

Abstract
De novo CD5+ Diffuse large B cell lymphoma (DLBCL) is a rare and aggressive subtype of DLBCL. It is a distinct clinicopathologic entity with complex molecular profile and poor prognosis. A 59 year old female presented with pyrexia of unknown origin since 1 month. On examination, there was severe pallor, hepatosplenomegaly and no palpable lymphadenopathy. Complete blood count revealed bicytopenia with normal total leucocyte count. Liver and renal function tests were normal. Ultrasonography abdomen revealed splenic enlargement with two focal lesions attributed to either splenic abscess or infarcts. Patient was being managed as splenic infarct but continued to have bicytopenia. Further investigation showed elevated serum ferritin, triglycerides and LDH. With a clinical suspicion of infection and haemophagocytic lymphohistiocytosis bone marrow aspiration (BMA) and biopsy (BMBx) was done. BMA showed extensive haemophagocytosis and ~7.4 % large lymphoma-like cells. On this basis PET-CT was suggested which showed enlarged spleen with diffuse uptake. BMBx showed nodular and intrasinusoidal collection of abnormal lymphoid cells. On immunohistochemistry, these cells were positive for CD20, CD5, MUM1, BCL-2, BCL-6 and negative for CD3, CD10 and CD23. CD34 highlighted focal intrasinusoidal pattern. The complete clinicopathological profile suggested the diagnosis of de novo CD5+ DLBCL, with primary hepatosplenic pattern of involvement. CD5+ DLBCL presenting as splenic infarct is very rare. This case was unusual as the diagnosis of a primary aggressive lymphoma with haemophagocytosis was established in a patient who presented with fever and splenic infarct without lymphadenopathy. This indicates the importance of good morphological assessment of a bone marrow aspirate and biopsy to make a correct diagnosis.
Keywords: DLBCL, Hemophagocytosis, De novo CD5+, Splenic infarct, Hepatosplenic
Introduction
De novo CD5+ DLBCL is a rare and aggressive subtype of DLBCL. This variant is a distinct clinicopathologic entity with complex molecular profile and poor prognosis as compared to CD5- DLBCL. Most of these cases are activated B cell subtype (ABC) of DLBCL. These patients have an extra-nodal presentation with frequent CNS disease and inferior treatment response [1].
CD5 molecule belongs to family of protein receptors called SRCR (scavenger receptor cysteinerich), expressed on T cells and a subset of normal naïve B cells. Its expression is mainly seen in cases of chronic lymphocytic leukemia, mantle cell lymphoma, marginal B-cell lymphoma and rarely in DLBCL [2]. In addition, ~40 % cases of intravascular large B-cell lymphoma also express CD5. CD5+ DLBCL with splenic infarct is a very rare presentation. Causes for splenic infarction include thromboembolic disorders, haematologic disease (haemoglobinopathies, leukaemia, lymphoma, myelofibrosis, polycythemia vera), splenic vascular disease, vasculitis, portal hypertension, infiltrative disorders, and pancreatic disease.
This case report highlights the role of CD5 in differential diagnosis of B-cell lymphoma along with a brief review of literature. Apart from CD5+ positivity, there was also the presence of marked hemophagocytosis which contributed to an unusual component in our case.
Case History
A 59 year old female presented with pyrexia of unknown origin since 1 month. On examination, there was hepatosplenomegaly and severe anemia. Patient had past history of Diabetes Mellitus Type II. There was no palpable lymphadenopathy. Complete blood count revealed bicytopenia with hemoglobin of 6.6 g/dl, platelets: 28,000/µl along with a normal total leucocyte count. Liver function and renal function tests were normal. An ultrasonography scan of the abdomen revealed splenic enlargement with presence of two focal lesions (?splenic abscess? Infarction). Serum ferritin and triglycerides were elevated (6196 ng/mL and 673 mg/dl respectively) LDH was also elevated (1516 IU/L).
With a clinical suspicion of infection and haemophagocytic lymphohistiocytosis bone marrow aspiration (BMA) and biopsy (BMBx) was done. BMA showed normoblastic erythroid hyperplasia, dyserythropoiesis, normal myeloid maturation and mild dysmegakaryopoiesis. In addition, there were ~7.4 % large lymphoid cells with scant basophilic cytoplasm, round nuclei with coarsely condensed nuclear chromatin. There was extensive hemophagocytosis (Fig. 1). Considering a possibility of hemophagocytic lymphohistiocytosis secondary to lymphoma, a positron emission tomography-computed tomography scan (PET-CT) and Epstein Barr virus (EBV) serology was suggested.
Fig. 1.
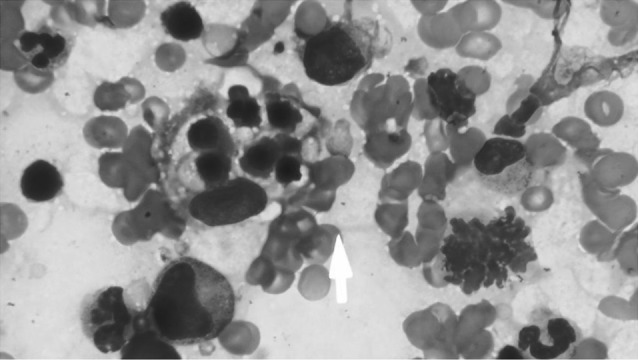
Bone marrow aspirate picture showing haemophagocytosis indicated by an arrow (Wright Giemsa, ×1000)
BMBx showed nodular collection of large lymphoid cells with few areas showing sinusoidal pattern (Fig. 2a, b). A panel of immunohistochemical markers was applied to characterize these cells. The lymphoid cells were positive for CD20 (Fig. 3a), CD5 (Fig. 3b), MUM1 (Fig. 3c), BCL-2, BCL-6 (weak positivity). CD31 highlighted the focal intrasinusoidal pattern. Ki-67 showed an index of 60 % (Fig. 3d). The lymphoid cells were negative for CD3, CD10 and CD23. PET-CT scan showed intense uptake by the vertebra and focal uptake by sternum and along with an enlarged spleen with diffuse uptake. (Figure 4a, b). On basis of above findings differential diagnosis were de novo CD5+ Diffuse large B-cell lymphoma not otherwise specified (DLBCL, NOS) because it is CD5+, CD10−, MUM1+ and presents with a primary extranodal disease in ~1/3rd cases. The cases with lack of lymphadenopathy, mass lesion, presence of bone marrow invasion and hepatosplenoemgaly have been called primary hepatosplenic CD5+ DLBCL. However, haemophagocytosis has not been previously reported in these cases. The third entity considered was Intravascular B cell Lymphoma which is an aggressive DLBCL with an extranodal presentation, CD5 expression in about 40 % cases and BMA can show haemophagocytosis. Considering all the features, the final diagnosis considered was a de novo CD5+ DLBCL, common variant with a primary hepatosplenic pattern of involvement. EBV quantitative polymerase chain reaction (EBV–QPCR) was negative thus ruling out EBV positive lymphoma of the elderly.
Fig. 2.

a Bone marrow biopsy showing nodular collection of abnormal large lymphoid cells marked by a solid arrow (Haematoxylin and eosin, ×400), b Bone marrow biopsy showing lymphoid cells in sinusoidal pattern marked by a dashed arrow (Haematoxylin and eosin, ×400)
Fig. 3.

a CD20 is positive in the large lymphoid cells (X200), b CD5 is positive in the large lymphoid cells (×200) c MUM1 positive lymphoid cells. (×400) d Ki67 positivity in 60 % lymphoid cells. (×400)
Fig. 4.

a PET-CT scan showing intense uptake by the vertebra and focal uptake by sternum. b PET-CT scan showing enlarged spleen with diffuse uptake
Discussion
DLBCL comprises approximately 40 % of the Non Hodgkin lymphoma [3]. De novo CD5+ DLBCL is a unique subgroup of DLBCL and accounts for about 5–10 % of all cases [4]. This rare entity is associated with onset in old age, female predominance, advanced stage at diagnosis, the presence of B symptoms, high lactate dehydrogenase levels, and the frequent involvement of extranodal sites. Genetically these lymphomas are believed to originate from somatically mutated CD5+ progenitor B-cells. Immunohistochemically, CD5+ DLBCL is mostly CD10−, BCL2+, and MUM1+ and falls into the category of activated B cell (ABC) DLBCL. Complex chromosomal aberrations, unmutated IgVH and characteristics similar to ABC–DLBCL could potentially explain the aggressive course of CD5+ DLBCL [5]. It is important to identify this entity as it has a poorer prognosis than other DLBCL, NOS. The main differentials include CLL in Richter’s transformation to DLBCL, mantle cell lymphoma (MCL), secondary CD5+ DLBCL (Non-Richter’s), intravascular B cell lymphoma and primary CNS lymphoma [6].
In addition to CD5 positivity our patient also showed presence of hemophagocytosis. Hemophagocytic syndrome (HPS) is a clinical syndrome which is a potentially fatal hyperinflammatory condition caused by a highly stimulated but ineffective immune response. Main signs of HPS include prolonged fever, hepatosplenomegaly with lab derangements comprising of cytopenias, hypertriglyceridemia, hypofibrinogenemia, and elevated ferritin. [7] HPS-associated malignancies are mainly T-cell and natural killer–cell lymphomas. B-cell lymphoma-associated HPSs (B-LAHS) have mainly been reported in the Asian population mostly with IVLBCL. [8] IVLBCL is an aggressive group within DLBCL. Patients mostly present with hepatosplenomegaly (77 %), anemia or thrombocytopenia (84 %), hemophagocytosis (61 %), and BM involvement of tumor cells (75 %). [9, 10].
Therapies directed against CD5 are lacking. Addition of regimens based on Rituximab has led to an improvement in the outcome of patients with CD5+ DLBCL but the outcome remains inferior in comparison to that seen in CD5-DLBCL. Combinations of newer anti CD20 monoclonal antibodies with other biological agents such as lenalidomide, or Bruton tyrosine kinase inhibitor agents such as ibrutinib can be studied in CD5+ DLBCL [5].
Compliance with Ethical Standards
Ethical Approval
This article does not contain any studies with animals performed by any of the authors. This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
None.
References
- 1.Yamaguchi Motoko, Seto Masao, Okamoto Masataka, Ichinohasama Ryo, Nakamura Naoya, Yoshino Tadashi, et al. De novo CD5 diffuse large B-cell lymphoma: a clinicopathologic study of 109 patients. Blood. 2002;99:815–821. doi: 10.1182/blood.V99.3.815. [DOI] [PubMed] [Google Scholar]
- 2.Ferry JA, Yang WI, Zukerberg LR, Wotherspoon AC, Arnold A, Harris NL. CD5+ extranodal marginal zone B-cell (MALT) lymphoma: a low grade neoplasm with a propensity for bone marrow involvement and relapse. Am J Clin Pathol. 1996;105:31–37. doi: 10.1093/ajcp/105.1.31. [DOI] [PubMed] [Google Scholar]
- 3.Campo E, Swerdlow SH, Harris NL. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117:5019–5032. doi: 10.1182/blood-2011-01-293050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harada S, Suzuki R, Uehira K. Molecular and immunological dissection of diffuse large B cell lymphoma: CD5+ and CD5− with CD10+ groups may constitute clinically relevant subtypes. Leukemia. 1999;13:1441–1447. doi: 10.1038/sj.leu.2401487. [DOI] [PubMed] [Google Scholar]
- 5.Niitsu N, Okamoto M, Tamaru JI, Yoshino T, Nakamura N, Nakamura S, et al. Clinicopathologic characteristics and treatment outcome of the addition of rituximab to chemotherapy for CD5- positive in comparison with CD-5 negative diffuse large B-cell lymphoma. Ann Oncol. 2010;21:2069–2074. doi: 10.1093/annonc/mdq057. [DOI] [PubMed] [Google Scholar]
- 6.Dong HY, Gorczyca W, Liu Z, Tsang P, Wu CD, Cohen P, et al. B-cell lymphomas with coexpression of CD5 and CD10. Am J Clin Pathol. 2003;119:218–230. doi: 10.1309/U98ADVKUC26R2RJA. [DOI] [PubMed] [Google Scholar]
- 7.Szyper-Kravitz M. The hemophagocytic syndrome/macrophage activation syndrome: a final common pathway of a cytokine storm. Isr Med Assoc J. 2009;11:633–634. [PubMed] [Google Scholar]
- 8.Jordan MB, Allen CE, Weitzman S, et al. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118:4041–4052. doi: 10.1182/blood-2011-03-278127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimazaki C, Inaba T, Okano A, et al. Clinical characteristics of B-cell lymphoma-associated hemophagocytic syndrome (B-LAHS): comparison of CD5+ with CD5- B-LAHS. Intern Med. 2001;40:878–882. doi: 10.2169/internalmedicine.40.878. [DOI] [PubMed] [Google Scholar]
- 10.Murase Takuhei, Yamaguchi Motoko, Suzuki Ritsuro, Okamoto Masataka, Sato Yumiko, Tamaru Jun-ichi, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109:478–485. doi: 10.1182/blood-2006-01-021253. [DOI] [PubMed] [Google Scholar]