.1600-0609.2004.00310.x. [DOI] [PubMed] [Google Scholar]
- 16.Virjee S, Hatton C. Congenital dyserythropoiesis type I and alpha-interferon therapy. Br J Haematol. 1996;94(3):581–582. [PubMed] [Google Scholar]
- 17.Wickramasinghe SN. Response of CDA type I to alpha-interferon. Eur J Haematol. 1997;58(2):121–123. doi: 10.1111/j.1600-0609.1997.tb00935.x. [DOI] [PubMed] [Google Scholar]
- 18.Parez N, Dommergues M, Zupan V, Chambost H, Fieschi JB, Delaunay J, Mielot F, Cramer EM, Dommergues JP, Wickramasinghe SN, Tchernia G. Severe congenital dyserythropoietic anaemia type I: prenatal management, transfusion support and alpha-interferon therapy. Br J Haematol. 2000;110(2):420–423. doi: 10.1046/j.1365-2141.2000.02168.x. [DOI] [PubMed] [Google Scholar]
- 19.Shamseddine A, Taher A, Jaafar H, Haidar JH, Nasr R, Arzoumanian V, Salem Z, Bazarbachi A. Interferon alpha is an effective therapy for congenital dyserythropoietic anaemia type I. Eur J Haematol. 2000;65(3):207–209. doi: 10.1034/j.1600-0609.2000.9c244.x. [DOI] [PubMed] [Google Scholar]
- 20.Roda L, Pasche J, Fournier A, Terorotua V, Wickramasinghe SN, Tamary H, Schischmanoff PO, Tchernia G, Delaunay J. Congenital dyserythropoietic anemia, type 1, in a polynesian patient: response to interferon alpha2b. J Pediatr Hematol Oncol. 2002;24(6):503–506. doi: 10.1097/00043426-200208000-00020. [DOI] [PubMed] [Google Scholar]
- 21.Yarali N, Fisgin T, Duru F, Atilla P, Muftuoglu SF, Kaymaz SF. Successful management of congenital dyserythropoietic anemia type I with interferon alpha in a child. Pediatr Hematol Oncol. 2005;22(4):265–270. doi: 10.1080/08880010590935149. [DOI] [PubMed] [Google Scholar]
- 22.Marwaha RK, Bansal D, Trehan A, Garewal G. Interferon therapy in congenital dyserythropoietic anemia type I/II. Pediatr Hematol Oncol. 2005;22(2):133–138. doi: 10.1080/08880010590907221. [DOI] [PubMed] [Google Scholar]
- 23.Heimpel H, Schwarz K, Ebnother M, Goede JS, Heydrich D, Kamp T, Plaumann L, Rath B, Roessler J, Schildknecht O, Schmid M, Wuillemin W, Einsiedler B, Leichtle R, Tamary H, Kohne E. Congenital dyserythropoietic anemia type I (CDA I): molecular genetics, clinical appearance, and prognosis based on long-term observation. Blood. 2006;107(1):334–340. doi: 10.1182/blood-2005-01-0421. [DOI] [PubMed] [Google Scholar]
- 24.Goede JS, Benz R, Fehr J, Schwarz K, Heimpel H. Congenital dyserythropoietic anemia type I with bone abnormalities, mutations of the CDAN I gene, and significant responsiveness to alpha-interferon therapy. Ann Hematol. 2006;85(9):591–595. doi: 10.1007/s00277-006-0143-z. [DOI] [PubMed] [Google Scholar]