

Abstract
Before the maturation of rod and cone photoreceptors, the developing retina relies on light detection by intrinsically photosensitive retinal ganglion cells (ipRGCs) to drive early light-dependent behaviors. ipRGCs are output neurons of the retina; however, they also form functional microcircuits within the retina itself. Whether ipRGC microcircuits exist during development and whether they influence early light detection remain unknown. Here, we investigate the neural circuit that underlies the ipRGC-driven light response in developing mice. We use a combination of calcium imaging, tracer coupling, and electrophysiology experiments to show that ipRGCs form extensive gap junction networks that strongly contribute to the overall light response of the developing retina. Interestingly, we found that gap junction coupling was modulated by spontaneous retinal waves, such that acute blockade of waves dramatically increased the extent of coupling and hence increased the number of light-responsive neurons. Moreover, using an optical sensor, we found that this wave-dependent modulation of coupling is driven by dopamine that is phasically released by retinal waves. Our results demonstrate that ipRGCs form gap junction microcircuits during development that are modulated by retinal waves; these circuits determine the extent of the light response and thus potentially impact the processing of early visual information and light-dependent developmental functions.
SIGNIFICANCE STATEMENT Light-dependent functions in early development are mediated by intrinsically photosensitive retinal ganglion cells (ipRGCs). Here we show that ipRGCs form an extensive gap junction network with other retinal neurons, including other ipRGCs, which shapes the retina's overall light response. Blocking cholinergic retinal waves, which are the primary source of neural activity before maturation of photoreceptors, increased the extent of ipRGC gap junction networks, thus increasing the number of light-responsive cells. We determined that this modulation of ipRGC gap junction networks occurs via dopamine released by waves. These results demonstrate that retinal waves mediate dopaminergic modulation of gap junction networks to regulate pre-vision light responses.
Keywords: amacrine, CNiFER, electrical synapses, nicotinic acetylcholine receptor
Introduction
Across the developing nervous system, immature networks generate correlated spontaneous activity between neighboring groups of cells (for review, see Blankenship and Feller, 2010; Wenner, 2012; Wang and Bergles, 2015). This phenomenon has been well studied in the retina, where, before eye-opening, retinal waves are mediated by cholinergic signaling and propagate throughout the developing visual system (Ackman et al., 2012). Retinal waves are critical for establishing retinotopic and eye-specific maps in both the superior colliculus and lateral geniculate nucleus of the thalamus (for review, see Huberman et al., 2008; Kirkby et al., 2013; Ackman and Crair, 2014).
The effect of waves on the development of early neural circuits within the retina is less well understood (Kerschensteiner, 2013). They are known to influence dendritic growth and synapse formation (Bansal et al., 2000; Wong and Wong, 2001; Lohmann et al., 2002). Additionally, they have been implicated in network plasticity of the developing retina, whereby in the absence of cholinergic retinal waves, “recovered waves” mediated by gap junctions emerge (Stacy et al., 2005; Sun et al., 2008; Stafford et al., 2009; Kirkby and Feller, 2013). These recovered waves propagate more rapidly than cholinergic waves, cover a larger area, and are modulated by dopaminergic signaling. We previously suggested that the gap junction networks underlying recovered waves are suppressed by cholinergic signaling (Kirkby and Feller, 2013), which highlights the dynamic interaction between these two different wave-generating circuits.
Interestingly, the circuits mediating recovered waves (Kirkby and Feller, 2013), and those of cholinergic waves (Renna et al., 2011), strongly interact with intrinsically photosensitive retinal ganglion cells (ipRGCs). In both cases, light stimulation of ipRGCs increases retinal wave activity, suggesting that intraretinal microcircuits respond to ipRGC inputs. ipRGCs express the photopigment melanopsin and contribute to non–image-forming functions of vision, such as entrainment of circadian rhythms (Berson et al., 2002; Hattar et al., 2003; Rollag et al., 2003). Being the first photoreceptor to mature, ipRGCs provide the first visual input to retinal circuits and their brain targets (Fahrenkrug et al., 2004; Sekaran et al., 2005; Tu et al., 2005; Schmidt et al., 2008). ipRGCs are output neurons of the retina; however, there is growing evidence that they additionally form functional microcircuits within the retina (L.P. Müller et al., 2010; Zhang et al., 2008, 2012; Reifler et al., 2015). The contribution of intraretinal ipRGC microcircuits to early light responses during development remains unknown.
Here we explore the neural circuits underlying the ipRGC-driven light responses of the developing retina and the mechanisms by which retinal waves regulate these circuits. We use both anatomical and physiological methods to demonstrate that ipRGCs are extensively gap junction coupled to each other during development and that the extent of coupling increases in the absence of cholinergic waves. We show that this coupling is regulated by dopamine released during retinal waves. Moreover, we demonstrate that, even in the presence of cholinergic waves, ipRGC gap junction microcircuits propagate light-driven signals, thus strongly contributing to the overall light response of the developing retina.
Materials and Methods
Animals.
All experiments were performed on mice aged postnatal day P4-P7 of either sex from C57BL/6 WT (Harlan Laboratories) or Opn4-EGFP (P. Kofuji, Minnesota University, Minneapolis) (Schmidt et al., 2008) lines. Animal procedures were approved by the University of California, Berkeley Institutional Animal Care and Use Committees and conformed to the National Institutes of Health Guide for the care and use of laboratory animals, the Public Health Service Policy, and the Society for Neuroscience Policy on the Use of Animals in Neuroscience Research. Animals were anesthetized with isoflurane and decapitated, and the eyes were enucleated. Retinas were removed from eyecups in 95% O2–5% (v/v) CO2 bicarbonate buffered ACSF (in mm as follows): 119 NaCl, 26.2 NaHCO3, 11 glucose, 2.5 KCl, 1 K2HPO4, 2.5 CaCl2, 1.3 MgCl2).
Whole-mount retinal preparations.
Isolated retinas were mounted RGC-side up on filter paper. Retinas were dark adapted for at least 30 min at room temperature in oxygenated ACSF until transfer to the recording chamber, where they were continually superfused (1–2 ml/min) with oxygenated ACSF at 29°C–32°C.
Electrophysiology and neurobiotin (Nb) fills.
Retinas were visualized through a window cut in the filter paper with differential interference contrast optics on a Zeiss Axioskop 2 FS plus microscope under an ACHROPLAN 40× water-immersion objective. ipRGCs were identified by GFP signal in Opn4-EGFP mice under epifluorescent illumination, at 470/40 excitation filter and 525/50 emission filter. A hole was pierced in the inner limiting membrane of the retina using a glass recording pipette to access the RGC layer. RGCs were targeted under control of a micromanipulator (MP-225, Sutter Instruments). Recording pipettes were pulled with a tip resistance of 6–7 mΩ (for Nb) or 4–5 mΩ (for voltage clamp) and filled with internal solution (Nb fills, in mm as follows): 116 K+ d-gluconate, 6 KCl, 2 NaCl, 20 HEPES, 0.5 EGTA, 4 ATP-Na2, 0.3 GTP-Na3, 10 phosphocreatine-Na2, 0.05 Nb; voltage-clamp recordings, in mm as follows: 110 CsMeSO4, 2.8NaCl, 20 HEPES, 4 EGTA, 5 TEA-Cl, 4 ATP-Mg, 0.3 GTP-Na3, 10 phosphocreatine-Na2, 5 QX 314-Br; cell-attached recordings, in mm as follows: 150 NaCl, containing 0.02 mm Alexa-594. Data were acquired using pCLAMP 10.2 recording software and an Axopatch 200B amplifier (Molecular Devices), sampled at 10 kHz and filtered between 160 and 2000 Hz.
For tracer coupling experiments, Nb tracer (0.5%, SP-1120, Vector Laboratories) was added to internal solution. Cells were voltage-clamped, and pipettes were removed after a 5 min diffusion of Nb internal solution. Retinas were incubated for 25 min in the recording chamber after pipette removal. Cell morphology was assessed after pipette removal to confirm good cell health. Tissue was subsequently fixed and immunolabeled for Nb and the marker of interest (e.g., GFP; see Fig. 2), and imaged on a confocal scanning microscope (Zeiss LSM 780 NLO AxioExaminer, Molecular Imaging Center at University of California–Berkeley). The depth series of optical slices (1 μm between slices) was acquired using a Zeiss 20× water-immersion objective. Cell counts were performed by hand on each optical slice, and stacks were reconstructed offline using ImageJ maximum intensity projections for figure presentation.
Figure 2.
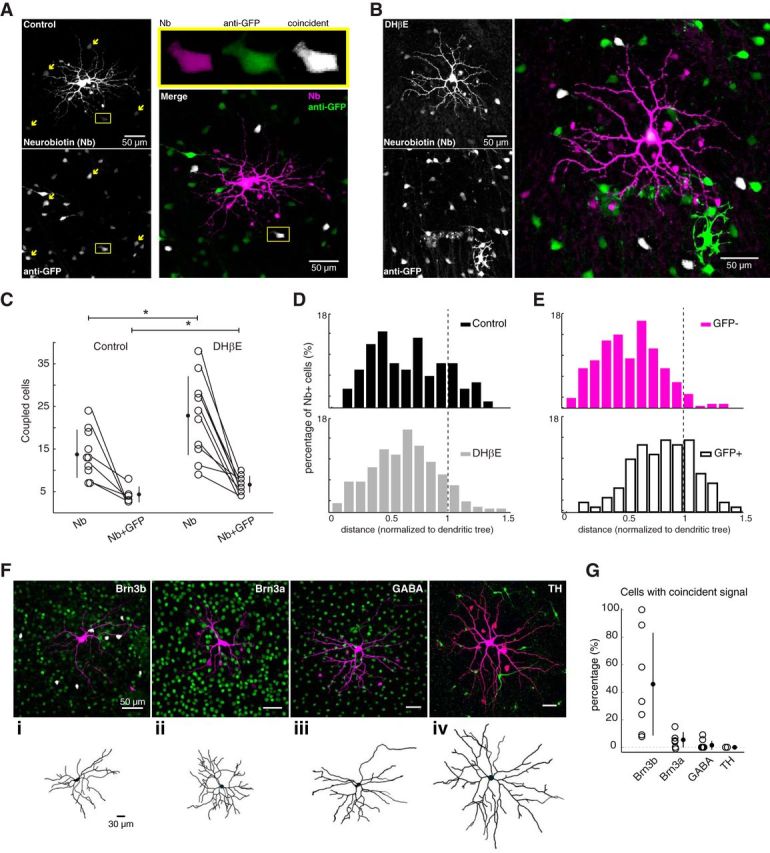
ipRGCs form intraretinal tracer-coupled networks. A, Fluorescent image of Nb-filled ipRGC targeted in Opn4-EGFP mouse, stained with a fluorescent streptavidin conjugate (left, top), and immunostained for GFP (left, bottom). Arrows indicate cells with coincident Nb and anti-GFP signals. Inset, Enlargement of one of the cells with coincident signals (yellow box) for the different color channels. Merged Nb (magenta) and anti-GFP (green) images (right) represent coincident Nb and anti-GFP signals (Nb + GFP), pseudocolored white for visualization. B, Fluorescent image of Nb-filled ipRGC targeted in Opn4-EGFP mouse after 60 min exposure to DHβE (8 μm). Details as in A. C, Total number of coupled cells (Nb) and coupled GFP+ cells (Nb + GFP) in control (ACSF) and after blocking retinal waves (DHβE, 60 min). Lines connect data points from the same Nb-filled ipRGC. Error bars indicate SD around the mean. *p < 0.05, two-tailed Student's t test (Nbcontrol vs NbDHβE, Nb+GFPcontrol vs NB+GFPDHβE). D, Histogram distribution of distances of all Nb+ cells to the soma of injected cell in control (top) and after 60 min exposure to DHβE (bottom). E, Histogram distribution of distances of all Nb+ cells to the soma of the injected cell that were either GFP− (top) or GFP+ (bottom). Coupled cells in the absence and presence of DHβE were included. F, Top, Fluorescent images of Nb-filled ipRGCs (magenta) colabeled (green) with antibodies against Brn3b (i), Brn3a (ii), GABA (iii), or TH (iv). White represents cells with coincident signals (Nb + marker). Bottom, Morphological reconstructions of cells (top) fixed to the same scale. Scale bar, 30 μm. G, Number of Nb + marker-labeled cells as a percentage of the total number of Nb-labeled cells, for the markers in C. Error bars indicate SD around the mean.
Whole-cell voltage-clamp recordings were obtained using glass microelectrodes of 4–5 mΩ (PC-10 pipette puller, Narishige). Holding voltage (Vh) for measuring photocurrents after correction of the liquid junction potential (−13 mV) was −60 mV. Spikelets and spikes were defined as events with amplitudes 2 SDs above the mean and with spacing >5 ms apart using a custom MATLAB protocol (MathWorks). Traces were analyzed 200 ms at a time to avoid artifacts from slow transient currents. Irradiance-response curves were performed in cell-attached mode. Light was delivered using a tungsten halogen lamp together with an optical filter at 480 ± 4 nm. Firing rates were measured in response to a 5 s pulse of full-field 480 nm illumination of increasing light intensity. Light intensity was adjusted using optical density (OD) filters. All firing rates were normalized to the maximal response at OD = 0 (no filter present), corresponding to an irradiance of ∼2.4 × 1014 photons s−1cm−2.
Alexa dye injections.
Retinas were visualized through a window cut in the filter paper with differential interference contrast optics, as described above. Injection pipettes were pulled with a tip resistance of 20–30 mΩ (Sutter Instruments) and back-filled with a 10 mm solution (in 200 mm KCl) of AlexaFluor-594 hydrazide. Cells were impaled with the pipette, and dye was injected with negative current of 0.1–0.9 nA, 200 ms long pulses at 2 Hz. Samples were fixed for immunoreactions 5 min after injection.
Immunoassays.
Whole-mount retinas were removed from recording chamber and transferred to a 4% PFA solution for 30 min at room temperature. Following fixation, retinas were washed in PBS for 20 min at room temperature and remounted onto a new piece of filter paper. They were incubated in blocking buffer (1.5% BSA, 0.2% Na-Azide, 0.2% Triton X-100) (3× 15 min) and then in primary immunoreaction solution. Concentrations of the different primary reactants in blocking buffer were 1:1000 goat (Abcam) or rabbit (Invitrogen) anti-GFP (24 h at 4°C), 1:2500 rabbit anti-melanopsin (48 h at 4°C, Advanced Targeting Systems) and 1:1000 rabbit anti-TH (24 h at 4°C, Abcam), 1:750 goat anti-Brn3a (Santa Cruz Biotechnology), 1:50 goat anti-Brn3b (Santa Cruz Biotechnology), 1:500 rabbit anti-GABA (Sigma-Aldrich). After primary reaction, retinas were washed in PBS (3× 15 min) and then incubated for 3 h at room temperature in 1:200 concentrations of secondary antibodies: donkey anti-rabbit or donkey anti-goat (Invitrogen). Nb was stained using a 1:800 streptavidin-594 (Invitrogen) solution in blocking buffer.
Calcium imaging and visual stimulation.
Retinas were bulk loaded with the calcium indicator Oregon Green BAPTA-1 AM (OGB-1) using the multicell bolus loading technique and epifluorescent imaging described previously (Blankenship et al., 2009). Excitation light was filtered with a 470/40 optical filter and yielded ∼3.4 × 1017 photons s−1 cm−2 to maximally stimulate the ipRGC intrinsic light response. Time series images of 30 or 40 s were acquired at 2 Hz with a 225 ms exposure time using a 40× water-immersion objective. Elliptical ROIs were manually drawn around cells displaying increases in fluorescence within the first 7 s after imaging onset. Cells were classified as light-responsive if they exhibited ΔF/F above threshold within 7 s of light onset in two consecutive trials spaced 10 min apart. Thresholds were determined for each experiment and ranged from ΔF/F = 1.2%–5%.
Multielectrode array (MEA) recordings.
Isolated pieces of retina were placed RGC-side down onto a 60-electrode commercial MEA that was arranged in an 8 × 8 grid, excluding the four corners, with 10 μm-diameter electrodes at a 100 μm interelectrode spacing (Multi Channel Systems). The retina was held in place using a dialysis membrane weighted with a ring of platinum wire. The recording chamber was superfused with oxygenated ACSF and maintained between 30°C and 34°C. Preparations were stimulated with unfiltered broad-band full-field light delivered by a tungsten halogen lamp with irradiance (in photons s−1 cm−2) of 2.4 × 1012 at 480 nm and 2.9 × 1013 at 600 nm. Raw data were filtered between 120 and 2000 Hz, and spikes were sorted offline to identify single units using Plexon Offline Sorter software. Spike-sorted data were analyzed in MATLAB. Units that showed an increase in firing rate following light onset were classified as light-responsive. Cross-correlograms (CCGs) of light-responsive units were calculated using MATLAB's cross-correlogram function. Cells were categorized as being coupled if their normalized CCG between ± 2.5 ms was 0.7 or lower than between 2.5 and 7.5 ms.
High performance liquid chromatography (HPLC).
Whole retinal eye cups were dissected from both eyes and flash frozen in liquid nitrogen. Liquid nitrogen vials were sent to the Vanderbilt Neurochemistry Core Laboratory for dopamine analysis using HPLC with electrochemical detection.
Fluorescence resonance energy transfer (FRET) imaging.
D2 cell-based neurotransmitter fluorescent engineered reporters (CNiFERs) were kindly provided by D. Kleinfeld and P. Slesinger (University of California–San Diego) (A. Müller et al., 2014). CNiFERs were maintained in a humidified incubator at 37°C with 5% (v/v) CO2 in growth media containing DMEM (containing 4.5 g/L glucose, l-glutamine and Na pyruvate; Invitrogen) supplemented with 10% heat-inactivated FBS (Invitrogen). Cells were trypsinized (0.05%), triturated, and seeded into new flasks at a density ratio of 1:5 upon confluence (approximately every 2–3 d).
Imaging of CNiFERs was based on methods using ACh-CNiFERS described previously (Ford et al., 2012). Before experiments, CNiFERs were removed from culture flasks using brief (30 s) application of trypsin (0.05%) and were concentrated in growth media. CNiFERs were deposited onto the inner limiting membrane by using a micropipette to transfer solution on top of a filter-mounted retinal piece, mounted ganglion cell side up, and then allowing them to settle onto the surface. Clusters of 2–3 cells were imaged at the focal plane ∼5–10 μm above the inner limiting membrane. We used 5 min imaging windows since CNiFER cells migrated out of the imaging field of view over time periods longer than this. We performed simultaneous patch-clamp recordings of RGCs ∼50–200 μm from the imaged CNiFERs. FRET images were acquired at 2 Hz using a 60× objective and an excitation wavelength of 435 nm. Individual FRET channel detection was accomplished using a Dual-View image splitter (Optical Insights) with appropriate yellow and cyan channel filters. Background fluorescence was subtracted from both channels. FRET ratios were computed as background-corrected YFP/CFP fluorescence averaged over a region of interest around a single CNiFER.
We quantified the time lag between each FRET transient and the closest wave by measuring the time from the peak of each FRET increase to the trough of the closest wave-associated EPSC (see Fig. 5). We compared these results to time-shuffled data to assess the likelihood of our observed temporal correlation occurring by chance. This involved first measuring the means and SDs of the wave and FRET rates from our dataset, then simulating recordings using the measured rates while assuming that the two events occurred independently of one another, and then calculating the time between each FRET transient and its closest wave.
Figure 5.

Retinal waves stimulate diffuse dopamine release. A, Left, Fluorescent image of dopaminergic amacrine cells labeled by anti-TH in a P4 whole-mount retina. Right, Concentrations of DA and its primary metabolite DOPAC in P4–P6 retinas detected using HPLC. Each data point indicates a different retina. B, Left, Schematic of CNiFER experimental setup: CNiFER cell deposited on surface of inner limiting membrane and simultaneous patch-clamp recording from nearby RGC (see Materials and Methods). TN-XXL, Calcium-dependent FRET sensor; D2R: Type 2 dopamine receptor. Right, Fluorescent and DIC images of CNiFERs on surface of whole-mount retina. C, Example traces of CNiFER imaging and simultaneous voltage-clamp recording from an RGC showing three consecutive FRET events: FRET ratio (black, YFP/CFP); current trace (gray) from nearby RGC. Large EPSCs in RGC trace are associated with retinal waves (arrows). D, Example traces of CNiFER imaging and simultaneous voltage-clamp recording from an RGC where retinal wave is not associated with a FRET event. Details as in C. E, Times of a transient FRET increase after the closest wave-associated EPSC for observed data (black) and for time-shuffled data (red; see Materials and Methods). F, Magnitude of a FRET response when subsequent wave is (as in C) versus is not (as in D) associated with a FRET event. When the FRET event is absent for a wave, the FRET event evoked by the previous wave is large in magnitude. *p < 0.05 (two-tailed Student's t test). G, Example FRET traces of CNiFER responses from four different cells in the same field of view to a 0.5 s puff of high-potassium solution (K-puff) applied to the inner nuclear layer to stimulate dopaminergic amacrine cells. Response to the second K-puff is not detected whether the first FRET increase is greater than around 10%. H, FRET increases of transient events recorded in ACSF (control) and DHβE. *p < 0.05 (two-tailed Student's t test). I, Dose–response curve of DA-CNiFERs deposited on the inner limiting membrane surface to nearby DA puffs of known concentration. Inset, Example responses. Gray region represents range of magnitudes of spontaneous FRET increases observed in our recordings. Error bars indicate SD around the mean.
Pharmacology.
Dihydro-β-erythroidine (DHβE, 8 μm), d-AP5 (50 μm), 6,7-dinitroquinoxaline-2,3-dione (DNQX, 20 μm), SR-95531 (gabazine, 5 μm), strychnine (4 μm), meclofenamic acid (MFA, 50 μm), and SCH23390 hydrochloride (SCH, 10 μm), raclopride (8 μm), TTX (1 mm) were added to perfusion media as stock solutions prepared in distilled water. QX 314 bromide (5 mm) was added to the internal solution. Antagonists were purchased from Tocris Bioscience. The synaptic mixture consisted of a mixture of gabazine, strychnine, d-AP5, DNQX, and, when specified, DHβE, at the above concentrations.
Results
Acutely blocking retinal waves increases the number of light-responsive cells
We first characterized the impact of acutely blocking waves on the overall light response of the developing retina. To characterize the ipRGC-mediated light response, we simultaneously stimulated melanopsin in ipRGCs and the calcium indicator OGB-1 with epifluorescent light, similar to previous studies (Sekaran et al., 2003, 2005; Bramley et al., 2011). This led to a transient increase in fluorescence in a subset of neurons in the ganglion cell layer (Fig. 1A,C) that corresponded to spiking activity (Fig. 1B). There are multiple subtypes of ipRGCs (M1-M5) distinguished by their light sensitivity, morphology, molecular identity, and projection targets in the brain (for review, see Schmidt et al., 2011). At least three of these subtypes emerge early in development (Tu et al., 2005; Schmidt et al., 2008; Sexton et al., 2015). Our intensity of imaging light (3.4 × 1017 photons s−1 cm−2 at 480 nm) should maximally activate all subtypes of ipRGCs at this age (Tu et al., 2005; Schmidt et al., 2008; Sexton et al., 2015). We then repeated these experiments after acutely blocking retinal waves with the nAChR antagonist DHβE (8 μm) for 60 min. Wave blockade produced a twofold increase in the number of cells that exhibited light-evoked calcium transients (Fig. 1C,D; DHβE/control = 2.11 ± 0.88, n = 12 retinas), agreeing with our previous work that describes the emergence of a light-sensitive network in the absence of cholinergic signaling (Kirkby and Feller, 2013). This increase in light responses was insensitive to combined blockade of GABAergic, glutamatergic, glycinergic, and cholinergic input, indicating that it was not due to a change in synaptic input from these neurotransmitters (Fig. 1D; n = 6).
Figure 1.

Blocking retinal waves increases the number of light-responsive neurons. A, Left, Fluorescent image of a retina loaded with OGB-1 (average of 30 sequential images, baseline). Middle/Right, Heat maps of maximal ΔF/F during the first 7 s of light stimulation in ACSF (control, middle) and after 60 min exposure to the nAChR antagonist DHβE (8 μm, right). Numbers indicate example light-responsive cells shown in C. B, Simultaneous calcium imaging and cell-attached recording from a light-responsive cell indicate that calcium imaging signal corresponds to an increase in firing rate. Top, Cell-attached recording shows cell's spiking response to light stimulation (indicated by blue bar). Middle, Firing rate of the same cell. Bottom, Fractional change of OGB-1 fluorescence (ΔF/F) of the cell. C, Time course of ΔF/F for the 10 cells of the ganglion cell layer indicated in A in response to light stimulation by the excitation light used for imaging. Traces of each of the 10 cells are shown in control (left) and after 60 min exposure to DHβE. D, Number of light-responsive cells after a manipulation divided by the number of light-responsive cells before the manipulation for three conditions: 60 min in ACSF (control/control); 60 min exposure to DHβE (DHβE/control); after DHβE application, 10 min exposure to a mixture containing DHβE (8 μm), glutamate receptor antagonists DNQX (20 μm), and AP5 (50 μm), GABA-A receptor antagonist gabazine (5 μm), and glycine receptor antagonist strychnine (4 μm) (synaptic block/control). Lines connect data points from the same retina. Dashed red line indicates the mean of control/control. Error bars indicate SD around the mean. *p < 0.05, compared with ratio of 1 (one-tailed Student's t test). E, Pseudo-colored images of light-responsive cells identified using calcium imaging, injected with the fluorescent molecule Alexa-594 (magenta), and stained with an anti-melanopsin antibody (green). Left, Example cell in control conditions where 12 of 13 light-responsive cells were positive for melanopsin. Right, Example cell that gained light response after DHβE exposure where 1 of 5 cells were positive for melanopsin.
In a subset of experiments, we targeted light-responsive neurons for intracellular dye injections and subsequently immunostained them for melanopsin (Fig. 1E). In control conditions, the majority of neurons that exhibited light-evoked calcium transients were positive for melanopsin immunoreactivity (n = 12 of 13). In contrast, after cholinergic blockade, the majority of neurons that had gained a light response tested negative for melanopsin immunoreactivity (n = 1 of 5) indicating that they themselves did not have a robust intrinsic light response but rather had inherited it through input from nearby ipRGCs. This high incidence of melanopsin expression among light-responsive cells in control conditions is in contrast with previous reports suggesting that, at P4-P5, only 56% of light-responsive cells are ipRGCs because 44% lose their light sensitivity in the presence of gap junction blockers (Sekaran et al., 2005). Our observed higher incidence of melanopsin-expressing ipRGCs could be attributed to two factors; first, our targeted injections were biased toward cells with larger somas; therefore, it might favor a particular subtype of ipRGCs with abundant melanopsin expression. Second, a subset of ipRGCs might require gap junction input to generate a robust light response, thus losing their light response in the presence of gap junction blockers. This possibility is explored below.
Increase in the number of light-responsive cells occurs via increased gap junction coupling
We hypothesized that the increased number of light-responsive cells after wave blockade is due to modulation of ipRGC gap junction coupling. In adult retina, ipRGCs are tracer-coupled (Müller et al., 2010) and electrically coupled via gap junctions (Reifler et al., 2015) to wide-field spiking GABAergic amacrine cells. Additionally, during development, calcium imaging experiments suggest that there is gap junction coupling between ipRGCs and other neurons (Sekaran et al., 2005).
Thus, we tested whether gap junction coupling underlies the increased number of light-responsive neurons in the absence of retinal waves using several approaches. We first performed tracer-coupling experiments. We filled GFP-expressing ipRGCs in the Opn4-EGFP mouse, which labels M1-M3 ipRGC subtypes (Schmidt et al., 2008), with the gap junction permeable tracer Nb using a patch pipette (6–7 mΩ tip resistance). After a 60 min blockade of retinal waves with DHβE, we found that the number of neurons tracer-coupled to ipRGCs increased significantly (Fig. 2A,B; mean ± SD; control: tracer-coupled cells = 13.90 ± 5.65, n = 10 injected neurons; DHβE: tracer-coupled cells = 22.82 ± 9.26, n = 11). Additionally, we discovered that a subset of the coupled neurons expressed GFP (Fig. 2A), and their number increased slightly but significantly after a 60 min blockade of retinal waves (Fig. 2B; control: GFP+ tracer-coupled cells = 4.33 ± 1.86, n = 6 cells; DHβE: GFP+ tracer-coupled cells = 6.73 ± 1.95, n = 11). Hence, our findings indicate that ipRGCs are coupled to other ipRGCs and to cells that do not express detectable GFP, which suggests that ipRGCs might propagate their light responses to neurons with low or nonexistent melanopsin expression.
We characterized the spatial arrangement of GFP+ and GFP− coupled cells both before and after 60 min blockade of retinal waves. Although the overall distribution of coupled cells did not change with wave blockade (Fig. 2D), we found that the distribution of soma locations of GFP− tracer-coupled cells was skewed toward inside of the dendritic field, whereas the distribution of GFP+ tracer-coupled cells was skewed toward outside the dendritic field (Fig. 2E; control and DHβE data grouped together).
We further explored the cell types comprising the gap junction networks of developing ipRGCs by conducting a series of co-labeling experiments in which the tracer-coupled cells were tested for molecular markers of retinal cell types. We found that Nb colocalized with the ganglion cell marker Brn3b, which predominantly labels the M2-M5 ipRGC subtypes, a subset of M1 ipRGCs, as well as a variety of other RGCs (Chen et al., 2011; Jain et al., 2012), but rarely colocalized with Brn3a, which does not label ipRGCs (Jain et al., 2012) (Fig. 2Fi,Fii,G). The presence of Brn3b and not Brn3a in the tracer-coupled cells indicates that ipRGCs are preferentially coupled to other ipRGCs and avoid conventional RGCs. We found that tracer-coupled cells rarely colocalized with GABA (Fig. 2Fiii,G), in contrast to the tracer-coupling pattern described for adult ipRGCs (Müller et al., 2010). Additionally, tracer-coupled cells did not colocalize with tyrosine hydroxylase (TH), which labels dopaminergic amacrine cells, a putative postsynaptic target of ipRGCs (Zhang et al., 2008, 2012) (Fig. 2Fiv,G). It is important to note that we did not observe correlations between ipRGC morphologies and coupling patterns; hence, we did not distinguish between ipRGC subtypes in our analyses (Fig. 2F, bottom). Our tracer-coupling observations show that ipRGCs are extensively tracer-coupled to other cells, including other ipRGCs, and that this coupling increases when cholinergic waves are blocked.
We next investigated whether there is functional coupling of ipRGCs to other retinal neurons via gap junctions. For our first test, we determined whether the depolarization of a single ipRGC (identified by its light-evoked calcium transient; see Fig. 1) propagates to the postjunctional retinal neurons. To isolate electrical signaling, we performed these experiments in a mixture of synaptic blockers (see Materials and Methods). Indeed, we found that short depolarizing steps in ipRGCs evoked calcium transients in nearby, although not adjacent, cells, indicating gap junction coupling (Singer et al., 2001) (Fig. 3A,B). A 60 min blockade of retinal waves with DHβE induced a significant increase in the number of postjunctional neurons (Fig. 3C; control: number of cells with evoked calcium transient = 2.12 ± 1.13 n = 8; DHβE: number of cells with evoked calcium transient = 4.0 ± 1.22, n = 5). Under both experimental conditions, a subset of these postjunctional neurons exhibited light-evoked calcium transients. (Fig. 3D; control: 5 of 17 cells; DHβE: 12 of 20 cells), which indicates that coupling occurs between both ipRGCs and non-ipRGCs and is consistent with tracer-coupling results (Figure 2). The amplitudes of depolarization-evoked calcium transients were similar in control and DHβE (Fig. 3E; ΔF/Fcontrol = 3.5 ± 2.3%; ΔF/FDHβE = 2.6 ± 1.2%), suggesting that the increase in coupled cells observed after blocking waves is dominated by formation of new connections or dramatic strengthening of pre-existing gap junction synapses.
Figure 3.

ipRGCs form functional intraretinal gap junction networks. A, Left, DIC image represents recording electrode targeting an ipRGC identified by light stimulation as in Figure 1. Right, Fluorescent image of same retina loaded with calcium indicator OGB-1. Cells indicated with blue and red dots correspond to traces shown in B. Recordings were conducted in DHβE, DNQX, AP5, gabazine, and strychnine to isolate electrical synapses. Top, Voltage-clamp protocol for simultaneous imaging and whole-cell voltage clamp: 500 ms voltage steps from −60 to 0 mV were injected into the recorded ipRGC (indicated by *). Bottom, Traces from cells indicated in A that either displayed responses (blue) or failed to display responses (red) when the voltage-clamped ipRGC (*) was depolarized. Blue bar indicates excitation light for calcium imaging. C, Number of cells in which both voltage steps evoked calcium transients for separated control and DHβE (60 min) groups. *p < 0.05 (two-tailed Student's t test). D, Percentage of cells with voltage-clamp-evoked responses that also showed light-evoked calcium transients for control and DHβE. E, Amplitudes (ΔF/F) of the calcium transients evoked by depolarizing steps for separated control and DHβE groups. Error bars indicate SD around the mean.
For our second test of physiological coupling, we used MEA recordings to determine whether light-evoked action potentials propagate via gap junctions. Similar to our calcium imaging results, we observed an increase in the number of light-responsive cells (seen as individual units after spike sorting) following DHβE application (DHβE/control = 1.39 ± 0.26, control/control = 1.10 ± 0.07, n = 8 retinas, data not shown; Fig. 4A, example raster plot). The new light-responsive cells displayed lower peak firing rates than the original light-responsive cells but similar latency-to-peak (Fig. 4B,C). Because ipRGC subtypes are classified in part by latency (Tu et al., 2005; Sexton et al., 2015), our observations suggest that these new light-responsive cells do not correspond to a distinct subtype of ipRGC, but rather are cells receiving input via gap junctions in the absence of an intrinsically driven component. After DHβE application, the original light-responsive cells displayed no significant changes in light-evoked firing rates, latency-to-peak or irradiance-response curves, suggesting that DHβE does not alter the intrinsic light response properties of these ipRGCs (Fig. 4C,D). To further confirm that the light-responsive cells are gap junction coupled, we computed CCGs of light-responsive units and found that several pairs displayed the characteristic double peak structure of direct electrical coupling (Brivanlou et al., 1998; DeVries, 1999), including cells that had gained a light response in DHβE (Fig. 4E; 220 of 816 possible pairs of light-responsive units, n = 6 retinas). Furthermore, we observed direct coupling in CCGs of some pairs in control conditions, albeit with a broader distribution that is likely indicative of common input (Fig. 4E; 54 pairs total, n = 6 retinas).
Figure 4.
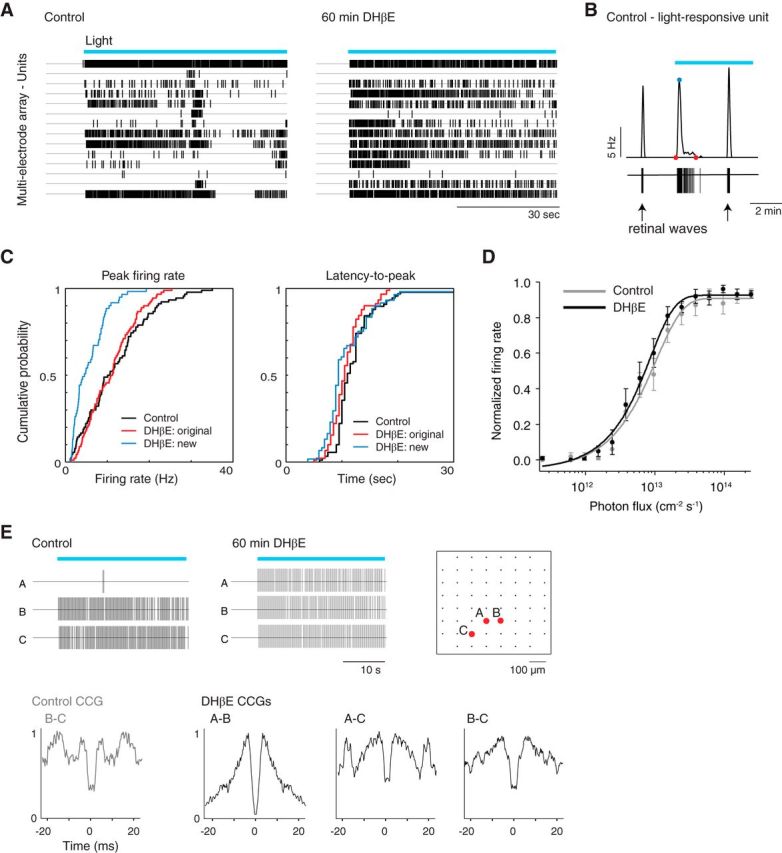
Spiking activity of ipRGCs indicates gap junction coupling before and after blocking retinal waves. A, Raster plots represent spike times of units isolated using MEA recordings following light stimulation in ACSF (control, left) and DHβE (right). B, Example of single-unit spike train (bottom) and instantaneous firing rate (top) during light stimulation in control. Red points indicate beginning and end of light-evoked activity. Blue point indicates peak firing rate. The two other depicted bursts of activity (one before and one during light stimulation) correspond to retinal waves. C, Cumulative probability distributions of the light response parameters of peak firing rate and latency-to-peak firing rate before (control, black) and after cholinergic block (DHβE, original (red) and new (blue) light-responsive cells). D, Irradiance-response curves of GFP-expressing ipRGCs in Opn4-EGFP mouse line in ACSF (control, gray) and DHβE (black). Error bars indicate SD around the mean. E, Top, Raster plots represent spike times of sample light-responsive units (their locations on the MEA are indicated in diagram on right) in ACSF (control, left) and DHβE (middle). Bottom, CCGs for pairs of the units shown in raster plot for control and DHβE. Blue bars represent 480 nm light stimulation.
Together, our findings demonstrate that, during development, ipRGCs are gap junction coupled to a variety of neurons, including other ipRGCs, and that the extent of this coupling increases in the absence of cholinergic wave-related signaling.
Cholinergic retinal waves regulate ipRGC coupling via dopamine release
How does blocking cholinergic waves increase the coupling of ipRGCs? In adult retina, the regulation of gap junction networks is mediated primarily by the neuromodulator dopamine (Witkovsky et al., 2004; Bloomfield and Völgyi, 2009), which is released from dopaminergic amacrine cells (DACs) upon ambient illumination (Zhang et al., 2007) and reaches ipRGCs, activating their Type 1 dopamine receptors (D1R) (Van Hook et al., 2012). Because this dopamine release is driven by photoreceptor activation (Zhang et al., 2007), it is unclear whether dopamine is released early in development before the maturation of photoreceptors. Our previous study suggests that this is indeed the case because we found that gap junction networks in the developing retina are modulated by dopaminergic signaling (Kirkby and Feller, 2013). Thus, we tested the hypothesis that retinal waves drive dopamine release, which in turn modulates ipRGC gap junction coupling during development.
First, we confirmed the presence of DACs (Yoshida et al., 2011) by immunostaining for TH between postnatal ages P4-P6 (Fig. 5A, left). Second, we showed that dopamine is produced and metabolized during these ages using HPLC with electrochemical detection (Fig. 5A, right). Third, we directly tested whether cholinergic waves correlate with the diffuse release of dopamine by using a cell-based dopamine sensor (CNiFER) technique (A. Müller et al., 2014) previously used for ACh-sensing (Nguyen et al., 2010; Ford and Feller, 2012) (Fig. 5B). We found that the sensor displayed transient FRET increases, indicating a diffuse release of dopamine (Fig. 5C,D). Simultaneous voltage-clamp recordings from nearby RGCs revealed that these FRET increases lagged wave-induced currents by 20–30 s (Fig. 5C,E). This lag is due to a variety of factors. First, the sensor itself has relatively slow responses. Direct application of dopamine to the sensor leads to time-to-peak of the FRET increase of ∼5–10 s, as previously reported (A. Müller et al., 2014) (Fig. 5I, inset). Second, the density of dopaminergic amacrine cells, which reside in the inner nuclear layer, is very low, ∼100 μm−2 (Fig. 5A), which corresponds to 1–2 cells within our imaging field. Hence, the dopamine likely needs to diffuse a long distance before reaching the sensor that resides on the surface of the inner-limiting membrane. In comparison, we used the ACh-CNiFER in the same location and in the presence of a high density of ACh-releasing interneurons, many of which reside in the ganglion cell layer, and we also saw long delays of ∼10–15 s (Ford et al., 2012).
We evaluated the likelihood of the observed 20–30 s correlation between a retinal wave and a FRET transient occurring and found that it was significantly higher than that expected by chance (see Materials and Methods; Fig. 5E), suggesting that retinal waves induce the FRET transients. However, not all waves were followed by a FRET transient (Fig. 5D); in these cases, the prior wave was linked to a large FRET increase (Fig. 5F). We could reproduce this phenomenon using consecutive puffs of high-potassium solution (K-puff) to the IPL (Fig. 5G), indicating that it might be a limitation in the response dynamics of the CNiFER and not a lack of wave-evoked dopamine release. To determine the impact of retinal waves on dopamine release, we blocked cholinergic waves with DHβE and found that there was a dramatic decrease in the frequency of FRET transients (control: 0.64 ± 0.15 transients/min; DHβE: 0.12 ± 0.07 transients/min, n = 10, p < 0.05) and their amplitude (Fig. 5H), corresponding to approximately a 10-fold decrease in dopamine concentration per transient (Fig. 5I). Thus, we conclude that cholinergic waves induce diffuse release of dopamine and that blocking them reduces this release.
To test whether dopamine signaling influences ipRGC gap junction coupling, we blocked D1Rs with the antagonist SCH 23390 (SCH, 10 μm). Blocking D1Rs, which has no effect on cholinergic retinal waves (Kirkby and Feller, 2013), induced a fivefold increase in the number of light-responsive neurons (Fig. 6; SCH/control = 5.53 ± 2.68, n = 8 retinas). This increase was greater than that induced by blocking cholinergic waves (Fig. 1), likely due to the residual dopamine that is still present during wave blockade (Fig. 5H). In contrast, blocking D2-type dopamine receptors (raclopride, 8 μm) or ionotropic glutamatergic receptors (DNQX, 20 μm), which are possible pathways for dopaminergic modulation (discussed below), did not change the number of light-responsive neurons (Fig. 6B; raclopride/control = 0.87 ± 0.23, n = 4, DNQX/control = 0.99 ± 0.23, n = 6), indicating that alternate dopaminergic pathways and intraretinal glutamatergic circuits of ipRGCs do not play a significant role in wave-driven dopaminergic modulation of ipRGC gap junctions. Our result of increased coupling in D1R antagonist agrees with several other studies showing that D1R antagonists increase coupling in retinal circuits via changes in gap junction phosphorylation (Bloomfield and Völgyi, 2009; Kothmann et al., 2009).
Figure 6.

Reduced dopamine signaling increases the number of light-responsive neurons. A, Left, Fluorescent image of a retina loaded with OGB-1 (average of 30 sequential images). Middle/Right, Heat maps of maximal ΔF/F during the first 7 s of light stimulation in control conditions (middle) and after 30 min bath application of the D1R antagonist SCH23390 (SCH, 10 μm, right). B, Number of light-responsive cells after a manipulation divided by the number of light-responsive cells before the manipulation for eight conditions: 60 min in ACSF (control/control); 60 min exposure to DHβE (DHβE/control); 30 min exposure to SCH (SCH/control) and 20 min washout of SCH (wash/control); 30 min exposure to D2R antagonist raclopride (8 μm, raclopride/control) and 20 min washout of raclopride (wash/control); 60 min exposure to DNQX (20 μm, DNQX/control) and 20 min washout of DNQX (wash/control). Lines connect data points from the same retina. Dashed red line indicates the mean of control/control. Error bars indicate SD around the mean. *p < 0.05, compared with ratio of 1 (one-tailed Student's t test). **p < 0.01, compared with ratio of 1 (one-tailed Student's t test).
Together, these data describe a putative mechanism by which waves regulate the extent of ipRGC gap junction networks via dopamine release that modulates gap junction coupling.
Gap junction coupling of ipRGCs contributes to the overall light response of the developing retina
Our tracer coupling and physiology experiments (Figs. 2, 3) indicate that ipRGC gap junction coupling is robust in the presence of cholinergic waves, which corresponds to a condition of high dopamine release. This leads to the question: does gap junction coupling of ipRGCs influence the light response in normal conditions, when wave-driven dopamine release diminishes gap junction connectivity? To address this, we investigated how gap junction coupling contributes to the light response of ipRGCs.
We conducted whole-cell voltage-clamp recordings of ipRGCs in response to blue light (λ = 450–490 nm, intensity = 3.4 × 1017 photons s−1 cm−2), which maximally stimulates all ipRGC subtypes (Berson et al., 2002; Do and Yau, 2010). Superimposed on the slow photocurrents that are characteristic of ipRGCs (Do and Yau, 2010), we detected small, inward, transient currents resembling spikelets (Fig. 7A,B; n = 10 cells). Spikelets are transient depolarizations that originate in prejunctional neurons and travel through gap junctions and via the dendrites to the soma of the postjunctional cell. They have been demonstrated to represent a physiological trademark of gap junction coupling between several different classes of neurons (Valiante et al., 1995; Landisman and Connors, 2005; Pereda, 2014), including the retina (Trenholm et al., 2013a, b). ipRGC spikelets were blocked by the sodium channel blocker TTX (1 μm) and exhibited shorter interspike intervals than light-evoked action potentials, suggesting that they originate from the spiking of multiple prejunctional cells (Fig. 7A–C; n = 7). It is important to note that the interspike intervals of action potentials during light responses are too long to be accounted for by action potential refractory periods, so refractory period was not used to compare action potentials and spikelets. Therefore, further study will be required to directly test whether spikelets emerge from multiple ipRGCs. Importantly, spikelets were recorded using intracellular solutions that contain the sodium channel blocker QX 314 (5 mm), indicating that they are not generated by spikes in the voltage-clamped neuron but rather in the prejunctional ipRGCs.
Figure 7.

Gap junction networks contribute to ipRGC photocurrents. A, B, Whole-cell voltage-clamp recording of ipRGC during 3 s blue light illumination in ACSF (control, black) and 1 μm TTX (red). B, Left, Shaded region is expanded. Right, Fifteen sample spikelets are displayed. C, Distribution of interspike intervals for spikelets during the 10 s following light onset (black) and of interspike intervals for spikes recorded in cell-attached configuration (gray). D, Top, Diagram of setup illustrates that wide-field illumination stimulates multiple gap junction-coupled ipRGCs in the field of view. Bottom, Example traces of ipRGC photocurrents in response to 3 s blue light illumination in control and in the gap junction blocker MFA (50 μm, 30 min). E, Peak photocurrent amplitude for control and MFA groups. Error bars indicate SD around the mean. *p < 0.05 (two-tailed Student's t test). Gray circles were considered a different cell population and thus were not included in the analysis.
Because gap junctions act as low pass filters, we might expect that action potentials originating in prejunctional neurons contribute only a small depolarization to a postjunctional neuron (Trenholm et al., 2013a). However, the slow photocurrents associated with activation of melanopsin should be less filtered as they pass through gap junctions. Thus, we tested how gap junction coupling contributes to ipRGC light responses by blocking gap junctions to effectively eliminate inputs from prejunctional ipRGCs during light stimulation. We compared photocurrents in the absence and presence of the gap junction blocker meclofenamic acid (MFA; 50 μm; Fig. 7D), and found that photocurrents recorded in MFA exhibited significantly smaller amplitudes than those recorded in control (ACSF: peak amplitude = 49.56 ± 16.29 pA, n = 11 cells; MFA: peak amplitude = 18.96 ± 9.06 pA, n = 12 cells; Fig. 7D,E). Compared with control, MFA did not cause a significant difference in the input resistance of ipRGCs, as previously described for this age (Schmidt et al., 2008). There still remains the possibility that MFA is impacting unclamped conductances that are contributing to photocurrents; however, given that some cells exhibited control-like photocurrent amplitudes in MFA whereas others exhibited a significant decrease (refer to Fig. 7E), we conclude that the primary effect of MFA was to block gap junction coupling. These results indicate that ipRGC photocurrents are readily transmitted through gap junctions. Therefore, the light response of an ipRGC integrates an intrinsic photocurrent with an extrinsic component from prejunctional ipRGCs.
To characterize the impact of ipRGC gap junction networks on the overall light response of the developing retina, we used calcium imaging to quantify the number of light-responsive cells after blocking gap junctions. Application of MFA led to a marked reduction in the number of light-responsive cells (Fig. 8A–C; control/control ratio = 1.06 ± 0.17, n = 11 retinas; MFA/control = 0.25 ± 0.14, n = 9). Importantly, several cells that maintained their light response did not exhibit a decreased response amplitude (Fig. 8D; n = 5 retinas, 101 cells); thus, the loss of light-evoked calcium transients cannot be explained by an off-target effect of MFA on the intrinsic light response or on voltage-gated calcium channels that underlie the calcium transient (Vessey et al., 2004; Bramley et al., 2011). These data cannot be directly compared with amplitude of photocurrents described above (Fig. 7) because there are not within-cell comparisons of the effect of MFA on photocurrent amplitudes. Indeed, there were many ipRGCs that exhibited strong photocurrents in the presence of MFA (Fig. 7E). Together, these data indicate that during development ipRGC gap junction networks provides a significant contribution to both the single-cell and the whole-retina light response.
Figure 8.

Gap junction networks shape the light response of the developing retina. A, Left, Fluorescent image of a retina loaded with OGB-1 (average of 30 sequential images). Middle/Right, Heat maps of maximal ΔF/F during the first 7 s of light stimulation in control conditions (middle) and after 30 min bath application of the gap junction blocker MFA (50 μm, right). Numbers indicate example cells shown in B. B, Time course of ΔF/F for the 10 cells of the ganglion cell layer indicated in A in response to light stimulation. Left traces, Control ACSF. Right traces, After 30 min of MFA exposure (50 μm). C, Number of light-responsive cells after a manipulation divided by the number of light-responsive cells before the manipulation for three conditions: 60 min in ACSF (control/control); 30 min exposure to MFA (MFA/control); and 20 min washout of MFA (wash/control). Lines connect data points from the same retina. Dashed red line indicates the mean of control/control. Error bars indicate SD around the mean. **p < 0.01, compared with ratio of 1 (one-sample Student's t test). D, Amplitude of light-evoked calcium transients for individual cells in control conditions and after application of MFA for 30 min (5 retinas, 101 cells). Black lines indicate cells that exhibited little or no change in their response amplitude.
Discussion
In this study, we demonstrate that cholinergic retinal waves regulate the early light response of the developing retina by modulating ipRGC gap junction networks. First, we show that acutely blocking cholinergic waves produces a marked increase in the number of light-responsive cells due to an increase in ipRGC gap junction coupling. Second, we provide evidence for a putative mechanism in which waves drive phasic release of dopamine that in turn regulates the extent of ipRGC gap junction coupling. Third, we demonstrate that ipRGC gap junction networks are active in the presence of cholinergic waves and contribute to the photocurrents of individual cells and to the overall light response of the developing retina. These findings directly demonstrate that ipRGCs connect to other neurons within the retina during development and that they do this via gap junctions rather than chemical synapses, consistent with a previous study (Sekaran et al., 2005). Furthermore, they provide insight into mechanisms of activity-dependent modulation of gap junction networks.
ipRGC gap junction networks are regulated by cholinergic waves
Previous studies have demonstrated that, in the absence of cholinergic waves, an alternative wave-generating circuit that depends on gap junctions emerges (Stacy et al., 2005; Sun et al., 2008; Anishchenko and Feller, 2009; Stafford et al., 2009; Kirkby and Feller, 2013). Here we have provided mechanistic insight into this network plasticity. First, we have demonstrated that ipRGCs are tracer coupled and electrically coupled to other ipRGCs and to non-ipRGCs (Figs. 2–4). Such an intraretinal microcircuit offers a pathway by which a projection neuron can feed back and affect the retinal network. This type of circuit has been shown in adult retina, with a growing body of literature indicating that ipRGCs not only signal to downstream brain targets but also exert widespread intraretinal influence (Müller et al., 2010; Zhang et al., 2008, 2012; Joo et al., 2013; Reifler et al., 2015). Second, we have shown that blocking cholinergic waves during development increases the extent of both tracer and electrical coupling from ipRGCs to other retinal neurons, implying that waves suppress ipRGC gap junction networks. Hence, the regulation of gap junction networks by cholinergic retinal waves determines the extent and reach of ipRGC-dependent light responses within the retina.
Wave-evoked dopamine release modulates ipRGC gap junctions
We previously hypothesized that high dopamine signaling may function to suppress gap junction networks during cholinergic waves (Kirkby and Feller, 2013). Here we show that dopamine is indeed released during waves, thus providing mechanistic insight into this network plasticity (Fig. 5). Similar to the effect of blocking waves, blocking dopaminergic signaling through D1Rs, but not D2Rs, also increases the number of light-responsive-cells (Fig. 6). These results suggest that D1R signaling suppresses ipRGC gap junction coupling. Thus, we propose a putative mechanism where spontaneous cholinergic waves evoke dopamine release that reduces ipRGC gap junction coupling.
Although dopamine is released episodically, we speculate that it provides a tonic modulation of gap junctions. This is consistent with our observation that it takes 60 min of wave blockade for gap junction coupling to increase. Although D1 receptors are low affinity and therefore only respond to high concentrations of dopamine (Dreyer et al., 2010), the resulting second messenger cascade and resulting phosphorylation of connexins are likely to integrate this signal. Studies conducted in heterologous expression systems indicate that PKA modulation of gap junctions occurs on the time scale of several minutes (Wang et al., 2015) (i.e., several waves).
The model emerging from the present study is that retinal waves suppress the spread of light-evoked intraretinal signals from ipRGCs by activating dopaminergic amacrine cells to stimulate release of dopamine, which, acting via D1 receptors, reduces the gap junctional coupling of ipRGCs. This finding is consistent with a previous circuit model proposed for the “recovered waves” observed in a knock-out mouse lacking the β2-subunit of nAChRs (Kirkby and Feller, 2013, their Fig. 6). Recovered waves are mediated by gap junction coupling; their frequency is increased by light stimulation, increased by D1 receptor antagonists and reduced by D2 receptor antagonists. In the proposed circuit model for recovered waves, there is an increase in ipRGC coupling to a yet to be identified neuron (pictured as an amacrine cell, Kirkby and Feller, 2013, their Fig. 6), which in turn increases coupling to other RGCs in a manner dependent on the balance of opposing effects of D1 and D2 receptors (Kothmann et al., 2009). In the presence of waves, there is high dopamine release. This favors activation of D1 receptors, which are of low affinity and more sensitive to phasic changes in DA levels, thus suppressing gap junction conductance. In the absence of waves, there is low dopamine. This favors activation of D2 receptors, which are of high affinity and more sensitive to tonic DA levels, thus increasing gap junction conductance.
The data presented here are consistent with this model. First, in response to wave blockade, we observed an increase in coupling to small-soma cells in the ganglion cell layer, which are likely to be an amacrine cell type, in a manner dependent on activation of D1 receptors. However, we did not observe a sensitivity of coupling to D2 receptor antagonists. Furthermore, we did not see the occurrence of recovered waves. Hence, prolonged blockade of retinal waves, such as that in the β2-nAChR knock-out mouse might be necessary for the upregulation of the D2R in this intervening amacrine cell to mediate the recovered waves.
How waves stimulate dopamine release remains to be elucidated. Two possibilities are that DACs are directly depolarized by nAChR activation, or that they are indirectly activated via ipRGCs that form glutamatergic synapses with DACs (Zhang et al., 2008, 2012). Our findings are inconsistent with the latter because blocking glutamatergic transmission did not increase the number of light-responsive cells (Fig. 6B), thus indicating that the extent of ipRGC gap junction coupling is not modulated by glutamatergic-dependent release of dopamine. Elucidating the mechanisms that mediate the interplay between chemical and electrical neural networks will require future studies that explore how signaling pathways activated by D1Rs produce changes in gap junctional conductance (O'Brien, 2014; Pereda, 2014).
Gap junction networks shape the light response of the retina in the presence of wave-evoked dopamine release
This study and previous studies have indicated that gap junctions are suppressed during retinal waves (Stacy et al., 2005; Akrouh and Kerschensteiner, 2013). However, we find that, even in the presence of waves, ipRGC gap junction networks continue to shape the light response of the retina. Our results indicate that ipRGCs in the developing retina form a syncytium that ensures the depolarization of one ipRGC will contribute to the depolarization of neighboring ipRGCs (Fig. 3). These findings are consistent with previous studies of developing ipRGCs where it was demonstrated that gap junction blockers decreased cell capacitance (Schmidt et al., 2008) and the number of light-responsive cells in the adult (Sekaran et al., 2003) and during development (Sekaran et al., 2005). Indeed, previous studies estimated that at P4-P5 only 56% of light-responsive cells were ipRGCs since the rest lost their light response in the presence of the gap junction blocker carbenoxolone (CBX) (Sekaran et al., 2005). Subsequent studies demonstrated that CBX has off-target effects that blocks light-evoked [Ca2+]i rise in isolated ipRGCs (Bramley et al., 2011), although the concentrations of carbenoxolone used in the developing retina (10 μm) appeared to show weaker off-target effects. Furthermore, multielectrode array recordings of ipRGC activity in the first postnatal week indicated that 100 μm CBX did not decrease the correlated firing between ipRGCs, indicating that either the coupling was not directly between ipRGCs or that CBX was not impacting functional coupling (Tu et al., 2005). Here we found that the gap junction blocker MFA does not affect the amplitude of light-evoked calcium transients in a subset of ipRGCs, indicating that MFA might not interfere with ipRGC calcium influx (Fig. 8).
Because gap junctions act as a low pass filter, the contribution of light-evoked currents from neighboring ipRGCs is likely dominated by the slow depolarization evoked by photoactivation of conductances rather than the small fast depolarizations induced by spikelets (Fig. 7). Indeed, blocking gap junction networks significantly decreases both the photocurrent amplitudes of ipRGCs and the overall number of light-sensitive cells in the retina (Figs. 7, 8). This scenario sharply contrasts with the function of coupling recently described for direction-selective ganglion cells (Trenholm et al., 2013a, b). For those cells, gap junction coupling combines with local synaptic input to generate correlated dendritic spikes that contribute to direction coding (Trenholm et al., 2014). However, for ipRGCs, coupling of photocurrents leads to more efficient detection and propagation of light information (Figs. 7, 8), and thus might hold implications for pre-vision light-dependent developmental functions, such as the development of retinal vasculature (Rao et al., 2013), and of light avoidance behaviors that are thought to contribute to pup survival (Johnson et al., 2010; Delwig et al., 2012).
In conclusion, our results show that, during development, ipRGCs form extensive gap junction microcircuits that shape the early retinal light response. Retinal waves exert a far-reaching, neuromodulatory influence on these circuits via dopaminergic modulation of gap junctions, thus potentially impacting the processing of early visual input. It is likely that this type of wave-dependent, dopaminergic modulation also impacts the development and fine-tuning of other gap junction networks in the immature retina.
Footnotes
This work was supported by the National Institutes of Health Grants R01EY013528 and P30EY003176 to M.B.F. and Grant F31EY024842 to D.A.A., and National Science Foundation Graduate Research Fellowship Program to L.A.K. and D.A.A. We thank all members of the M.B.F. laboratory for comments on the manuscript; Andrew Huberman and Rana El-Danaf (University of California–San Diego) for assistance with the dye injection experiments; David Kleinfeld and Paul Slesinger (University of California–San Diego) for providing CNiFERs; and Raymond Johnson (Vanderbilt Neurochemistry Core) for performing HPLC dopamine analysis.
The authors declare no competing financial interests.
References
- Ackman JB, Crair MC. Role of emergent neural activity in visual map development. Curr Opin Neurobiol. 2014;24:166–175. doi: 10.1016/j.conb.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackman JB, Burbridge TJ, Crair MC. Retinal waves coordinate patterned activity throughout the developing visual system. Nature. 2012;490:219–225. doi: 10.1038/nature11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akrouh A, Kerschensteiner D. Intersecting circuits generate precisely patterned retinal waves. Neuron. 2013;79:322–334. doi: 10.1016/j.neuron.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anishchenko A, Feller MB. Go with the flow, but only in one direction. Neuron. 2009;64:152–154. doi: 10.1016/j.neuron.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Bansal A, Singer JH, Hwang BJ, Xu W, Beaudet A, Feller MB. Mice lacking specific nicotinic acetylcholine receptor subunits exhibit dramatically altered spontaneous activity patterns and reveal a limited role for retinal waves in forming ON and OFF circuits in the inner retina. J Neurosci. 2000;20:7672–7681. doi: 10.1523/JNEUROSCI.20-20-07672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295:1070–1073. doi: 10.1126/science.1067262. [DOI] [PubMed] [Google Scholar]
- Blankenship AG, Feller MB. Mechanisms underlying spontaneous patterned activity in developing neural circuits. Nat Rev Neurosci. 2010;11:18–29. doi: 10.1038/nrn2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship AG, Ford KJ, Johnson J, Seal RP, Edwards RH, Copenhagen DR, Feller MB. Synaptic and extrasynaptic factors governing glutamatergic retinal waves. Neuron. 2009;62:230–241. doi: 10.1016/j.neuron.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield SA, Völgyi B. The diverse functional roles and regulation of neuronal gap junctions in the retina. Nat Rev Neurosci. 2009;10:495–506. doi: 10.1038/nrn2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramley JR, Wiles EM, Sollars PJ, Pickard GE. Carbenoxolone blocks the light-evoked rise in intracellular calcium in isolated melanopsin ganglion cell photoreceptors. PLoS One. 2011;6:e22721. doi: 10.1371/journal.pone.0022721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brivanlou IH, Warland DK, Meister M. Mechanisms of concerted firing among retinal ganglion cells. Neuron. 1998;20:527–539. doi: 10.1016/S0896-6273(00)80992-7. [DOI] [PubMed] [Google Scholar]
- Chen SK, Badea TC, Hattar S. Photoentrainment and pupillary light reflex are mediated by distinct populations of ipRGCs. Nature. 2011;476:92–95. doi: 10.1038/nature10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delwig A, Logan AM, Copenhagen DR, Ahn AH. Light evokes melanopsin-dependent vocalization and neural activation associated with aversive experience in neonatal mice. PLoS One. 2012;7:e43787. doi: 10.1371/journal.pone.0043787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries SH. Correlated firing in rabbit retinal ganglion cells. J Neurophysiol. 1999;81:908–920. doi: 10.1152/jn.1999.81.2.908. [DOI] [PubMed] [Google Scholar]
- Do MT, Yau KW. Intrinsically photosensitive retinal ganglion cell. Physiol Rev. 2010;90:1547–1581. doi: 10.1152/physrev.00013.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer JK, Herrik KF, Berg RW, Hounsgaard JD. Influence of phasic and tonic dopamine release on receptor activation. J Neurosci. 2010;30:14273–14283. doi: 10.1523/JNEUROSCI.1894-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrenkrug J, Nielsen HS, Hannibal J. Expression of melanopsin during development of the rat retina. Neuroreport. 2004;15:781–784. doi: 10.1097/00001756-200404090-00008. [DOI] [PubMed] [Google Scholar]
- Ford KJ, Feller MB. Assembly and disassembly of a retinal cholinergic network. Vis Neurosci. 2012;29:61–71. doi: 10.1017/S0952523811000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford KJ, Félix AL, Feller MB. Cellular mechanisms underlying spatiotemporal features of cholinergic retinal waves. J Neurosci. 2012;32:850–863. doi: 10.1523/JNEUROSCI.5309-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattar S, Lucas RJ, Mrosovsky N, Thompson S, Douglas RH, Hankins MW, Lem J, Biel M, Hofmann F, Foster RG, Yau KW. Melanopsin and rod-cone photoreceptive systems account for all major accessory visual functions in mice. Nature. 2003;424:76–81. doi: 10.1038/nature01761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberman AD, Feller MB, Chapman B. Mechanisms underlying development of visual maps and receptive fields. Annu Rev Neurosci. 2008;31:479–509. doi: 10.1146/annurev.neuro.31.060407.125533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain V, Ravindran E, Dhingra NK. Differential expression of Brn3 transcription factors in intrinsically photosensitive retinal ganglion cells in mouse. J Comp Neurol. 2012;520:742–755. doi: 10.1002/cne.22765. [DOI] [PubMed] [Google Scholar]
- Johnson J, Wu V, Donovan M, Majumdar S, Rentería RC, Porco T, Van Gelder RN, Copenhagen DR. Melanopsin-dependent light avoidance in neonatal mice. Proc Natl Acad Sci U S A. 2010;107:17374–17378. doi: 10.1073/pnas.1008533107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo HR, Peterson BB, Dacey DM, Hattar S, Chen SK. Recurrent axon collaterals of intrinsically photosensitive retinal ganglion cells. Vis Neurosci. 2013;30:175–182. doi: 10.1017/S0952523813000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner D. Spontaneous network activity and synaptic development. Neuroscientist. 2013;20:272–290. doi: 10.1177/1073858413510044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby LA, Feller MB. Intrinsically photosensitive ganglion cells contribute to plasticity in retinal wave circuits. Proc Natl Acad Sci U S A. 2013;110:12090–12095. doi: 10.1073/pnas.1222150110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby LA, Sack GS, Firl A, Feller MB. A role for correlated spontaneous activity in the assembly of neural circuits. Neuron. 2013;80:1129–1144. doi: 10.1016/j.neuron.2013.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothmann WW, Massey SC, O'Brien J. Dopamine-stimulated dephosphorylation of connexin 36 mediates AII amacrine cell uncoupling. J Neurosci. 2009;29:14903–14911. doi: 10.1523/JNEUROSCI.3436-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landisman CE, Connors BW. Long-term modulation of electrical synapses in the mammalian thalamus. Science. 2005;310:1809–1813. doi: 10.1126/science.1114655. [DOI] [PubMed] [Google Scholar]
- Lohmann C, Myhr KL, Wong RO. Transmitter-evoked local calcium release stabilizes developing dendrites. Nature. 2002;418:177–181. doi: 10.1038/nature00850. [DOI] [PubMed] [Google Scholar]
- Müller A, Joseph V, Slesinger PA, Kleinfeld D. Cell-based reporters reveal in vivo dynamics of dopamine and norepinephrine release in murine cortex. Nat Methods. 2014;11:1245–1252. doi: 10.1038/nmeth.3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller LP, Do MT, Yau KW, He S, Baldridge WH. Tracer coupling of intrinsically photosensitive retinal ganglion cells to amacrine cells in the mouse retina. J Comp Neurol. 2010;518:4813–4824. doi: 10.1002/cne.22490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen QT, Schroeder LF, Mank M, Müller A, Taylor P, Griesbeck O, Kleinfeld D. An in vivo biosensor for neurotransmitter release and in situ receptor activity. Nat Neurosci. 2010;13:127–132. doi: 10.1038/nn.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien J. The ever-changing electrical synapse. Curr Opin Neurobiol. 2014;29C:64–72. doi: 10.1016/j.conb.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereda AE. Electrical synapses and their functional interactions with chemical synapses. Nat Rev Neurosci. 2014;15:250–263. doi: 10.1038/nrn3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Chun C, Fan J, Kofron JM, Yang MB, Hegde RS, Ferrara N, Copenhagen DR, Lang RA. A direct and melanopsin-dependent fetal light response regulates mouse eye development. Nature. 2013;494:243–246. doi: 10.1038/nature11823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifler AN, Chervenak AP, Dolikian ME, Benenati BA, Li BY, Wachter RD, Lynch AM, Demertzis ZD, Meyers BS, Abufarha FS, Jaeckel ER, Flannery MP, Wong KY. All spiking, sustained ON displaced amacrine cells receive gap-junction input from melanopsin ganglion cells. Curr Biol. 2015;25:2763–2773. doi: 10.1016/j.cub.2015.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renna JM, Weng S, Berson DM. Light acts through melanopsin to alter retinal waves and segregation of retinogeniculate afferents. Nat Neurosci. 2011;14:827–829. doi: 10.1038/nn.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollag MD, Berson DM, Provencio I. Melanopsin, ganglion-cell photoreceptors, and mammalian photoentrainment. J Biol Rhythms. 2003;18:227–234. doi: 10.1177/0748730403018003005. [DOI] [PubMed] [Google Scholar]
- Schmidt TM, Taniguchi K, Kofuji P. Intrinsic and extrinsic light responses in melanopsin-expressing ganglion cells during mouse development. J Neurophysiol. 2008;100:371–384. doi: 10.1152/jn.00062.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TM, Chen SK, Hattar S. Intrinsically photosensitive retinal ganglion cells: many subtypes, diverse functions. Trends Neurosci. 2011;34:572–580. doi: 10.1016/j.tins.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekaran S, Foster RG, Lucas RJ, Hankins MW. Calcium imaging reveals a network of intrinsically light-sensitive inner-retinal neurons. Curr Biol. 2003;13:1290–1298. doi: 10.1016/S0960-9822(03)00510-4. [DOI] [PubMed] [Google Scholar]
- Sekaran S, Lupi D, Jones SL, Sheely CJ, Hattar S, Yau KW, Lucas RJ, Foster RG, Hankins MW. Melanopsin-dependent photoreception provides earliest light detection in the mammalian retina. Curr Biol. 2005;15:1099–1107. doi: 10.1016/j.cub.2005.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton TJ, Bleckert A, Turner MH, Van Gelder RN. Type I intrinsically photosensitive retinal ganglion cells of early post-natal development correspond to the M4 subtype. Neural Dev. 2015;10:17. doi: 10.1186/s13064-015-0042-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer JH, Mirotznik RR, Feller MB. Potentiation of l-type calcium channels reveals nonsynaptic mechanisms that correlate spontaneous activity in the developing mammalian retina. J Neurosci. 2001;21:8514–8522. doi: 10.1523/JNEUROSCI.21-21-08514.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacy RC, Demas J, Burgess RW, Sanes JR, Wong RO. Disruption and recovery of patterned retinal activity in the absence of acetylcholine. J Neurosci. 2005;25:9347–9357. doi: 10.1523/JNEUROSCI.1800-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford BK, Sher A, Litke AM, Feldheim DA. Spatial-temporal patterns of retinal waves underlying activity-dependent refinement of retinofugal projections. Neuron. 2009;64:200–212. doi: 10.1016/j.neuron.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Warland DK, Ballesteros JM, van der List D, Chalupa LM. Retinal waves in mice lacking the β2 subunit of the nicotinic acetylcholine receptor. Proc Natl Acad Sci U S A. 2008;105:13638–13643. doi: 10.1073/pnas.0807178105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenholm S, McLaughlin AJ, Schwab DJ, Awatramani GB. Dynamic tuning of electrical and chemical synaptic transmission in a network of motion coding retinal neurons. J Neurosci. 2013a;33:14927–14938. doi: 10.1523/JNEUROSCI.0808-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenholm S, Schwab DJ, Balasubramanian V, Awatramani GB. Lag normalization in an electrically coupled neural network. Nat Neurosci. 2013b;16:154–156. doi: 10.1038/nn.3308. [DOI] [PubMed] [Google Scholar]
- Trenholm S, McLaughlin AJ, Schwab DJ, Turner MH, Smith RG, Rieke F, Awatramani GB. Nonlinear dendritic integration of electrical and chemical synaptic inputs drives fine-scale correlations. Nat Neurosci. 2014;17:1759–1766. doi: 10.1038/nn.3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu DC, Zhang D, Demas J, Slutsky EB, Provencio I, Holy TE, Van Gelder RN. Physiologic diversity and development of intrinsically photosensitive retinal ganglion cells. Neuron. 2005;48:987–999. doi: 10.1016/j.neuron.2005.09.031. [DOI] [PubMed] [Google Scholar]
- Valiante TA, Perez Velazquez JL, Jahromi SS, Carlen PL. Coupling potentials in CA1 neurons during calcium-free-induced field burst activity. J Neurosci. 1995;15:6946–6956. doi: 10.1523/JNEUROSCI.15-10-06946.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hook MJ, Wong KY, Berson DM. Dopaminergic modulation of ganglion-cell photoreceptors in rat. Eur J Neurosci. 2012;35:507–518. doi: 10.1111/j.1460-9568.2011.07975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessey JP, Lalonde MR, Mizan HA, Welch NC, Kelly ME, Barnes S. Carbenoxolone inhibition of voltage-gated Ca channels and synaptic transmission in the retina. J Neurophysiol. 2004;92:1252–1256. doi: 10.1152/jn.00148.2004. [DOI] [PubMed] [Google Scholar]
- Wang HC, Bergles DE. Spontaneous activity in the developing auditory system. Cell Tissue Res. 2015;361:65–75. doi: 10.1007/s00441-014-2007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Lin YP, Mitchell CK, Ram S, O'Brien J. Two-color fluorescent analysis of connexin 36 turnover: relationship to functional plasticity. J Cell Sci. 2015;128:3888–3897. doi: 10.1242/jcs.162586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenner P. Motor development: activity matters after all. Curr Biol. 2012;22:R47–R48. doi: 10.1016/j.cub.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Witkovsky P, Veisenberger E, Haycock JW, Akopian A, Garcia-Espana A, Meller E. Activity-dependent phosphorylation of tyrosine hydroxylase in dopaminergic neurons of the rat retina. J Neurosci. 2004;24:4242–4249. doi: 10.1523/JNEUROSCI.5436-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WT, Wong RO. Changing specificity of neurotransmitter regulation of rapid dendritic remodeling during synaptogenesis. Nat Neurosci. 2001;4:351–352. doi: 10.1038/85987. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Feng L, Grimbert F, Rangarajan KV, Buggele W, Copenhagen DR, Cang J, Liu X. Overexpression of neurotrophin-3 stimulates a second wave of dopaminergic amacrine cell genesis after birth in the mouse retina. J Neurosci. 2011;31:12663–12673. doi: 10.1523/JNEUROSCI.1100-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DQ, Zhou TR, McMahon DG. Functional heterogeneity of retinal dopaminergic neurons underlying their multiple roles in vision. J Neurosci. 2007;27:692–699. doi: 10.1523/JNEUROSCI.4478-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DQ, Wong KY, Sollars PJ, Berson DM, Pickard GE, McMahon DG. Intraretinal signaling by ganglion cell photoreceptors to dopaminergic amacrine neurons. Proc Natl Acad Sci U S A. 2008;105:14181–14186. doi: 10.1073/pnas.0803893105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DQ, Belenky MA, Sollars PJ, Pickard GE, McMahon DG. Melanopsin mediates retrograde visual signaling in the retina. PLoS One. 2012;7:e42647. doi: 10.1371/journal.pone.0042647. [DOI] [PMC free article] [PubMed] [Google Scholar]