

Abstract
Citrobacter freundii causes opportunistic infections in humans and animals, which are becoming difficult to treat due to increased antibiotic resistance. The aim of this study was to explore phages as potential antimicrobial agents against this opportunistic pathogen. We isolated and characterized five new virulent phages, SH1, SH2, SH3, SH4, and SH5 from sewage samples in Tunisia. Morphological and genomic analyses revealed that the five C. freundii phages belong to the Caudovirales order, Podoviridae family, and Autographivirinae subfamily. Their linear double-stranded DNA genomes range from 39,158 to 39,832 bp and are terminally redundant with direct repeats between 183 and 242 bp. The five genomes share the same organization as coliphage T7. Based on genomic comparisons and on the phylogeny of the DNA polymerases, we assigned the five phages to the T7virus genus but separated them into two different groups. Phages SH1 and SH2 are very similar to previously characterized phages phiYeO3-12 and phiSG-JL2, infecting, respectively, Yersinia enterocolitica and Salmonella enterica, as well as sharing more than 80% identity with most genes of coliphage T7. Phages SH3, SH4, and SH5 are very similar to phages K1F and Dev2, infecting, respectively, Escherichia coli and Cronobacter turicensis. Several structural proteins of phages SH1, SH3, and SH4 were detected by mass spectrometry. The five phages were also stable from pH 5 to 10. No genes coding for known virulence factors or integrases were found, suggesting that the five isolated phages could be good candidates for therapeutic applications to prevent or treat C. freundii infections. In addition, this study increases our knowledge about the evolutionary relationships within the T7virus genus.
Keywords: Citrobacter freundii, pathogen, phages, virulent, T7virus, therapeutic applications
Introduction
Members of the Gram-negative Enterobacteriaceae have caused significant diseases throughout human history. They are responsible for many human infections in the intestine, urinary tract, bloodstream, and wounds (Abbott, 2011; Shanks et al., 2012). The genus Citrobacter belongs to this bacterial family, although it was originally classified within the genus Salmonella due to biochemical and serological similarities (Harhoff, 1949; Ewing and Davis, 1972). Citrobacter freundii is the type species of this genus, with a genome size of ~5 Mb and a G+C content of 50 to 52% (Kumar et al., 2013; Kimura et al., 2014). C. freundii is commonly found in soil, water, foods, and the intestinal tracts of animals and humans (Drelichman and Band, 1985). Some strains of C. freundii can also cause opportunistic infections in humans and animals, which are becoming more difficult to treat due to increased antibiotic resistance. As such, C. freundii infections have become a public health concern (Samonis et al., 2008; Antonelli et al., 2015; Campos et al., 2015) and alternatives or adjuncts to antibiotic treatment are required.
In this context, lytic/virulent phages are being re-investigated as potential antimicrobial agents to either combat bacterial diseases or to stop the dissemination of multi-resistant bacteria. The potential of phages to control or treat bacterial diseases has been previously demonstrated (Smith and Huggins, 1982; Slopek et al., 1983). However, their use was mostly abandoned for several well-documented reasons including the inability to purify phage preparations from bacterial components, the lack of understanding of basic phage biology, the inability to differentiate temperate from lytic phages, narrow host ranges, the development of phage-resistant bacterial mutants, and the inherent difficulties of patenting phages and their use. It is believed that progress has been made to overcome most, if not all, these difficulties (Carlton, 1999; Loc-Carrillo and Abedon, 2011).
Several phages infecting various strains of C. freundii have been recently characterized. Six of them belong to the Myoviridae family [double-stranded DNA genome (dsDNA), contractile tail] and were isolated from water samples in Texas. Their genomic characterization indicated that three of these phages (Moon, Miller, Merlin) are related to the T4virus genus (Edwards et al., 2015; Hwang et al., 2015; LeSage et al., 2015) while the other three (Mordin, Michonne, Moogle) are related to the Felixo1virus genus (Bernal et al., 2015; Guan et al., 2015; Nguyen et al., 2015). The complete genomic sequence of the C. freundii phage Stevie is also available (Shaw et al., 2015). This Siphoviridae phage (dsDNA, noncontractile tail), which was isolated from a dirt sample in Texas, is related to the T1virus genus. Phages of the Podoviridae family (dsDNA, short tail) can also infect C. freundii strains as the podophage LK1 was isolated from sewage and its genome size was estimated to be 20–23 kb (Chaudhry et al., 2014). The podophage phiCFP-1 was isolated from sewage in China and classified as a T7virus with a genome of 38,625 bp with 43 orfs and direct terminal repeats of 229 bp (Zhao et al., 2015).
Phages belonging to the T7virus genus are particularly interesting for therapeutic applications as they are usually easy to culture and have a short lytic cycle. They also have smaller genomes and a conserved organization, which facilitates their in-depth analysis. Their genomes can be divided into three transcriptional regions including early-, middle-, and late-expressed genes (Scholl and Merril, 2005; Zhu et al., 2010). As for the prototype coliphage T7, the genes of these phages can be transcribed due to an efficient phage-encoded RNA polymerase that specifically recognizes a set of conserved promoters dispersed throughout the phage genome (Chen and Schneider, 2005; Huang et al., 2012).
Here, we describe five lytic Podoviridae phages infecting C. freundii isolated from sewage samples in Tunisia. Their analyses showed that they belong to the Autographivirinae subfamily and they share similarities with phages infecting other Enterobacteriaceae.
Materials and methods
Bacterial strains, phage isolation, and culture conditions
Five bacterial isolates were obtained by plating Tunisian wastewater samples on Salmonella-Shigella agar (Biokar) and incubating the plates for 24 h at 37°C. The species of each bacterial isolate was determined by 16S rRNA sequencing and API 20 E strip (BioMérieux). C. freundii strains were genotyped using multi-locus sequence typing (MLST) of seven housekeeping genes (aspC, clpX, fadD, mdh, arcA, dnaG, and lysP) as described previously (Bai et al., 2012). The allelic profile and sequence type (ST) of each strain was identified using the MLST database website (http://pubmlst.org/cfreundii/). Evolutionary analyses were conducted with MEGA7 (Kumar et al., 2016). The neighbor-joining phylogenetic tree (Saitou and Nei, 1987) of the five strains was generated from the concatenated sequences of the seven loci. The evolutionary distances were computed using the Maximum Composite Likelihood method (Tamura et al., 2004) and are in the units of the number of base substitutions per site.
Two C. freundii isolates were used as hosts for phage isolation. Water samples were obtained from four different areas in Tunis (Table 1). One millilitre of the filtered water samples was mixed with 1 ml of an overnight bacterial culture in 3 ml of Brain Heart Infusion broth (BHI) (Biokar or BD). After incubation for 24 h at 37°C, the mixtures were centrifuged and 4 μl of each filtered-supernatant was spotted on a fresh bacterial lawn. After incubation at 37°C for 24 h, phage lysis zones were picked with a sterile truncated tip and amplified in the presence of their respective host in BHI for 24 h at 37°C. Then, the mixtures were centrifuged and the supernatants filtered. Isolated plaques were obtained using the double-layer agar method and picked with a sterile truncated tip. This step was repeated three times to ensure phage purity. Phages and bacterial strains were deposited at the Félix d'Hérelle Reference Center for Bacterial Viruses of the Université Laval (www.phage.ulaval.ca) under the following names: phages SH1 (HER 516), SH2 (HER 517), SH3 (HER 518), SH4 (HER 519), and SH5 (HER 520) as well as C. freundii strains CF3 (HER 1518) and CF5 (HER 1516).
Table 1.
Origins of phages and their host strains.
| Phage | Origins of phages | Host strain | Origins of strain |
|---|---|---|---|
| SH1 | Wadi of Khaznadar | CF5 | Entry water treatment plant of Menzah1 |
| SH2 | Wastewater from Mellassine | CF5 | |
| SH3 | Office of national sanitation of Ksar Said | CF3 | Entry water treatment plant of Gammarth |
| SH4 | Wastewater from Mellassine | CF3 | |
| SH5 | Wadi of Ezzouhour city | CF3 |
Microbiological assays
The host range of the five phages was determined by spotting 4 μl of various serial dilutions (100 to 10−7) of a phage lysates on BHI soft agar (0.75% agar) containing one bacterial strain. After overnight incubation at 37°C, plates were examined for the presence of isolated plaques in the spotted areas, which indicated a full phage lytic cycle on the host. The host range was tested on 5 C. freundii (this study), one Cronobacter turicensis (290708/07) and 25 bacterial strains available at the Félix d'Hérelle Reference Center for Bacterial Viruses of the Université Laval: 10 Escherichia coli (HER1024, HER1040, HER1144, HER1255, HER1462, HER1155, HER1290, HER1022, HER1213, and HER1445), two Shigella dysenteriae (HER1020 and HER1031), one Shigella sonnei (HER1043), two Salmonella Paratyphi (HER1045, HER1220), one Salmonella Typhi (HER1038), two Salmonella Typhimurium (HER1023, HER1095), two Salmonella Newport (HER1185 and HER1019), one Salmonella Heidelberg (HER1428), one Salmonella Senftenberg (HER1397), and 3 Yersinia enterocolitica (HER1249, HER1071, HER1072). Phage susceptibility to pH (2 to 10) was also determined in BHI broth with the pH adjusted using hydrochloric acid (HCl) or sodium hydroxide (NaOH). One hundred microlitre of each phage lysate at >109PFU/ml were mixed with 900 μl of media for each pH condition and incubated at 37°C for 60 min. Phage titer was then determined using the double-layer agar method.
Electron microscopy
Phages were prepared and observed as described previously (Fortier and Moineau, 2007). The reported dimensions are the means of at least ten virions stained with uranyl acetate (2%).
Phage structural proteins
Phages were precipitated from lysates (1L) with 10% polyethylene glycol (PEG) 8000 and 2922g of sodium chloride then concentrated using a discontinuous CsCl gradient followed by a continuous CsCl gradient, as described previously (Chibani Azaïez et al., 1998; Sambrook and Russel, 2001). A purified phage sample was sent directly for structural protein identification by liquid chromatography/tandem mass spectrometry (LC-MS/MS) at the Plateforme Protéomique, Centre de Génomique de Québec (Université Laval). A custom database was generated using the putative predicted proteins. Results were analyzed using Scaffold Proteome software version 4.4.5.
Genome sequencing and bioinformatics analyses
Phage DNA was extracted from high titer phage lysates using a Plasmid Maxi Kit (Qiagen) with modifications described elsewhere (Deveau et al., 2002). Phage DNA was prepared for sequencing using the Nextera XT DNA library preparation kit (Illumina) according to the manufacturer's instructions. The libraries were then sequenced on a MiSeq system using a MiSeq reagent kit v2 (Illumina, 500 cycles). De novo assembly was performed with Ray assembler version 2.2.0 using k-mer sizes of 21, 51, 96, 31, and 51 and we obtained mean coverage depths for each single phage contig of 2717, 1643, 3804, 134, and 2431 for SH1, SH2, SH3, SH4, and SH5, respectively. Coverage was calculated with Samtools. Open reading frames (ORFs) were identified using ORF Finder (Rombel et al., 2002) and GeneMark (Lukashin and Borodovsky, 1998) then confirmed by visual inspection for the presence of a Shine-Dalgarno sequence close to a start codon (AUG, UUG or GUG) using BioEdit 7.2.0 (Hall, 1999). ORFs were considered if they contained at least 30 amino acids (aa). Similarities with known proteins were searched with BLAST. Hits were considered when the E-value was lower than 10−3. The percentage of identity between proteins was calculated by dividing the number of identical residues by the size of the smallest protein. The theoretical molecular weight (MW) and isoelectric point (pI) of the ORFs were calculated using the Compute pI/MW tool (http://web.expasy.org/compute_pi/).
Determination of genome ends
To confirm the direct terminal repeats, primers adjacent to the predicted terminal ends were designed using Primer-BLAST at NCBI. The putative ends were established by aligning the genome termini with similar phage genomes using ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The primers were used to sequence directly from the phage DNA at the sequencing and genotyping platform of the Université Laval using the ABI data 3730XL DNA analyzer. The primers used are described in Table 2. Terminal repeat sequences were determined using Staden software (version 1.7.0) (Staden, 1996).
Table 2.
Primers used to determine the terminal repeats.
| Phage | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| SH1 | GCCTCACTGTTCCGTCATTT | CAACTGAAAGGAGGTGGCTC |
| SH2 | TGTCTCAGGGAGTGGCTTTA | GCTCAATGTTACGCTTGCTG |
| SH3 | GCCCTACCCCAGTCTATCAT | CTATCCCTACGCCATCTTGC |
| SH4/SH5 | CTGCTGTTCTACTTGCTGCT | GCTATGGTCCCTGACTGCTA |
DNA polymerase phylogeny
The DNA polymerase sequence dataset used for phylogeny included phage proteins from different families and genera (Labrie et al., 2013). The sequences were aligned using MAFFT with the E-INS-i parameter (Katoh and Standley, 2013). The alignment was then processed to generate the tree as previously described (Mercanti et al., 2015). Briefly, the best amino-acid substitution model implemented in PhyML 3.0 to calculate the best tree was predicted with ProtTest 3.2 (Darriba et al., 2011). The Shimodaira-Hasegawa-like procedure was used to determine the branch support values (Shimodaira, 2002). Finally, Newick utility package (Junier and Zdobnov, 2010) and ITOL (Letunic and Bork, 2011) were used to render the tree.
Nucleotide sequence accession numbers
The annotated phage genomic sequences were deposited in GenBank under the numbers KU687347 (SH1), KU687348 (SH2), KU687349 (SH3), KU687350 (SH4), KU687351 (SH5).
Results
Isolation of bacteria and phages
Five bacterial strains were isolated from different wastewater samples. Gram staining showed Gram negative bacilli. Sequencing of 16S rRNA and API 20E strip identification revealed that they belong to the C. freundii species. MLST analyses showed that the five strains also belong to different genotypes, CF5 belong to ST19 and the four other strains belong to four novel and different ST. Phylogenetic analyses (Figure 1) revealed that CF3, CF4, and CF7 belonged to a different branch from CF5 and CF8. Two C. freundii isolates (CF3 and CF5) were selected from each branch and used as host organisms to isolate phages.
Figure 1.
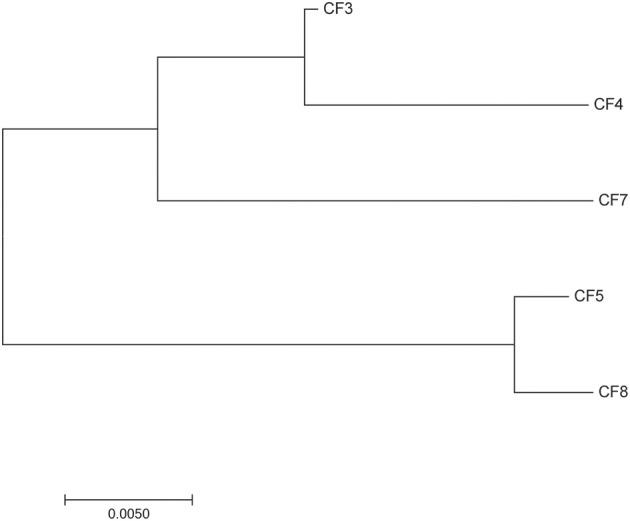
Neighbor-joining phylogenetic tree of the five strains of Citrobacter freundii.
A total of five virulent phages, SH1, SH2, SH3, SH4, and SH5, were isolated from four sewage samples (Table 1). For phages SH1 and SH2, plaques of 2 mm in diameter appeared after only 3 h of incubation at 37°C and the plaques became larger with diameters ranging from 4 to 6 mm after overnight incubation, as shown in Figure 2. Phage SH3 produced smaller plaques of 1 mm in diameter while phages SH4 and SH5 produced plaques of about 3 mm in diameter.
Figure 2.

Plaques formed by phages SH1, SH2, SH3, SH4, and SH5, respectively, from left to right on their host strains of C. freundii after an overnight incubation at 37°C.
The host range of the five phages was determined using the 31 Gram-negative bacterial strains described in the Materials and Methods section. Phages SH1 and SH2 were able to lyse their host strain, C. freundii CF5, and S. Typhi HER1038. Phage SH3 was able to lyse its host strain, C. freundii CF3 and C. freundii CF4. Phages SH4 and SH5 lysed their host strain, C. freundii CF3, as well as C. freundii CF4 and C. turicensis 290708/7.
Sensitivity to pH
The five phages were tested for their susceptibility to different pH conditions. They were exposed to pHs ranging from 2 to 10 for 1 h at 37°C. All phages were completely inactivated when exposed to pH 2 and pH 3. A 10-fold reduction in phage titer was also noticed at pH 4. All phage suspensions were stable from pH 5 to pH 10.
Morphological characteristics
Negatively stained purified phages were observed with an electron microscope and all five possessed an icosahedral capsid and small non-contractile tail (Figure 3, Table 3). However, the tips of the tails differed which led us to divide them into two morphological groups. The first group included phages SH1 and SH2, which had a narrower base plate compared to the second group, which included phages SH3, SH4, and SH5 (Figure 3). Nonetheless, their overall morphology allowed us to classify the five phages into the Caudovirales order and the Podoviridae family.
Figure 3.
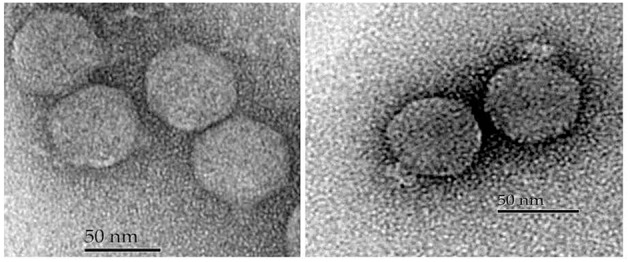
Electron micrographs of phages SH1 (left) and SH3 (right).
Table 3.
Morphological and genomic characteristics of the five isolated phages and phage T7.
| Phage | Capsid (nm) | Tail (nm) | Genome size (bp) | GC% | Terminal repeat (bp) | |
|---|---|---|---|---|---|---|
| Width | Length | |||||
| SH1 | 61 ± 1.6 | 14 ± 0.9 | 12 ± 1.0 | 39,434 | 51.0 | 230 |
| SH2 | 58 ± 2.5 | 14 ± 1.4 | 10 ± 1.1 | 39,158 | 50.7 | 242 |
| SH3 | 65 ± 1.4 | 21 ± 1.1 | 13 ± 1.0 | 39,444 | 50.6 | 183 |
| SH4 | 67 ± 1.3 | 29 ± 2.4 | 16 ± 1.2 | 39,274 | 52.6 | 190 |
| SH5 | 65 ± 1.1 | 27 ± 1.8 | 16 ± 2.0 | 39,832 | 52.5 | 190 |
| T7* | 56 | 14 | 9 | 39,936 | 50 | 160 |
Genomic characteristics
The double-stranded DNA of the five phages was extracted and sequenced. The genome size of these phages ranged from 39,158 to 39,832 bp, which was similar to that of coliphage T7 (39,936 bp) (Table 3). The GC contents of the phage genomes were similar to that of their C. freundii hosts, 50 to 51% (Frederiksen, 2015). After genome alignments with similar phages, primers adjacent to the predicted terminal ends were used to directly sequence the phage genomic DNA. As expected, the sequencing signal dropped at the end of the genome (Figure 4) and this was used to determine the position of the terminal ends and their sequences. The last adenine at the end of the repeated sequences was not considered because it is added by the polymerase (Clark, 1988; Garneau et al., 2010). Our analyses revealed that the five Podoviridae phage (podophage) genomes contained direct terminal repeats at both ends (Table 3). The length of the direct terminal repeats of phages SH1 (230 bp) and SH2 (242 bp) were similar to that of Yersinia phage phiYeO3-12 (232 bp; Pajunen et al., 2001), Salmonella phage phiSG-JL2 (230 bp; Kwon et al., 2008), and Citrobacter phage phiCFP-1 (229 bp; Zhao et al., 2015). Terminal repeat lengths of SH3 (183 bp), SH4 (190 bp), and SH5 (190 bp) were close to the length of coliphage K1F (179 bp; Scholl and Merril, 2005).
Figure 4.

SH5 genome sequencing with both reverse and forward primers.
Genome organization
Analyses of the predicted orfs in the genomes of the five newly isolated podophages revealed that they all have the same transcriptional orientation and use only ATG as an initiation codon (Tables 4, 5). Comparative genome analyses also indicated that these phages were affiliated with the Autographivirinae subfamily and the T7virus genus. Similar to the morphological groupings, we could also divide the five phage genomes into subgroups (Figure 5). The first group included phages SH1 and SH2, which had high identity (80%) to genes of Yersinia phage phiYeO3-12 as well as coliphages T7 and T3. The second phage group (SH3, SH4, and SH5) could be divided into two subgroups. Group 2A included phage SH3, which was close to coliphage K1F, while group 2B was comprised of phages SH4 and SH5, which are similar to Cronobacter phage Dev2.
Table 4.
Features of the ORFs of phage SH2, identity with SH1, predicted functions of proteins, and best matches with database.
| Predicted protein | Aligned protein | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ORF | SH1 ORF,% | Start (bp) | Stop (bp) | Size (aa) | MW(kDa) | pI | SD sequence TAAGGAGGT (5′-3′)a | Predicted function | BLAST (extent; %aa identity)b | E Value | Size (aa) | Accession number |
| 1 | ORF1, 99 | 948 | 1406 | 152 | 17.0 | 7.6 | TGAGGTAACaccaaATG | S-adenosyl-l-methionine hydrolase | gp0.3 [Yersinia phage phiYeO3-12] (152/152; 100%) | 4.00E-110 | 152 | NP_052065.1 |
| 2 | ORF2, 87 | 1479 | 1679 | 66 | 7.5 | 6.8 | ATAGGACTAacaccATG | gp0.45 [Salmonella phage phiSG-JL2] (65/66; 98%) | 4.00E-41 | 66 | YP_001949746.1 | |
| 3 | ORF3, 74 | 1699 | 1857 | 52 | 6.0 | 9.7 | ACAGGAGGAttagcaATG | hypothetical protein [Enterobacter phage E-4] (49/52; 94%) | 3.00E-26 | 52 | AKA61646.1 | |
| 4 | ORF4, 94 | 1854 | 2051 | 65 | 7.8 | 10.5 | TGGTGAAACacgcATG | hypothetical protein [Enterobacter phage E-4] (63/65; 97%) | 2.00E-38 | 65 | AKA61645.1 | |
| 5 | ORF5, 95 | 2073 | 3182 | 369 | 42.3 | 7.1 | TAAGGACACactgaaATG | Protein kinase | gp0.7 [Yersinia phage phiYeO3-12] (346/369; 94%) | 0.0 | 369 | NP_052070.1 |
| 6 | ORF6,99 | 3253 | 5907 | 884 | 98.8 | 7.1 | CAATGAGGTaagcaATG | RNA polymerase | DNA-directed RNA polymerase [Enterobacter phage E-2] (881/884; 99%) | 0.0 | 884 | AKA61565.1 |
| 7 | ORF7, 90 | 6008 | 6502 | 164 | 19.6 | 9.2 | TAAGAGGATtactttATG | gp1.05 [Salmonella phage phiSG-JL2] (76/165; 46%) | 3.00E-38 | 169 | YP_001949751.1 | |
| 8 | ORF8, 100 | 6593 | 6733 | 46 | 5.9 | 10.9 | TAAGATACTATG | gp1.1 [Yersinia phage phiYeO3-12] (46/46; 100%) | 2.00E-23 | 46 | NP_052073.1 | |
| 9 | ORF9, 69 | 6736 | 7008 | 91 | 10.3 | 7.9 | AGTGGAACTaatgATG | Deoxyguanosine triphospho-hydrolase inhibitor | gp1.2 [Salmonella phage phiSG-JL2] (90/91; 99%) | 1.00E-160 | 92 | YP_001949753.1 |
| 10 | ORF10, 98 | 7103 | 8119 | 338 | 38.4 | 5.0 | TGAGGAACAaccgtATG | DNA ligase | gp1.3 [Salmonella phage phiSG-JL2] (333/338; 99%) | 0.0 | 338 | YP_001949754.1 |
| 11 | ORF11, 94 | 8291 | 8548 | 85 | 9.9 | 11.2 | TAAGGAGACaacatcATG | LysR family transcriptional regulator | gp1.6 [Yersinia phage phiYeO3-12] (85/85; 100%) | 2.00E-53 | 85 | NP_052078.1 |
| 12 | ORF12, 66 | 8548 | 9132 | 194 | 21.6 | 9.1 | TAAGGAGGTgctgtaATG | Nucleotide kinase | phiYe-F10_00014 [Yersinia phage phiYe-F10] (163/190; 86%) | 6.00E-115 | 190 | AKQ06773.1 |
| 13 | ORF14, 85 | 9119 | 9256 | 45 | 5.3 | 5.2 | TAAGGGGCTgtgctATG | AVU28_gp19 [Enterobacter phage E-3] (42/45; 93%) | 1.00E-21 | 45 | AKA61598.1 | |
| 14 | ORF15, 62 | 9253 | 9489 | 78 | 8.8 | 4.8 | TAAGGAGGCcaataaATG | Bacterial RNA polymerase inhibitor | ORF13 [Yersinia phage vB_YenP_AP5] (77/78; 99%) | 1.00E-50 | 78 | AIM40358.1 |
| 15 | ORF16, 98 | 9542 | 10240 | 232 | 26.0 | 4.8 | AAAGGAGAAacatcATG | Single-stranded DNA-binding | phiYe-F10_00017 [Yersinia phage phiYe-F10] (227/232; 98%) | 4.00E-166 | 232 | AKQ06776.1 |
| 16 | ORF18, 73 | 10240 | 10701 | 153 | 17.6 | 9.5 | CGAGGACTTctaATG | Endonuclease | gp3 [Yersinia phage phiYeO3-12] (152/153; 99%) | 2.00E-105 | 153 | NP_052083.1 |
| 17 | ORF19, 99 | 10694 | 11149 | 151 | 16.9 | 9.0 | TAAAGAAAAATG | N-acetylmuramoyl-l-alanine amidase | AVU28_gp15 [Enterobacter phage E-3] (151/151; 100%) | 2.00E-108 | 151 | AKA61594.1 |
| 18 | ORF20, 100 | 11154 | 11261 | 35 | 4.2 | 8.5 | GAGGGTGATaccATG | 3.7 protein [Yersinia phage phiYeO3-12] (35/35; 100%) | 2.00E-15 | 35 | NP_052086.1 | |
| 19 | ORF21,99 | 11328 | 13028 | 566 | 6.3 | 5.2 | TAAGGAATGtacaATG | Primase/Helicase | gp4A [Salmonella phage phiSG-JL2] (560/566; 99%) | 0.0 | 566 | YP_001949764.1 |
| 19.1 | ORF21.1, 94 | 11362 | 11517 | 51 | 5.6 | 6.5 | TCTTTCTGTttcATG | hypothetical protein [Enterobacteria phage T3] (49/51; 96%) | 1.00E-26 | 51 | AGM10719.1 | |
| 19B | ORF21B,99 | 11514 | 13028 | 504 | 55.9 | 5.1 | GGAGGCAGTaaccctATG | Primase/Helicase | Primase/Helicase protein [Enterobacter phage E-3] (501/504; 99%) | 0.0 | 504 | AKA61593.1 |
| 19.2 | ORF21.2, 97 | 12748 | 13077 | 109 | 12.1 | 6.7 | GAAGGGAAAaccacATG | gp4.2 [Enterobacteria phage T3] (103/109; 94%) | 2.00E-66 | 109 | NP_523317.1 | |
| 20 | ORF22, 97 | 13124 | 13336 | 70 | 7.7 | 10.0 | ATAGGAGACacatcATG | gp4.3 [Yersinia phage phiYeO3-12] (68/70; 97%) | 4.00E-29 | 70 | NP_052091.1 | |
| 21 | ORF23, 100 | 13349 | 13633 | 94 | 10.7 | 9.9 | TAAGGAGCGaacactATG | gp4.5 [Yersinia phage phiYeO3-12] (94/94; 100%) | 2.00E-62 | 94 | NP_052092.1 | |
| 22 | ORF24, 98 | 13701 | 15815 | 704 | 79.8 | 6.5 | AAAGGAGGGcattATG | DNA polymerase | gp5 [Yersinia phage phiYeO3-12] (700/704; 99%) | 0.0 | 704 | NP_052093.1 |
| 23 | 15825 | 16157 | 110 | 13.0 | 9.5 | TAAGGAGGAtttATG | Homing Endonuclease | gp5.3 [Yersinia phage phiYeO3-12] (110/110; 100%) | 2.00E-75 | 110 | NP_052095.1 | |
| 24 | ORF25, 99 | 16135 | 16437 | 101 | 11.1 | 6.3 | AAAGGAGAAacattATG | HNS binding | gp5.5 [Yersinia phage phiYeO3-12] (100/101; 99%) | 4.00E-66 | 101 | NP_052097.1 |
| 25 | ORF26, 100 | 16553 | 16762 | 69 | 7.3 | 9.8 | TTGGGAGGTactctaATG | gp5.7 [Yersinia phage phiYeO3-12] (69/69; 100%) | 8.00E-42 | 69 | NP_052098.1 | |
| 26 | ORF27, 99 | 16705 | 16941 | 60 | 8.8 | 4.2 | CAATGGTGGagcATG | gp5.9 [Yersinia phage phiYeO3-12] (60/60; 100%) | 1.00E-34 | 60 | NP_072071.1 | |
| 27 | ORF28, 99 | 16938 | 17849 | 303 | 34.7 | 4.9 | GGAGGATGAcgaATG | Exonuclease | AVU28_gp07 [Enterobacter phage E-3] (301/303; 99%) | 0.0 | 303 | AKA61586.1 |
| 28 | ORF29, 100 | 17831 | 17944 | 37 | 4.1 | 9.7 | CAAGGAGATttacttATG | gp6.3 [Yersinia phage phiYeO3-12] (37/37; 100%) | 1.00E-15 | 37 | NP_052102.1 | |
| 29 | ORF30, 96 | 18039 | 18284 | 81 | 9.3 | 5.9 | TTAAGAGGTgaatttATG | gp6.5 [Yersinia phage phiYeO3-12] (79/81; 98%) | 4.00E-51 | 81 | NP_052103.1 | |
| 30 | ORF31, 69 | 18289 | 18540 | 83 | 8.8 | 9.1 | ACAGGAGTAattatATG | Head | gp6.7 [Yersinia phage vB_YenP_AP5] (83/83; 100%) | 2.00E-49 | 83 | YP_009102822.1 |
| 31 | ORF32, 96 | 18568 | 18888 | 106 | 11.0 | 9.8 | TAGGGAGAAacatcATG | Host specificity protein B | gp7.3 [Salmonella phage phiSG-JL2] (105/106; 99%) | 1.00E-62 | 106 | YP_001949779.1 |
| 32 | ORF33, 99 | 18899 | 20506 | 535 | 58.6 | 4.5 | TAAGGAGGActgaATG | Head-to-tail joining | gp8 [Yersinia phage phiYeO3-12] (535/535; 100%) | 0.0 | 535 | NP_052106.1 |
| 33 | ORF34, 97 | 20608 | 21540 | 310 | 33.8 | 4.3 | TTAGGAGATttaacaATG | Capsid assembly | ORF30 [Citrobacter phage phiCFP-1] (303/310; 98%) | 0.0 | 310 | AKA62148.1 |
| 34 | ORF35, 95 | 21697 | 22740 | 347 | 36.8 | 6.2 | TAAGGAGATtcaacATG | Minor and Major capsid | 10A [Yersinia phage phiYe-F10] (344/346; 99%) | 0.0 | 347 | AKQ06793.1 |
| 35 | 22779 | 23012 | 77 | 7.4 | 4.5 | TCAGAAGACtATG | Minor capsid | AVU28_gp35 [Enterobacter phage E-3] (73/77; 95%) | 1.00E-39 | 77 | AKA61614.1 | |
| 36 | ORF36, 99 | 23125 | 23715 | 196 | 22.2 | 4.5 | ACAGGAGGTaacatcATG | Tail tubular A | gp11 [Yersinia phage phiYeO3-12] (196/196; 100%) | 1.00E-141 | 196 | NP_052110.1 |
| 37 | ORF37, 99 | 23731 | 26136 | 801 | 89.8 | 5.9 | CAAGGAGGCtctATG | Tail tubular B | gp12 [Salmonella phage phiSG-JL2] (797/801; 99%) | 0.0 | 801 | YP_001949785.1 |
| 38 | ORF38, 98 | 26209 | 26619 | 136 | 15.8 | 5.6 | TAAAGCATTATG | Internal virion A | AXI78_gp37 [Enterobacter phage E-2] (134/136; 99%) | 4.00E-95 | 136 | AKA61575.1 |
| 39 | ORF39, 100 | 26622 | 27215 | 197 | 21.2 | 9.4 | GTAGGAGGTaactATG | Internal virion B | gp14 [Yersinia phage phiYeO3-12] (194/197; 98%) | 4.00E-136 | 197 | NP_052114.1 |
| 40 | ORF40, 80 | 27218 | 29461 | 747 | 84.6 | 6.1 | CCGGGAGGTaataATG | Internal virion C | ORF37 [Citrobacter phage phiCFP-1] (711/747; 95%) | 0.0 | 747 | AKA62155.1 |
| 41 | ORF41, 83 | 29484 | 33452 | 1322 | 144.2 | 6.7 | TAAGGAGGCtccATG | Internal virion D | ORF38 [Citrobacter phage phiCFP-1] (1296/1322; 98%) | 0.0 | 1322 | AKA62156.1 |
| 42 | ORF42, 92 | 33524 | 35500 | 658 | 69.9 | 6.0 | AAAGGAGGTcacATG | Tail fiber | gp17 [Salmonella phage phiSG-JL2] (593/658; 90%) | 0.0 | 658 | YP_001949790.1 |
| 43 | ORF43, 99 | 35511 | 35714 | 67 | 7.4 | 6.1 | TAAGGAGGAcataATG | Lysis | gp17.5 [Yersinia phage phiYeO3-12] (66/67; 99%) | 5.00E-39 | 67 | NP_052118.1 |
| 44 | ORF44, 100 | 35718 | 35984 | 88 | 9.9 | 4.7 | CAAGGAGTAacctATG | DNA packaging A | gp18 [Salmonella phage phiSG-JL2] (88/88; 100%) | 1.00E-55 | 88 | YP_001949792.1 |
| 45 | ORF45, 99 | 36062 | 36526 | 150 | 17.3 | 9.2 | ATGGGAGGTgttATG | Endopeptidase Rz | ORF42 [Citrobacter phage phiCFP-1] (152/154; 99%) | 1.00E-107 | 154 | AKA62160.1 |
| 45.7 | ORF45.7, 99 | 36189 | 36443 | 84 | 9.3 | 9.8 | TAATCCAAAATG | gp18.7 [Salmonella phage phiSG-JL2] (83/84; 99%) | 9.00E-52 | 84 | YP_001949794.1 | |
| 46 | ORF46, 99 | 36501 | 38264 | 587 | 66.6 | 5.3 | TAAGGAGATgcagaATG | DNA packaging B | gp19 [Salmonella phage phiSG-JL2] (581/587; 99%) | 0.0 | 587 | YP_001949795.1 |
| 46.2 | ORF46.2, 93 | 37213 | 37383 | 56 | 6.1 | 10.0 | GAAGACTTGtactATG | 19.2 protein [Yersinia phage phiYeO3-12] (56/56; 100%) | 2.00E-28 | 77 | NP_052123.1 | |
| 46.3 | ORF46.3, 95 | 37687 | 37815 | 42 | 4.7 | 11.9 | TGGCGGGTTccgcgATG | 19.3 protein [Yersinia phage phiYeO3-12] (42/42; 100%) | 1.00E-19 | 42 | NP_052124.1 | |
| 47 | ORF47, 96 | 38509 | 38658 | 49 | 5.5 | 7.9 | AAAGGAGGTggctcAATG | AVU28_gp23 [Enterobacter phage E-3] (48/49; 98%) | 9.00E-25 | 49 | AKA61602.1 | |
Start codon indicated in boldface; Match to SD sequence is indicated by underlining; SD position is indicated in uppercase.
The number of identical amino acids/The total number of amino acids of smallest protein.
Table 5.
Features of the ORFs of phage SH5, identity with SH4 and SH3, predicted functions of proteins, and best matches with database.
| Predicted protein | Aligned protein | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ORF | SH4 ORF,% | SH3 ORF, % | Start | Stop | Size(aa) | MW(kDa) | pI | SD sequence (TAAGGAGGT) (5′-3′)a | Predicted function | Blast (extent; %aa identityb | E Value | Size (aa) | Accession number |
| 1 | ORF1, 100 | 934 | 1137 | 68 | 7.9 | 6.1 | ATAGGATAAacaagATG | metaG-MbCM1_078 [Synechococcus phage metaG-MbCM1] (28/59; 47%) | 3.00E-11 | 59 | YP_007001569.1 | ||
| 2 | ORF2, 100 | 1134 | 1658 | 175 | 20.2 | 5.7 | TAAGGAACTacaatcATG | CPT_Seurat66 [Escherichia phage Seurat](87/167; 52%) | 6.00E-48 | 167 | YP_009152010.1 | ||
| 3 | ORF3, 100 | ORF3, 84 | 1652 | 1807 | 52 | 5.9 | 9.4 | AGGTGAGGTcatcaagATG | gp0.35 [Enterobacteria phage EcoDS1] (47/50; 94%) | 2.00E-27 | 50 | YP_002003737.1 | |
| 4 | ORF4, 100 | 2006 | 2173 | 56 | 5.9 | 8.3 | ATAGGAGTTaactATG | PE3_004 [Escherichia phage PE3-1] (48/55; 87%) | 2.00E-24 | 55 | YP_009044252.1 | ||
| 5 | ORF5, 100 | ORF5, 99 | 2177 | 2374 | 66 | 7.5 | 11.0 | GCGGGATAAaccATG | gp0.6 [Enterobacteria phage EcoDS1](64/65; 98%) | 4.00E-37 | 65 | YP_002003739.1 | |
| 6 | ORF6, 100 | ORF6, 56 | 2374 | 2700 | 108 | 12.1 | 9.3 | TTGGGAGCAaactgtaATG | PE3_006 [Escherichia phage PE3-1] (76/108; 70%) | 8.00E-48 | 130 | YP_009044254.1 | |
| 7 | ORF7, 100 | ORF7, 93 | 2798 | 5479 | 894 | 100.5 | 7.6 | CAAGGACTTtaagtATG | RNA polymerase | gp1 [Cronobacter phage Dev2] (883/893; 99%) | 0.0 | 893 | YP_009005115.1 |
| 8 | ORF8, 100 | ORF8, 78 | 5492 | 5692 | 67 | 7.3 | 9.7 | TAAGGAGGCatctacATG | gp1.1 [Cronobacter phage Dev2] (66/66; 100%) | 6.00E-38 | 66 | YP_009005116.1 | |
| 9 | ORF9, 100 | 5771 | 6250 | 160 | 18.6 | 9.3 | AGAGGTTGAcactATG | gp1.06 [Cronobacter phage Dev2](155/159; 97%) | 1.00E-110 | 159 | YP_009005117.1 | ||
| 10 | ORF10, 100 | ORF10, 98 | 6339 | 6518 | 60 | 6.8 | 10.2 | ACTGGAGATttaaccATG | gp1.15 [Cronobacter phage Dev2](58/59; 98%) | 4.00E-33 | 59 | YP_009005118.1 | |
| 11 | ORF11, 100 | ORF11, 22 | 6522 | 6809 | 96 | 11.2 | 6.3 | GTAGGAGCGtaagacATG | PE3_010 [Escherichia phage PE3-1](81/95; 85%) | 5.00E-56 | 95 | YP_009044258.1 | |
| 12 | ORF12, 99 | ORF12, 75 | 6827 | 7897 | 357 | 40.2 | 5.5 | TCTGGAGACattaacgATG | DNA ligase | gp1.3 [Enterobacteria phage EcoDS1] (318/357; 89%) | 0.0 | 365 | YP_002003747.1 |
| 13 | ORF13, 100 | ORF13, 49 | 8019 | 8273 | 85 | 9.85 | 9.9 | AGAGGAGAAaccttATG | gp1.6 [Enterobacteria phage EcoDS1] (73/84; 87%) | 6.00E-47 | 84 | YP_002003748.1 | |
| 14 | ORF14, 100 | ORF14, 67 | 8273 | 8593 | 107 | 12.2 | 6.9 | CAAGGAGGAgttctaATG | gp1.7 [Cronobacter phage Dev2] (85/107; 79%) | 5.00E-50 | 116 | YP_009005122.1 | |
| 15 | ORF15, 100 | ORF16, 65 | 8672 | 8887 | 72 | 8.2 | 4.5 | GAAGGAGAAaggactATG | Bacterial RNA polymerase inhibitor | gp2 [Cronobacter phage Dev2] (48/54; 89%) | 3.00E-25 | 54 | YP_009005123.1 |
| 16 | ORF16, 100 | ORF17, 85 | 8935 | 9633 | 233 | 25.4 | 4.8 | CTAGGAGATttacaccgATG | Helix-destabilizing protein | gp2.5 [Cronobacter phage Dev2] (229/232; 99%) | 2.00E-164 | 232 | YP_009005124.1 |
| 17 | ORF17, 100 | ORF18, 43 | 9870 | 10088 | 72 | 8.3 | 9.9 | TAAGAAGCAtATG | Endonuclease | gp3 [Cronobacter phage Dev2] (72/72; 100%) | 2.00E-44 | 139 | YP_009005125.1 |
| 18 | ORF18, 100 | ORF19, 81 | 10085 | 10309 | 75 | 8.6 | 9.7 | AAAGGAGCTaagaaATG | gp3.2 [Cronobacter phage Dev2] (74/74; 100%) | 3.00E-45 | 74 | YP_009005126.1 | |
| 19 | ORF19, 98 | ORF20, 90 | 10299 | 10757 | 153 | 16.9 | 8.8 | GCTGGTGGTgtacaATG | N-acetylmuramoyl-l-alanine amidase | gp3.5 [Cronobacter phage Dev2] (152/152; 100%) | 8.00E-109 | 152 | YP_009005127.1 |
| 20 | ORF20, 100 | ORF21, 61 | 10772 | 10984 | 71 | 7.4 | 10.1 | CAAGGAGTAttaacATG | gp3.7 [Cronobacter phage Dev2] (69/70; 99%) | 1.00E-28 | 70 | YP_009005128.1 | |
| 21 | 11142 | 11507 | 122 | 13.9 | 10.1 | GCGGGATAAaccATG | HNH endonuclease | gp3.8 [Enterobacteria phage T7] (65/121; 54%) | 1.00E-40 | 121 | NP_041974.1 | ||
| 22 | ORF21, 97 | ORF22, 90 | 11482 | 13179 | 566 | 62.2 | 5.1 | TAAGGAGGCtcATG | Primase/Helicase | gp4 [Cronobacter phage Dev2] (545/566; 96%) | 0.0 | 567 | YP_009005129.1 |
| 22B | ORF21B, 98 | ORF22B, 92 | 11782 | 13179 | 465 | 51.3 | 5.2 | TTGGGTAGGcATG | Primase/Helicase | gp4 [Cronobacter phage Dev2] (475/465; 98%) | 0.0 | 567 | YP_009005129.1 |
| 22.2 | ORF21.2, 61 | ORF22.2, 62 | 12908 | 13255 | 115 | 13.0 | 9.0 | AAAGGTAAGtctcATG | gp4.2 [Enterobacteria phage K1F] (63/107; 56%) | 2.00E-25 | 107 | CAJ29367.1 | |
| 23 | ORF22, 66 | ORF23, 69 | 13182 | 13766 | 194 | 21.2 | 4.7 | CAACGACTTctgaccATG | gp4.1 [Cronobacter phage Dev2] (168/177; 95%) | 1.00E-117 | 177 | YP_009005130.1 | |
| 24 | ORF23, 99 | ORF24, 90 | 13837 | 16008 | 724 | 80.9 | 7.0 | ATAGGAGACattATG | DNA polymerase | gp5 [Cronobacter phage Dev2] (717/723; 99%) | 0.0 | 723 | YP_009005131.1 |
| 25 | ORF24, 97 | ORF26, 96 | 16008 | 16292 | 95 | 10.5 | 5.2 | GAAGGAGTGtcactaATG | HNS binding protein | gp5.5 [Cronobacter phage Dev2] (92/94; 98%) | 4.00E-58 | 94 | YP_009005133.1 |
| 26 | ORF25, 100 | ORF27, 100 | 16289 | 16498 | 70 | 7.4 | 9.0 | ATTCGAGGTcaaacgATG | gp21 [Enterobacteria phage K1F] (69/69; 100%) | 5.00E-43 | 69 | YP_338112.1 | |
| 27 | ORF26, 100 | 16495 | 16770 | 92 | 9.9 | 5.3 | GGAGGCTGTctATG | ASC_0027 [Klebsiella phage K11](37/68; 54%) | 2.00E-19 | 68 | YP_002003815.1 | ||
| 28 | ORF27, 99 | ORF28, 90 | 16763 | 17629 | 289 | 32.9 | 5.4 | AAAGGAGGTctgcgggATG | Exonuclease | gp6 [Cronobacter phage Dev2] (284/288; 99%) | 0.0 | 288 | YP_009005135.1 |
| 29 | ORF28, 100 | ORF29, 56 | 17837 | 18109 | 91 | 9.9 | 5.2 | AGAGGAGACtttaagATG | gp6.5 [Cronobacter phage Dev2] (90/90; 100%) | 8.00E-58 | 90 | YP_009005136.1 | |
| 30 | ORF29, 100 | ORF30, 92 | 18120 | 18344 | 75 | 7.6 | 6.2 | AAAGGAGGGactATG | Head protein | gp6.7 [Cronobacter phage Dev2] (74/74; 100%) | 1.00E-42 | 74 | YP_009005137.1 |
| 31 | ORF30, 100 | ORF31, 91 | 18348 | 18752 | 135 | 15.5 | 6.1 | ACATGGGGTAAGacATG | gp34 [Citrobacter phage CR44b] (127/135; 94%) | 7.00E-89 | 185 | YP_009007168.1 | |
| 32 | ORF31, 100 | ORF32, 96 | 19011 | 20579 | 523 | 57.2 | 4.6 | GCAGGAGGTgacaaATG | Head to tail connector protein | gp8 [Cronobacter phage Dev2] (522/522; 100%) | 0.0 | 522 | YP_009005140.1 |
| 33 | ORF32, 99 | ORF33, 81 | 20684 | 21565 | 294 | 31.7 | 4.4 | AAAGGAGAAcgactcaATG | Capsid assembly protein | gp9 [Cronobacter phage Dev2] (290/293; 99%) | 0.0 | 293 | YP_009005141.1 |
| 34 | ORF33, 99 | ORF34, 94 | 21697 | 22746 | 350 | 36.4 | 5.8 | ATAGGAGAAttatcatATG | Major capsid protein | gp10 [Cronobacter phage Dev2] (347/349; 99%) | 0.0 | 349 | YP_009005142.1 |
| 35 | ORF34, 100 | ORF35, 97 | 23067 | 23633 | 189 | 21.3 | 4.4 | TAAGGAGGGcctATG | Tail tube protein A | gp11 [Cronobacter phage Dev2] (187/188; 99%) | 3.00E-135 | 188 | YP_009005144.1 |
| 36 | ORF35, 99 | ORF36, 87 | 23645 | 26014 | 790 | 87.6 | 5.8 | ATAGGAGGTgatATG | Tail tube protein B | gp12 [Cronobacter phage Dev2] (779/789; 99%) | 0.0 | 789 | YP_009005145.1 |
| 37 | ORF36, 97 | ORF37, 83 | 26090 | 26548 | 153 | 17.5 | 6.9 | ATAGGAGACtttATG | Internal virion protein A | gp13 [Cronobacter phage Dev2] (150/152; 99%) | 6.00E-107 | 152 | YP_009005146.1 |
| 38 | ORF37, 98 | ORF38, 91 | 26669 | 27256 | 196 | 20.4 | 6.8 | CCGGGAGGTgaaagATG | Internal virion protein B | gp14 [Cronobacter phage Dev2] (194/195; 99%) | 1.00E-136 | 195 | YP_009005147.1 |
| 39 | ORF38, 99 | ORF39, 89 | 27268 | 29550 | 761 | 85.3 | 5.5 | ATAGGAGGAccaATG | Internal virion protein C | gp15 [Cronobacter phage Dev2] (749/760; 99%) | 0.0 | 760 | YP_009005148.1 |
| 40 | ORF39, 99 | ORF40, 92 | 29556 | 33452 | 1299 | 141.0 | 5.8 | TAAGGAGTAataacaATG | Internal virion protein D | gp16 [Cronobacter phage Dev2] (1282/1298; 99%) | 0.0 | 1298 | YP_009005149.1 |
| 41 | ORF40, 100 | ORF41, 79 | 33520 | 36018 | 832 | 91.3 | 6.4 | TAAGGAGGCccaATG | Tail fibers | gp17 [Cronobacter phage Dev2] (805/832; 97%) | 0.0 | 832 | YP_009005150.1 |
| 42 | ORF41, 100 | ORF42, 95 | 36065 | 36259 | 65 | 6.9 | 8.0 | AACGGAGGTattATG | Lysis protein | gp17.5 [Cronobacter phage Dev2] (64/64; 100%) | 4.00E-37 | 64 | YP_009005151.1 |
| 43 | ORF42, 100 | ORF43, 97 | 36256 | 36519 | 87 | 10.1 | 4.8 | AGTGGAGGTaagacATG | DNA packaging protein | gp18 [Cronobacter phage Dev2] (87/87; 100%) | 1.00E-54 | 87 | YP_009005152.1 |
| 44 | ORF43, 100 | ORF44, 73 | 36624 | 37073 | 150 | 16.9 | 8.8 | CGAGGAGGGcaactATG | Endopeptidase Rz | gp18.5 [Cronobacter phage Dev2] (147/149; 99%) | 3.00E-101 | 149 | YP_009005153.1 |
| 44.7 | ORF43.7, 100 | ORF44.7, 72 | 36727 | 36999 | 90 | 9.8 | 9.6 | GAAGGTAAGcaATG | Endopeptidase Rz1 | gp18.7 [Enterobacteria phage EcoDS1] (60/89; 67%) | 2.00E-31 | 91 | YP_002003785.1 |
| 45 | ORF44, 100 | ORF45, 94 | 37103 | 38833 | 577 | 65.1 | 5.3 | TCAGGCGCTtATG | Maturation protein | gp19 [Cronobacter phage Dev2] (574/577; 99%) | 0.0 | 587 | YP_009005154.1 |
| 45.2 | ORF44.2, 100 | 37752 | 37877 | 41 | 4.8 | 12.1 | TATCCTCGTgATG | gp19.2 [Enterobacteria phage K1F] (27/55; 49%) | 7.00E-09 | 55 | CAJ29396.1 | ||
| 46 | ORF45, 100 | ORF46, 94 | 39124 | 39282 | 53 | 5.5 | 9.3 | GTATGTAGCATG | gp19.5 [Cronobacter phage Dev2] (52/52; 100%) | 2.00E-27 | 52 | YP_009005155.1 | |
Start codon indicated in boldface; Match to SD sequence is indicated by underlining; SD position is indicated in uppercase.
bThe number of identical amino acids/The total number of amino acids of smallest protein.
Figure 5.

Schematic representation of the genomic organization of phage T7 (NC_001604.1) compared to phages SH1, SH2, phiYeO3-12 (NC_001271.1), K1F (NC_007456.1), SH3, SH4, SH5, and Dev2 (NC_023558.1). Each arrow represents an ORF. Deduced ORFs sharing 95% amino acid identity are represented with the same color. Gray shading indicates ORFs whose translated products share 80% identity with the ones of phage T7. Finally, arrows with thick outlines and bold numbers represent structural proteins detected by LC-MS/MS.
The genomes of the five isolated phages are co-linear and share the same genomic organization as phage T7 with what seems to be early-, middle-, and late-expressed regions. The early genes are usually involved in host takeover and conversion of the host metabolism for the benefit of phage production (Pajunen et al., 2001). This region is also characterized by the presence of an RNA polymerase responsible for the transcription of all the middle- and late-expressed genes. The middle-expressed region includes genes responsible for DNA metabolism while the late region contains genes coding for structural proteins.
Proteomic analyses
The structural proteome of one phage representing each of the three subgroups (phage SH1 for group 1, SH3 for group 2A and SH4 for group 2B) was analyzed. Purified phages were analyzed by LC-MS/MS and the results are presented in Table 6. For phage SH1, 11 proteins were detected with an amino acid coverage ranging from 12 to 65%. Ten of the 11 genes coding for these proteins were located in the presumably late-expressed module, as expected for genes coding for structural proteins. The other protein (ORF19) was a N-acetylmuramoyl-L-alanine amidase probably involved in host lysis and it had the lowest coverage (12%). Its gene was located in the middle-expressed region. It is unclear if this protein is in the phage structure or if it is a non-structural phage protein that was carried over from the phage purification process.
Table 6.
Identified peptides for phages SH1, SH3 and SH4 and their predicted functions.
| Phage | Start | Stop | ORF | Predicted function | Mass (kDa) | Exclusive unique peptide | Coverage (%) |
|---|---|---|---|---|---|---|---|
| SH1 | 11614 | 12069 | 19 | N-acetylmuramoyl-L-alanine amidase | 17 | 2 | 12 |
| 18713 | 19039 | 31 | Capsid protein | 12 | 2 | 36 | |
| 19067 | 19387 | 32 | Host specficity protein B | 11 | 3 | 29 | |
| 19398 | 21005 | 33 | Capsid to tail joining protein | 59 | 29 | 65 | |
| 22196 | 23236 | 35 | Major capsid protein | 37 | 22 | 60 | |
| 23426 | 24016 | 36 | Tail tubular protein A | 22 | 5 | 23 | |
| 24032 | 26437 | 37 | Tail tubular protein B | 90 | 32 | 44 | |
| 26923 | 27516 | 39 | Internal virion protein B | 21 | 12 | 65 | |
| 27519 | 29762 | 40 | Internal virion protein C | 85 | 36 | 59 | |
| 29781 | 33743 | 41 | Internal virion protein D | 144 | 63 | 57 | |
| 33815 | 35791 | 42 | Tail fibers protein | 70 | 21 | 48 | |
| SH3 | 18963 | 20531 | 32 | Capsid to tail connector protein | 57 | 26 | 67 |
| 20676 | 21560 | 33 | Capsid assembly protein | 32 | 7 | 21 | |
| 21687 | 22730 | 34 | Major capsid protein | 36 | 8 | 50 | |
| 22925 | 23491 | 35 | Tail tube protein A | 21 | 5 | 32 | |
| 23503 | 25872 | 36 | Tail tube protein B | 88 | 24 | 37 | |
| 26404 | 26991 | 38 | Internal virion protein B | 20 | 10 | 65 | |
| 27003 | 29285 | 39 | Internal virion protein C | 85 | 25 | 42 | |
| 29290 | 33177 | 40 | Internal virion protein D | 141 | 45 | 45 | |
| 33243 | 35747 | 41 | Tail fibers protein | 91 | 20 | 33 | |
| SH4 | 18577 | 20145 | 31 | Capsid to tail connector protein | 57 | 5 | 39 |
| 21263 | 22312 | 33 | Major capsid protein | 36 | 15 | 40 | |
| 23211 | 25580 | 35 | Tail tube protein B | 88 | 10 | 24 | |
| 26111 | 26698 | 37 | Internal virion protein B | 20 | 2 | 32 | |
| 26710 | 28992 | 38 | Internal virion protein C | 85 | 14 | 29 | |
| 28998 | 32894 | 39 | Internal virion protein D | 141 | 14 | 20 | |
| 32962 | 35460 | 40 | Tail fibers protein | 91 | 9 | 18 |
For phage SH3, 9 structural proteins were detected with coverage ranging from 21 to 67%, while for phage SH4, 7 structural proteins were identified with coverage ranging from 18 to 40%. For these two phages, all the proteins detected were structural proteins from the capsid, head-tail joining, tail, tail tube, and tail fibers.
DNA polymerase phylogeny
Because the five Citrobacter podophages belong to the T7virus genus, we compared in greater detail their relationships with other characterized similar phages available in public database (Figure 6). The T7 DNA polymerase is a conserved protein often used to study the global distribution and diversity of podophages, in a manner analogous to the 16S rRNA in bacteria (Breitbart et al., 2004). Based on DNA polymerase phylogeny, the five phages were confirmed to belong to the T7virus genus in the subfamily Autographivirinae. However, they mapped at two different sub-branches. Phages SH1 and SH2 were similar to Yersinia phages phiYeO3-12 and vBYenP AP5, Salmonella phage phiSG-JL2, Citrobacter phage phiCFP-1, and Enterobacter phages E3 and E4. They were also closer to the prototype phage T7 than the other three phages characterized here. Phages SH3, SH4, and SH5 were part of the same clade of t7viruses as SH1 and SH2, but clustered in different subgroups. Phage SH3 was related to Enterobacteria phages K1F and EcoDS1, and Escherichia phage PE3-1. Phages SH4 and SH5 were more related to Cronobacter phage Dev2. Taken altogether, despite the differences between these two groupings, SH1/SH2 and SH3/SH4/SH5 seem to be derived from a common ancestor.
Figure 6.

Phylogenetic relationship between selected phage DNA polymerase sequences.
Comparison between phages SH1/SH2, phiYeO3-12, and coliphage T7 (Group 1)
Pairwise analyses between the deduced proteomes of phages SH1 and SH2 revealed 31 proteins (out of 53) with more than 95% identity (Table 4). Among them, seven (ORF8, ORF18, ORF23, ORF26, ORF29, ORF39, ORF44) were 100% identical, including two proteins with predicted functions, ORF39/internal virion protein B and ORF44/DNA packaging protein A. Phage SH1 also shared more than 95% identity with 31 proteins of Yersinia phage phiYeO3-12 including seven proteins with 100% identity (ORF8, ORF20, ORF22, ORF23, ORF25, ORF26, ORF29). Phage SH2 shared more than 95% identity with 34 proteins of Yersinia phage phiYeO3-12 including 13 proteins with 100% identity (Table 4). Phage SH2 seems more related to phage phiYeO3-12 than phage SH1. One of the most notable differences between phages SH1 and SH2/phiYeO3-12 was mobile elements. Phage SH1 is missing the homing endonuclease encoded on phages SH2 and phiYeO3-12 (ORF23SH2/ORF5.3 phiYeO3-12). However, phage SH1 has another homing endonuclease (ORF17), which is absent in both genomes of SH2 and phiYeO3-12. ORF17SH1 is homologous to a homing endonuclease found on C. rodentium phage CR44b (46%). Phage SH1 is also missing ORF35SH2/ORF10BphiYeO3-12 a minor capsid protein (Condron et al., 1991). On the other hand, the tail fiber protein of phage phiYeO3-12 shares only 68 and 67% identity with the tail fiber proteins of SH2 and SH1, respectively, which could explain the divergent host ranges between SH1/SH2 and phiYeO3-12. In fact, phages SH1 and SH2 were not able to infect the host strain Yersinia enterocolitica 6471/76-c (HER1249) of phage phiYeO3-12.
In addition, phages SH1 and SH2 shared 11 proteins with more than 80% amino acid identity with coliphage T7, including the RNA polymerase (ORF1T7 and ORF6SH1/SH2). The T7 RNA polymerase initiates transcription by exclusively recognizing its own promoters to ensure fast and efficient transcription of phage DNA. It is also involved in DNA replication, maturation and packaging (Studier and Moffatt, 1986; Zhang and Studier, 2004).
Another T7 protein homologous to SH1/SH2 proteins was ORF2.5T7 (homologous to ORF16SH1 and ORF15SH2), which is a single-stranded DNA binding protein. The orf2.5T7 gene is essential for phage DNA replication and recombination (Scaltriti et al., 2009, 2013). The N-acetylmuramoyl-L-alanine amidase ORF3.5T7 was also related to ORF19SH1 and ORF17SH2. This lysozyme is involved in cell lysis but may also inhibit transcription by binding to the RNA polymerase to ensure a controlled burst of late transcription (Inouye et al., 1973; Moffatt and Studier, 1987). ORF21SH1 and ORF19SH2 were similar to the T7 primase/helicase, ORF4T7. This primase/helicase activity is essential for DNA replication (Rosenberg et al., 1992) as the helicase catalyzes strand displacement during DNA replication while the primase is involved in the synthesis of the DNA lagging-strand (Mendelman et al., 1992).
The ORF5.7 protein of phage T7 shared a high level of identity with ORF26SH1 and ORF25SH2. ORF5.7 stimulates the expression of gene 5.5 which encodes a H-NS binding protein (Zhu et al., 2012). When gene 5.5 is missing, the phage plaque and the burst sizes are reduced (Owen-Hughes et al., 1992; Liu and Richardson, 1993). The H-NS binding protein inhibits the function of the highly conserved host histone-like nucleoid structuring (H-NS) protein, which influences gene expression, recombination and transcription.
A notable difference between phage T7 and phages SH1/SH2 was in their antirestriction proteins (gp0.3T7/ORF1SH1/SH2). Restriction-modification (R-M) systems are well-known resistance mechanisms used by bacteria to block phage replication (Labrie et al., 2010). Phages also have several means to bypass these systems (Samson et al., 2013). The Phage T7Ocr (overcoming classical restriction, ORF0.3) protein mimics the DNA phosphate backbone, interacting directly with the type R-MEcoKI enzyme, and interfering with the activity of this system (Atanasiu et al., 2002; Stephanou et al., 2009). At the same genomic location (Figure 5), the phage SH1 and SH2 orf1 genes code for a putative S-adenosyl-l-methionine hydrolase, homologous to gp0.3phiYeO3-12, which destroys S-adenosyl-l-methionine, an essential R–M cofactor (Studier and Movva, 1976). The Ocr protein of phage T7 does not have the hydrolase activity. However, the Ocr protein of E. coli podophage T3, whose gene is located at the same genomic position, possesses this hydrolase activity.
Comparison between phages SH3 and K1F (Group 2A)
The deduced proteome of phage SH3 (49 ORFs) ranged from 30 to 75% identity to the proteins of phages SH1 and SH2. However, phage SH3 had eight proteins with more than 95% identity to proteins of phages SH4 and SH5, including 100% identity between ORF27SH3 and ORF25SH4/ORF26SH5 (Table 5). Otherwise, the closest phage to SH3 was coliphage K1F with 23 proteins sharing more than 95% identity. Of these, four proteins are 100% identical, including two with a known function (lysis protein and DNA packaging protein). Genetic differences were noted between Citrobacter phage SH3 and E. coli phage K1F and the most important difference lies in tail fibers (Gp17K1F/ORF41SH3) that consist of two domains. The N-terminal domain is responsible for attachment to the phage tail and the C-terminal domain is involved in the recognition of and adsorption to the host LPS (Kajsík et al., 2014). The N-terminal parts of the tail fibers of both K1F and SH3 shared a region with the phage T7 tail fiber. However, the central catalytic portion of Gp17K1F encodes an endosialidase to penetrate the host polysaccharide capsule (Scholl and Merril, 2005) while ORF41SH3 contains a domain of the SGNH hydrolase superfamily like the tail fibers of phages Dev2, SH4, and SH5. However, the C-terminal part of ORF41SH3 is different than the tail fibers of phages SH4, SH5, and Dev2, which explains its different host range. The SH3 genome is also missing the putative group I intron present within the DNA polymerase of K1F (gp5.3) which encodes a homing endonuclease.
Comparison between phages SH4/SH5 and Dev2 (Group 2B)
Of the 45 genes of phage SH5, 33 were 100% identical to genes of phage SH4. Ten of these genes are also 100% identical to the T7virus Cronobacter phage Dev2 genes. These conserved genes suggest that the three phages may be derived from a common ancestor. In addition, phages SH4 and SH5 have more than 95% aa identity with almost all of the phage Dev2 structural proteins. Interestingly, the putative tail fiber proteins ORF40SH4 and ORF41SH5 were 99% identical to tail fiber gp17 of phage Dev2, suggesting a similar host range. We received phage Dev2 and tested its host range in parallel with phages SH4 and SH5 on the 31 bacterial strains available. The three phages were able to lyse the same strains, C. freundii CF3, C. freundii CF4, and C. turicensis 290708/07.
Phages SH4 and SH5 are missing the genes coding for gp5.1- and gp10.1-like located in the late-expressed region, found in Dev2 (Kajsík et al., 2014). Most genomic differences between SH4/SH5 and Dev2 were located in the early-expressed region. ORF21 of phage SH5, which encodes an HNH endonuclease with a zinc-binding motif involved in different steps of phage development (Anba et al., 2002), was missing from phages SH4 and Dev2. However, ORF21 shares 54% identity with T7 gp3.8.
The SH4 and SH5 proteins with the lowest similarity were ORF22SH4 (132 aa) and ORF23SH5 (194 aa) but these were still 66% identical. Their amino acid sequences could be aligned perfectly at the C-terminal end but ORF22SH4 is missing the N-terminal portion of ORF23SH5. A mutation may have occurred as we noticed the lack of a T base at the ATG codon of ORF22SH4. ORF23SH5 had 95% identity to gp4.1 of phage Dev2 but its function is unknown.
Discussion
In this study, we isolated and characterized five virulent Podoviridae phages infecting C. freundii, an emerging pathogenic bacterial species (Samonis et al., 2008). Genome analyses showed that the five newly isolated phages belong to the Autographivirinae subfamily and the T7virus genus. Their morphological and genomic properties allowed us to separate them into two different groups, group 1 (phages SH1 and SH2) and group 2 (phages SH3, SH4, and SH5). However, the two groups are co-linear and share conserved genomic organization. They are flanked by terminal repeats involved in concatemer formation, DNA packaging, and particle maturation (Chung et al., 1990). Despite their small size (close to 40 kb), the five phage genomes encode the usual modules with genes coding for proteins involved in DNA replication, transcription regulation, morphological proteins, lysis proteins, as well as DNA maturation and packaging. As such, they have very compact genomes with overlapping genes (Mendelman et al., 1992) as more than 90% of the five genome sequences were predicted to encode proteins. For phages SH1, SH3, and SH4 almost all the predicted structural proteins were detected by LC-MS/MS, showing that they are indeed transcribed and translated.
Another reason for sequencing the new phage genomes is to provide a clearer view about the dynamics of phage populations over space and time. Based on genomic and proteomic identification, we could define evolutionary relationships between these podophages (Brüssow and Hendrix, 2002). For example, phage T7 was isolated in 1945 (Delbrück, 1945), phage phiYeO3-12 from sewage in 1988 in Finland (Al-Hendy et al., 1991), phage K1F from sewage in 1984 in the USA (Scholl and Merril, 2005), and phage Dev2 was recently isolated from sewage in Slovakia (Kajsík et al., 2014). All five C. freundii phages characterized in this study were isolated from different sewage samples collected in Tunisia in 2014. These phages are geographically and temporally distant but from an evolutionary perspective, these phages likely shared a common ancestor.
As phages tend to coevolve with their bacterial hosts (Skurnik and Strauch, 2006) and C. freundii can produce enterotoxins (Guarino et al., 1987), we inspected the five phage genomes for the presence of host related genes, particularly those coding for known virulence-factors or integrase. No such genes were found, indicating that they are truly lytic phages as well as suggesting that they may be safe for therapeutic or prevention applications. Moreover, it was relatively easy to purify them and we obtained highly concentrated phage preparations. Conversely, these phages were inactivated at very acidic pH (2–3), suggesting that they may not survive in high numbers after passage through the gastrointestinal tract or in highly acidic foods. Others have shown that microencapsulation in alginate-chitosan microspheres significantly improved the survival and stability of phages under harsh acidic conditions (Ma et al., 2008). Finally, their limited host range suggests that they should be used in combination to maximize strain coverage. Of note, no CRISPR-Cas systems were found in the C. freundii genomes analyzed.
Taken altogether, the newly characterized Podoviridae phages SH1, SH2, SH3, SH4, and SH5 have appealing properties for prophylactic or therapeutic use to control the proliferation of C. freundii infections. The analyses of these Citrobacter phages also provided new evolutionary relationships with the expanding group of phages belonging to the T7virus genus, including with phages infecting Cronobacter and Yersinia species of the Enterobacteriaceae family.
Author contributions
SM, KS, RK conceived and designed the study and afforded the materials. SH performed the experiments, analyzed the data and drafted the manuscript. GR participated in the data analysis and helped in the coordination of the experiments. DT did the sequencing and the electron microscopy. SL designed the figures and helped in the bioinformatics analysis. SM critically revised the manuscript. All authors read and approved the manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Prof. Hana Drahovska for Cronobacter phage Dev2. We would also like to thank Barbara-Ann Conway (Medical Writer & Editor) for editorial assistance. SM holds a Tier 1 Canada Research Chair in Bacteriophages.
References
- Abbott S. L. (2011). Klebsiella, Enterobacter, Citrobacter, Serratia, Plesiomonas, and Other Enterobacteriaceae, in Manual of Clinical Microbiology, 10th Edn., eds Versalovic J., Jorgensen J. H., Funke G., Warnock D. W., Landry M. L., Carroll K. C. (Washington, DC: American Society of Microbiology Press; ), 639–657. 10.1128/9781555816728.ch37 [DOI] [Google Scholar]
- Ackermann H. W., Nguyen T. M. (1983). Sewage coliphages studied by electron microscopy. Appl. Environ. Microbiol. 45, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hendy A., Toivanen P., Skurnik M. (1991). Expression cloning of Yersinia enterocolitica 0 : 3 rfb gene cluster in Escherichia coli K12. Microb. Pathog. 10, 47–59. 10.1016/0882-4010(91)90065-I [DOI] [PubMed] [Google Scholar]
- Anba J., Cesselin B., Commissaire J., Crutz-Le Coq A.-M. (2002). Sequence analysis of the lactococcal bacteriophage bIL170: insights into structural proteins and HNH endonucleases in dairy phages. Microbiology 148, 985–1001. 10.1099/00221287-148-4-985 [DOI] [PubMed] [Google Scholar]
- Antonelli A., D'Andrea M. M., Vaggelli G., Docquier J.-D., Rossolini G. M. (2015). OXA-372, a novel carbapenem-hydrolysing class D β-lactamase from a Citrobacter freundii isolated from a hospital wastewater plant. J. Antimicrob. Chemother. 70, 2749–2756. 10.1093/jac/dkv181 [DOI] [PubMed] [Google Scholar]
- Atanasiu C., Su T.-J., Sturrock S. S., Dryden D. T. F. (2002). Interaction of the ocr gene 0.3 protein of bacteriophage T7 with EcoKI restriction/modification enzyme. Nucleic Acids Res. 30, 3936–3944. 10.1093/nar/gkf518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L., Xia S., Lan R., Liu L., Ye C., Wang Y., et al. (2012). Isolation and characterization of cytotoxic, aggregative Citrobacter freundii. PLoS ONE 7:e33054. 10.1371/journal.pone.0033054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal C. L., Berkowitz V. E., Cahill J. L., Rasche E. S., Kuty Everett G. F. (2015). Complete genome sequence of Citrobacter freundii myophage Michonne. Genome Announc. 3:e01134-15. 10.1128/genomeA.01134-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M., Miyake J. H., Rohwer F. (2004). Global distribution of nearly identical phage-encoded DNA sequences. FEMS Microbiol. Lett. 236, 249–256. 10.1111/j.1574-6968.2004.tb09654.x [DOI] [PubMed] [Google Scholar]
- Brüssow H., Hendrix R. W. (2002). Phage genomics: small is beautiful. Cell 108, 13–16. 10.1016/S0092-8674(01)00637-7 [DOI] [PubMed] [Google Scholar]
- Campos M. J., Palomo G., Hormeño L., Rodrigues A. P., Sánchez-Benito R., Píriz S., et al. (2015). Detection of QnrB54 and its novel genetic context in Citrobacter freundii isolated from a clinical case. Antimicrob. Agents Chemother. 59, 1375–1376. 10.1128/AAC.03895-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton R. M. (1999). Phage therapy: past history and future prospects. Arch. Immunol. Ther. Exp. (Warsz.) 47, 267–274. [PubMed] [Google Scholar]
- Chaudhry W. N., Haq I. U., Andleeb S., Qadri I. (2014). Characterization of a virulent bacteriophage LK1 specific for Citrobacter freundii isolated from sewage water. J. Basic Microbiol. 54, 531–541. 10.1002/jobm.201200710 [DOI] [PubMed] [Google Scholar]
- Chen Z., Schneider T. D. (2005). Information theory based T7-like promoter models: classification of bacteriophages and differential evolution of promoters and their polymerases. Nucleic Acids Res. 33, 6172–6187. 10.1093/nar/gki915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibani Azaïez S. R., Fliss I., Simard R. E., Moineau S. (1998). Monoclonal antibodies raised against native major capsid proteins of lactococcal c2-Like bacteriophages. Appl. Environ. Microbiol. 64, 4255–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y.-B., Nardone C., Hinkle D. C. (1990). Bacteriophage T7 DNA packaging: III. A “Hairpin” end formed on T7 concatemers may be an intermediate in the processing reaction. J. Mol. Biol. 216, 939–948. 10.1016/S0022-2836(99)80012-6 [DOI] [PubMed] [Google Scholar]
- Clark J. M. (1988). Novel non-templated nucleotide addition reactions catalyzed by procaryotic and eucaryotic DNA polymerases. Nucleic Acids Res. 16, 9677–9686. 10.1093/nar/16.20.9677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condron B. G., Atkins J. F., Gesteland R. F. (1991). Frameshifting in gene 10 of bacteriophage T7. J. Bacteriol. 173, 6998–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D., Taboada G. L., Doallo R., Posada D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. 10.1093/bioinformatics/btr088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbrück M. (1945). The burst size distribution in the growth of bacterial viruses (bacteriophages). J. Bacteriol. 50, 131–135. [DOI] [PubMed] [Google Scholar]
- Deveau H., Van Calsteren M.-R., Moineau S. (2002). Effect of exopolysaccharides on phage-host interactions in Lactococcus lactis. Appl. Environ. Microbiol. 68, 4364–4369. 10.1128/AEM.68.9.4364-4369.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drelichman V., Band J. (1985). Bacteremias due to Citrobacter diversus and Citrobacter freundii: incidence, risk factors, and clinical outcome. Arch. Intern. Med. 145, 1808–1810. 10.1001/archinte.1985.00360100068010 [DOI] [PubMed] [Google Scholar]
- Dunn J. J., Studier F. W., Gottesman M. (1983). Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol. 166, 477–535. 10.1016/S0022-2836(83)80282-4 [DOI] [PubMed] [Google Scholar]
- Edwards G. B., Luna A. J., Hernandez A. C., Kuty Everett G. F. (2015). Complete genome sequence of Citrobacter freundii myophage Moon. Genome Announc. 3:e01427-14. 10.1128/genomeA.01427-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing W. H., Davis B. R. (1972). Biochemical characterization of Citrobacter diversus (Burkey) Werkman and Gillen and designation of the neotype Strain. Int. J. Syst. Evol. Microbiol. 22, 12–18. 10.1099/00207713-22-1-12 [DOI] [Google Scholar]
- Fortier L.-C., Moineau S. (2007). Morphological and genetic diversity of temperate phages in Clostridium difficile. Appl. Environ. Microbiol. 73, 7358–7366. 10.1128/AEM.00582-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiksen W. (2015). “Citrobacter,” in Bergey's Manual of Systematics of Archaea and Bacteria, 2nd Edn (New York, NY: John Wiley & Sons, Ltd; ), 651–656. [Google Scholar]
- Garneau J. E., Dupuis M.-È., Villion M., Romero D. A., Barrangou R., Boyaval P., et al. (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. 10.1038/nature09523 [DOI] [PubMed] [Google Scholar]
- Guan J., Snowden J. D., Cahill J. L., Rasche E. S., Kuty Everett G. F. (2015). Complete genome sequence of Citrobacter freundii myophage Mordin. Genome Announc. 3:e01203-15. 10.1128/genomeA.01203-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarino A., Capano G., Malamisura B., Alessio M., Guandalini S., Rubino A. (1987). Production of Escherichia coli STa-like heat-stable enterotoxin by Citrobacter freundii isolated from humans. J. Clin. Microbiol. 25, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall T. A. (1999). BioEdit: a user-friendly biological sequence alignement editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98. [Google Scholar]
- Harhoff N. (1949). Studies on bacteria of the Ballerup group. Acta Pathol. Microbiol. Scand. 26, 167–174. 10.1111/j.1699-0463.1949.tb03152.x [DOI] [PubMed] [Google Scholar]
- Huang Y., Fan H., Pei G., Fan H., Zhang Z., An X., et al. (2012). Complete genome sequence of IME15, the first T7-like bacteriophage lytic to pan-antibiotic-resistant Stenotrophomonas maltophilia. J. Virol. 86, 13839–13840. 10.1128/JVI.02661-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang K., Luna A. J., Hernandez A. C., Kuty Everett G. F. (2015). Complete genome sequence of Citrobacter freundii myophage Miller. Genome Announc. 3:e01425-14. 10.1128/genomeA.01425-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inouye M., Arnheim N., Sternglanz R. (1973). Bacteriophage T7 lysozyme Is an N-Acetylmuramyl-l-alanine Amidase. J. Biol. Chem. 248, 7247–7252. [PubMed] [Google Scholar]
- Junier T., Zdobnov E. M. (2010). The Newick utilities: high-throughput phylogenetic tree processing in the Unix shell. Bioinformatics 26, 1669–1670. 10.1093/bioinformatics/btq243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajsík M., Oslanecová L., Szemes T., Hýblová M., Bilková A., Drahovská H., et al. (2014). Characterization and genome sequence of Dev2, a new T7-like bacteriophage infecting Cronobacter turicensis. Arch. Virol. 159, 3013–3019. 10.1007/s00705-014-2173-5 [DOI] [PubMed] [Google Scholar]
- Katoh K., Standley D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K., Kumar S., Takeo M., Mayilraj S. (2014). Genome sequencing, annotation of Citrobacter freundii strain GTC 09479. Genomics Data 2, 40–41. 10.1016/j.gdata.2013.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Kaur C., Kimura K., Takeo M., Raghava G. P. S., Mayilraj S. (2013). Draft genome sequence of the type species of the genus Citrobacter, Citrobacter freundii MTCC 1658. Genome Announc. 1:e00120-12. 10.1128/genomeA.00120-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Tamura K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H.-J., Cho S.-H., Kim T.-E., Won Y.-J., Jeong J., Park S. C., et al. (2008). Characterization of a T7-like lytic bacteriophage (φSG-JL2) of Salmonella enterica Serovar gallinarum biovar gallinarum. Appl. Environ. Microbiol. 74, 6970–6979. 10.1128/AEM.01088-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie S. J., Frois-Moniz K., Osburne M. S., Kelly L., Roggensack S. E., Sullivan M. B., et al. (2013). Genomes of marine cyanopodoviruses reveal multiple origins of diversity. Environ. Microbiol. 15, 1356–1376. 10.1111/1462-2920.12053 [DOI] [PubMed] [Google Scholar]
- Labrie S. J., Samson J. E., Moineau S. (2010). Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8, 317–327. 10.1038/nrmicro2315 [DOI] [PubMed] [Google Scholar]
- LeSage K. C., Hargrove E. C., Cahill J. L., Rasche E. S., Kuty Everett G. F. (2015). Complete genome sequence of Citrobacter freundii myophage Merlin. Genome Announc. 3:e01133-15. 10.1128/genomeA.01133-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Bork P. (2011). Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478. 10.1093/nar/gkr201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Richardson C. C. (1993). Gene 5.5 protein of bacteriophage T7 inhibits the nucleoid protein H-NS of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 90, 1761–1765. 10.1073/pnas.90.5.1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loc-Carrillo C., Abedon S. T. (2011). Pros and cons of phage therapy. Bacteriophage 1, 111–114. 10.4161/bact.1.2.14590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashin A. V., Borodovsky M. (1998). GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26, 1107–1115. 10.1093/nar/26.4.1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y., Pacan J. C., Wang Q., Xu Y., Huang X., Korenevsky A., et al. (2008). Microencapsulation of bacteriophage Felix O1 into chitosan-alginate microspheres for oral delivery. Appl. Environ. Microbiol. 74, 4799–4805. 10.1128/AEM.00246-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelman L. V., Notarnicola S. M., Richardson C. C. (1992). Roles of bacteriophage T7 gene 4 proteins in providing primase and helicase functions in vivo. Proc. Natl. Acad. Sci. U.S.A. 89, 10638–10642. 10.1073/pnas.89.22.10638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercanti D. J., Rousseau G. M., Capra M. L., Quiberoni A., Tremblay D. M., Labrie S. J., et al. (2015). Genomic diversity of phages infecting probiotic strains of Lactobacillus paracasei. Appl. Environ. Microbiol. 82, 95–105. 10.1128/AEM.02723-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt B. A., Studier F. W. (1987). T7 lysozyme inhibits transcription by T7 RNA polymerase. Cell 49, 221–227. 10.1016/0092-8674(87)90563-0 [DOI] [PubMed] [Google Scholar]
- Nguyen Q. T., Luna A. J., Hernandez A. C., Kuty Everett G. F. (2015). Complete genome sequence of Citrobacter freundii myophage Moogle. Genome Announc. 3:e01426-14. 10.1128/genomeA.01426-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen-Hughes T. A., Pavitt G. D., Santos D. S., Sidebotham J. M., Hulton C. S. J., Hinton J. C. D., et al. (1992). The chromatin-associated protein H-NS interacts with curved DNA to influence DNA topology and gene expression. Cell 71, 255–265. 10.1016/0092-8674(92)90354-F [DOI] [PubMed] [Google Scholar]
- Pajunen M. I., Kiljunen S. J., Söderholm M. E.-L., Skurnik M. (2001). Complete genomic sequence of the lytic bacteriophage φYeO3-12 of Yersinia enterocolitica serotype O:3. J. Bacteriol. 183, 1928–1937. 10.1128/JB.183.6.1928-1937.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombel I. T., Sykes K. F., Rayner S., Johnston S. A. (2002). ORF-FINDER: a vector for high-throughput gene identification. Gene 282, 33–41. 10.1016/S0378-1119(01)00819-8 [DOI] [PubMed] [Google Scholar]
- Rosenberg A. H., Patel S. S., Johnson K. A., Studier F. W. (1992). Cloning and expression of gene 4 of bacteriophage T7 and creation and analysis of T7 mutants lacking the 4A primase/helicase or the 4B helicase. J. Biol. Chem. 267, 15005–15012. [PubMed] [Google Scholar]
- Saitou N., Nei M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. [DOI] [PubMed] [Google Scholar]
- Sambrook J. F., Russel D. W. (2001). Molecular Cloning: A Laboratory Manual, Vols 1,2 and 3, 3rd Edn. New York, NY: Cold Spring Harbor Laboratory Press; soft cover | Sigma-Aldrich. [Google Scholar]
- Samonis G., Karageorgopoulos D. E., Kofteridis D. P., Matthaiou D. K., Sidiropoulou V., Maraki S., et al. (2008). Citrobacter infections in a general hospital: characteristics and outcomes. Eur. J. Clin. Microbiol. Infect. Dis. 28, 61–68. 10.1007/s10096-008-0598-z [DOI] [PubMed] [Google Scholar]
- Samson J. E., Magadán A. H., Sabri M., Moineau S. (2013). Revenge of the phages: defeating bacterial defences. Nat. Rev. Microbiol. 11, 675–687. 10.1038/nrmicro3096 [DOI] [PubMed] [Google Scholar]
- Scaltriti E., Polverini E., Grolli S., Eufemi E., Moineau S., Cambillau C., et al. (2013). The DNA binding mechanism of a SSB protein from Lactococcus lactis siphophage p2. Biochim. Biophys. Acta 1834, 1070–1076. 10.1016/j.bbapap.2013.02.014 [DOI] [PubMed] [Google Scholar]
- Scaltriti E., Tegoni M., Rivetti C., Launay H., Masson J.-Y., Magadan A. H., et al. (2009). Structure and function of phage p2 ORF34(p2), a new type of single-stranded DNA binding protein. Mol. Microbiol. 73, 1156–1170. 10.1111/j.1365-2958.2009.06844.x [DOI] [PubMed] [Google Scholar]
- Scholl D., Merril C. (2005). The genome of bacteriophage K1F, a T7-like phage that has acquired the ability to replicate on K1 strains of Escherichia coli. J. Bacteriol. 187, 8499–8503. 10.1128/JB.187.24.8499-8503.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks R. M. Q., Dashiff A., Alster J. S., Kadouri D. E. (2012). Isolation and identification of a bacteriocin with antibacterial and antibiofilm activity from Citrobacter freundii. Arch. Microbiol. 194, 575–587. 10.1007/s00203-012-0793-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. P., Aviles Medina C. A., Chen Y., Luna A. J., Hernandez A. C., Kuty Everett G. F. (2015). Complete genome of Citrobacter freundii siphophage Stevie. Genome Announc. 3:e01434-14. 10.1128/genomeA.01434-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira H. (2002). An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51, 492–508. 10.1080/10635150290069913 [DOI] [PubMed] [Google Scholar]
- Skurnik M., Strauch E. (2006). Phage therapy: facts and fiction. Int. J. Med. Microbiol. 296, 5–14. 10.1016/j.ijmm.2005.09.002 [DOI] [PubMed] [Google Scholar]
- Slopek S., Durlakowa I., Weber-Dabrowska M., Dabrowski M., Kucharewicz-Krukowska A. (1983). Results of bacteriophage treatment of suppurative bacterial infections. III. Detailed evaluation of the results obtained in further 150 cases. Arch. Immunol. Ther. Exp. (Warsz.) 32, 317–335. [PubMed] [Google Scholar]
- Smith H. W., Huggins M. B. (1982). Successful treatment of experimental Escherichia coli infections in mice using phage: its general superiority over antibiotics. J. Gen. Microbiol. 128, 307–318. 10.1099/00221287-128-2-307 [DOI] [PubMed] [Google Scholar]
- Staden R. (1996). The Staden sequence analysis package. Mol. Biotechnol. 5, 233–241. 10.1007/BF02900361 [DOI] [PubMed] [Google Scholar]
- Stephanou A. S., Roberts G. A., Tock M. R., Pritchard E. H., Turkington R., Nutley M., et al. (2009). A mutational analysis of DNA mimicry by ocr, the gene 0.3 antirestriction protein of bacteriophage T7. Biochem. Biophys. Res. Commun. 378, 129–132. 10.1016/j.bbrc.2008.11.014 [DOI] [PubMed] [Google Scholar]
- Studier F. W., Moffatt B. A. (1986). Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130. 10.1016/0022-2836(86)90385-2 [DOI] [PubMed] [Google Scholar]
- Studier F. W., Movva N. R. (1976). SAMase gene of bacteriophage T3 is responsible for overcoming host restriction. J. Virol. 19, 136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Nei M., Kumar S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U.S.A. 101, 11030–11035. 10.1073/pnas.0404206101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Studier F. W. (2004). Multiple roles of T7 RNA polymerase and T7 lysozyme during bacteriophage T7 infection. J. Mol. Biol. 340, 707–730. 10.1016/j.jmb.2004.05.006 [DOI] [PubMed] [Google Scholar]
- Zhao X., Huang S., Zhao J., He X., Li E., Li H., et al. (2015). Characterization of phiCFP-1, a virulent bacteriophage specific for Citrobacter freundii. J. Med. Virol. 88, 895–905. 10.1002/jmv.24401 [DOI] [PubMed] [Google Scholar]
- Zhu B., Lee S.-J., Tan M., Wang E.-D., Richardson C. C. (2012). Gene 5.5 protein of bacteriophage T7 in complex with Escherichia coli nucleoid protein H-NS and transfer RNA masks transfer RNA priming in T7 DNA replication. Proc. Natl. Acad. Sci. U.S.A. 109, 8050–8055. 10.1073/pnas.1205990109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Rao X., Tan Y., Xiong K., Hu Z., Chen Z., et al. (2010). Identification of lytic bacteriophage MmP1, assigned to a new member of T7-like phages infecting Morganella morganii. Genomics 96, 167–172. 10.1016/j.ygeno.2010.06.001 [DOI] [PubMed] [Google Scholar]