

Abstract
Rhodopsin is a prototypical G-protein-coupled receptor (GPCR) that is activated when its 11-cis-retinal moiety is photoisomerized to all-trans retinal. This step initiates a cascade of reactions by which rods signal changes in light intensity. Like other GPCRs, rhodopsin is deactivated through receptor phosphorylation and arrestin binding. Full recovery of receptor sensitivity is then achieved when rhodopsin is regenerated through a series of steps that return the receptor to its ground state. Here, we show that dephosphorylation of the opsin moiety of rhodopsin is an extremely slow but requisite step in the restoration of the visual pigment to its ground state. We make use of a novel observation: isolated mouse retinae kept in standard media for routine physiologic recordings display blunted dephosphorylation of rhodopsin. Isoelectric focusing followed by Western blot analysis of bleached isolated retinae showed little dephosphorylation of rhodopsin for up to 4 h in darkness, even under conditions when rhodopsin was completely regenerated. Microspectrophotometeric determinations of rhodopsin spectra show that regenerated phospho-rhodopsin has the same molecular photosensitivity as unphosphorylated rhodopsin and that flash responses measured by trans-retinal electroretinogram or single-cell suction electrode recording displayed dark-adapted kinetics. Single quantal responses displayed normal dark-adapted kinetics, but rods were only half as sensitive as those containing exclusively unphosphorylated rhodopsin. We propose a model in which light-exposed retinae contain a mixed population of phosphorylated and unphosphorylated rhodopsin. Moreover, complete dark adaptation can only occur when all rhodopsin has been dephosphorylated, a process that requires >3 h in complete darkness.
SIGNIFICANCE STATEMENT G-protein-coupled receptors (GPCRs) constitute the largest superfamily of proteins that compose ∼4% of the mammalian genome whose members share a common membrane topology. Signaling by GPCRs regulate a wide variety of physiological processes, including taste, smell, hearing, vision, and cardiovascular, endocrine, and reproductive homeostasis. An important feature of GPCR signaling is its timely termination. This normally occurs when, after their activation, GPCRs are rapidly phosphorylated by specific receptor kinases and subsequently bound by cognate arrestins. Recovery of receptor sensitivity to the ground state then requires dephosphorylation of the receptor and unbinding of arrestin, processes that are poorly understood. Here we investigate in mouse rod photoreceptors the relationship between rhodopsin dephosphorylation and recovery of visual sensitivity.
Keywords: dark adaptation, GPCR, photoreceptor, phototransduction, retina, rhodopsin
Introduction
A key feature of G-protein-coupled receptor (GPCR) biology shared by most GPCRs is their phosphorylation by one of a family of receptor kinases and their subsequent binding to arrestin (Carman and Benovic, 1998; Lefkowitz, 2004). These two sequential steps result in rapid quenching of receptor activity. This feature allows GPCRs to detect and transmit rapid changes in the environment. This is an important advantage for rod and cone photoreceptors that must detect fleeting images of varying intensity that rapidly traverse the retina. In rod photoreceptors, G-protein receptor kinase-1 (GRK1) sequentially phosphorylates a cluster of six to seven serine/threonine residues located near the C terminus of rhodopsin (Kühn, 1974; Chen et al., 1995; Hurley et al., 1998; Mendez et al., 2000). This is followed by binding of arrestin-1 (Arr1), resulting in complete termination of receptor activation (Wilden et al., 1986). The number of phosphorylation reactions and arrestin binding provide independent steps that tightly control the active lifetime of rhodopsin, which is manifested in highly reproducible single quantal responses (SQRs; Doan et al., 2006; Azevedo et al., 2015). This feature is important in rods, because SQRs represent a large proportion of the functional range of the rod.
Complete regeneration of rhodopsin to the dark-adapted state after exposure to light requires that rhodopsin dephosphorylates and that Arr1 decouples from the receptor. This is particularly important after the exposure of the eye to bright light in which substantial phosphorylation persists on six phosphorylatable residues on mouse and human rhodopsin (Kennedy et al., 2001). The rhodopsin dephosphorylation reaction appears to be slow in vivo (Kennedy et al., 2001; Lee et al., 2010). Moreover, regenerated phospho-rhodopsin has been measured to exhibit lower catalytic activity in in vitro assays (Miller et al., 1986; Wilden et al., 1986; Bennett and Sitaramayya, 1988; Wilden, 1995). However, these results reflect the summed activity from a pool of rhodopsin molecules on transduction gain but provide no information regarding the physiological consequences of the presence of these phosphorylated rhodopsin pigments, the nature of the light responses that these pigments generate, and the effect they have for the recovery of sensitivity in the intact retina.
Almost 40 years ago, Miller et al. (1977) measured the rate of opsin dephosphorylation in frog rod photoreceptors after bright light exposure. However, no electrophysiological measurements were made at that time that would allow correlation with the state of adaptation. The most convincing studies to date that support a link between dark adaptation and the state of rhodopsin phosphorylation came from in vivo experiments on wild-type (WT) mice (Kennedy et al., 2001; Lee et al., 2010). These studies correlated the rate of rhodopsin dephosphorylation with the rate of dark adaptation but fell short of demonstrating a causal link between these processes. Moreover, they demonstrated that a significant amount of rhodopsin is phosphorylated during prolonged light exposure.
The primary focus of our work here is to demonstrate the relationship between rhodopsin dephosphorylation and dark adaptation in mouse retina. Inhibition of rhodopsin dephosphorylation by genetic means in mice is problematic because the rhodopsin phosphatase gene has not been definitively identified (Ramulu et al., 2001). In this study, we have identified a methodology by which rhodopsin dephosphorylation is reduced to undetectable levels. We use this methodology to test the hypotheses that rhodopsin dephosphorylation is prerequisite to the recovery of sensitivity during dark adaptation. We find that regenerated phosphorylated rhodopsin can be photoactivated to initiate phototransduction but with reduced efficiency compared with the unphosphorylated form. Furthermore, we demonstrate that, when up to 90% of rhodopsin remains phosphorylated after a bleach, subsequent dark adaptation results in smaller quantal responses whose kinetics are indistinguishable from those elicited in rods containing the unphosphorylated form. Our findings, together with those of previous in vivo (Kennedy et al., 2001; Lee et al., 2010) and in vitro (Miller et al., 1977) studies, document that opsin dephosphorylation is responsible for a novel very long-lasting form of rod dark adaptation that affects vision at threshold.
Materials and Methods
Preparation.
All procedures were performed according to protocols approved by the Animal Care and Use Committees of Boston University School of Medicine and the University of Southern California and in accordance with the standards set forth in the Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act. Three strains of mice, WT, cone photoreceptor function loss-3 (cpfl3−/−), and rhodopsin kinase knock-out (Grk1−/−) were used in coordinated electrophysiological, biochemical, and spectrophotometric studies. All strains were on a C57BL/6J background. WT and cpfl3−/− mice were obtained from The Jackson Laboratory. The cpfl3−/− strain is one in which the α subunit of the cone transducin gene guanine nucleotide-binding protein, α-transducing activity polypeptide 2 (GNAT2) harbors a missense mutation that prevents activation of cone phototransduction, while otherwise preserving rod function and retinal morphology (Chang et al., 2006). This allows electrophysiological measurements of rod photoresponses from whole retina in the absence of a cone component. Mutant cpfl3−/− mice are hereafter referred to by their conventional designation as GNAT2−/− mice. Grk1−/− mice were obtained from the laboratory of Dr. Jason Chen (Baylor College of Medicine, Houston, TX). WT mice were females and 6–10 weeks of age at the time of experimentation; GNAT2−/− and Grk1−/− mice were of either sex and 3–12 weeks of age at the time of experiment. Mice of all strains were maintained on a 12 h light/dark cycle and were dark adapted for 12 h before all experiments.
At the beginning of each experiment, mice were killed in dim red light by cervical dislocation, followed by decapitation. After the animals were killed, eyes were removed and placed in a 3 cm plastic Petri dish containing physiological solution. From this point, all dissection and manipulations of the tissue were performed under infrared illumination with the aid of an infrared-sensitive video system. The eyes were hemisected, and the retinae were removed and dissected free of the retinal pigment epithelium (RPE). Retinae were then transferred to a light-tight container containing buffered (1.9 g/L NaHCO3, bubbled with 95% O2, 5% CO2) Ames medium (Sigma).
Bleaching and incubation of isolated retinae.
A number of experimental procedures required bleaching of a substantial fraction of the rhodopsin, followed by incubation in darkness for variable periods of time. In such cases, retinae were bleached using an optical source that consisted of an adjustable intensity tungsten/halogen filament, a 500 nm interference filter, and multiple neutral density filters. The light output provided a spatially uniform circular spot 5 mm in diameter that was focused on the retina. The bleached fraction, F, was calculated from the following relation:
where t is the duration of the exposure to light, I is the bleaching light intensity (5.5 × 105 photons μm−2 s−1), and P is the photosensitivity of mouse rhodopsin (5.7 × 10−9 μm2) measured in situ at the wavelength of peak absorbance (Woodruff et al., 2004). Retinae were bleached at a uniform rate of 0.3% per second. The plot illustrated in Figure 1 shows the relation between the fraction of rhodopsin bleached, as measured microspectrophotometrically, and predicted according to Equation 1. The fit of the straight line to the data suggests good correspondence between calculated and measured amounts of bleaching over the range of 0–95%. After bleaching, retinae were incubated for variable periods in darkness at 37°C in Ames culture medium (Ames and Nesbett, 1981) to which 1.9 g/L NaHCO3 and 3.33 g/L (0.3%) fatty-acid free bovine serum albumin (BSA; Sigma) had been added. The solution was equilibrated with 95% O2 and 5% CO2 at pH 7.4. To facilitate rhodopsin regeneration, 10 μm 11-cis-retinal was also added to the solution (unless otherwise stated).
Figure 1.

Comparison of fractional rhodopsin bleaching estimated by calculation and bleaching measured by MSP (n = 3). Fractional bleaching was calculated according to Equation 1. Measurements of the extent of bleaching were made by measuring MSP spectra of a few tens of rod outer segments projecting from the edge of a flat-mounted retina. Measurements were made before and after exposure to a calibrated bleaching light. The amount of rhodopsin bleached by each light exposure was then calculated as the difference in absorbance at 500 nm from spectra measured before and after bleach exposure, as described in Materials and Methods. Error bars are ±SEM. The solid line drawn at a slope of 1 indicates perfect correspondence of calculated to measured values.
Incubation in darkness took place in a custom-fabricated light-tight Delrin enclosure maintained at 37°C in a water bath. This enclosure was designed to accommodate a 3 ml glass embryo dish, in which the retina was bathed in buffered Ames solution. Moist gas (95% O2/5% CO2) was blown across the top of the culture dish in the enclosure to maintain pH at 7.4 and O2 in the solution at desired levels, unless otherwise noted.
Determination of rhodopsin phosphorylation by isoelectric focusing.
We measured the extent of phosphorylation on the six Ser/Thr sites on the C terminus of rhodopsin after different periods of incubation in darkness in retinae that had been exposed to calibrated bleaches. Experiments were performed both in situ in intact mice after pupillary dilation or in retinae that had been isolated from the RPE and then incubated for various times in darkness. In experiments involving isolated retinae, mice were first dark adapted overnight and killed by cervical dislocation and decapitation. The retinae were then dissected from the eye cup and RPE under infrared light and incubated in Ames bicarbonate buffer solution equilibrated with 95% O2/5% CO2. Phosphorylation determinations were made under two conditions: (1) in retinae that were dark adapted; or (2) in retinae that were first bleached and then incubated in darkness in the presence of 11-cis-retinal for various periods. The extent of bleaching was varied over the range 20–90%. The retinae, either bleached or dark adapted, were transferred to Eppendorf tubes, frozen in liquid nitrogen, wrapped in foil, and stored at −80°C before isoelectric focusing (IEF) analysis, as described below. For the in vivo experiments, mice were dark-adapted overnight. They were then kept in darkness or exposed to light that resulted in a 90% bleach. The retinae were then either frozen immediately after the bleach or were incubated in darkness for the indicated times, then frozen, and processed for IEF analysis.
All steps for IEF determinations were performed under infrared light or dim red light by methods similar to those described previously (Kühn and McDowell, 1977; Adamus et al., 1993; Concepcion and Chen, 2010). Frozen retinae were homogenized in 400 μl of buffer A [in mm: 25 HEPES, pH 7.5, 100 EDTA, 50 NaF, 5 adenosine, 1 phenylmethylsulfonyl fluoride, and a mixture of protease inhibitors (Roche)] with a Polytron (∼30 s). Samples were then centrifuged for 15 min (19,000 × g, 4°C), and the supernatant was discarded. The pellet was washed three times with 1 ml of 10 mm HEPES, pH 7.5, solution and resuspended in 1 ml of buffer B [in mm: 10 HEPES, pH 7.5, 1 MgCl2, 0.1 EDTA, 2% BSA, 50 NaF, 5 adenosine, and protease inhibitors (Roche)], and excess exogenous 11-cis-retinal was added to all samples (∼900–1200 pmol). The samples were incubated overnight at 4°C on a nutator. The samples were again centrifuged and washed with 10 mm HEPES and solubilized in 100 μl of buffer C (10 mm HEPES, pH 7.5, 0.1 mm EDTA, 1 mm MgCl2, 10 mm NaCl, 1% dodecyl-maltoside, and 1 mm dithiothreitol) for 3 h at 4°C. Finally, the samples were centrifuged, and glycerol was added to the supernatant to 5% final concentration and loaded into acrylamide gel [5% acrylamide, 0.01% dodecyl maltoside, 13.33% glycerol, Pharmalyte pH range 3–8 (GE Healthcare)] on a Pharmacia FBE 300 flatbed apparatus and focused for 2 h at 25 W. The proteins were transferred onto nitrocellulose membrane by capillary action, and the membrane was probed with the monoclonal antibody R2-12N that binds the N-terminal residues of rhodopsin. The bands were visualized with the ECL system or LI-COR Odyssey imaging system (LI-COR Biosciences). The intensity of the bands was quantified using NIH ImageJ. The values were obtained from at least three independent experiments.
Rod outer segment preparation/Western blot.
Rod outer segment (ROS) were isolated as described previously (Tsang et al., 1998; Moaven et al., 2013). Four retinae from two mice were pooled, frozen in liquid nitrogen, and stored in foil-wrapped 1.5 ml microfuge tubes at −80°C until use. Tissues were thawed and suspended in Ringer's buffer (in mm: 130 NaCl, 3.6 KCl, 2.4 MgCl2, 1.2 CaCl2, 10 HEPES, and 0.02 EDTA, pH 7.4) containing 8% OptiPrep (Sigma) and vortexed for 2 min. Tissue pieces were then allowed to settle for 1 min. The supernatant containing severed ROS was removed and collected, then 8% OptiPrep was again added, and the process was repeated four more times. The pooled supernatant was centrifuged on a discontinuous gradient containing 10% and 18% OptiPrep in Ringer's buffer at 70,000 rpm in a Beckman TLA100 rotor for at least 1 h. The ROS band was collected, diluted in 3 vol of Ringer's buffer, and centrifuged at 50,000 rpm. The pelleted ROS was resuspended in Ringer's buffer containing 1% n-dodecyl-β-maltoside and protease inhibitor mixture (Roche) and incubated at room temperature for 30 min. The ROS sample was combined with an equal amount of 2× sample buffer, and the proteins were separated on SDS-PAGE, blotted onto nitrocellulose, and probed with the following antibodies: rabbit anti-Gβ5 at 1:3000 dilution (CT-215; M. Simon, Professor Emeritus, Division of Biology, California Institute of Technology, Pasadena, CA), rabbit anti-rod arrestin (Chen et al., 2006), and mouse anti-GNAT1 (TF-15, 1:5,000; CytoSignal). Signals were detected using an LI-COR Odyssey Infrared Imaging system and quantified using ImageJ software.
trans-Retinal electroretinogram.
The trans-retinal electroretinogram (ERG) of isolated whole retinae provides a macroscopic measurement of the flash response amplitude and sensitivity. It is an ideal method for determining overall retinal responsiveness under conditions that are identical to those under which levels of rhodopsin phosphorylation were determined by IEF. Measurements were performed in a two-part chamber. In the top half of the chamber, the retina was placed with the photoreceptor side up, so as to expose rods to continuously circulating bicarbonate-buffered Ames solution via a perfusion system. This solution was maintained at 37°C and contained 1.9 g/L NaHCO3 (equilibrated with 95% O2/5% CO2, pH 7.4). It also contained 40 μm dl-AP-4 (Tocris Bioscience) to prevent rod to bipolar glutamatergic synaptic transmission and 100 μm BaCl2 to suppress the response of Müller cells (Nymark et al., 2005; Heikkinen et al., 2012). One recording electrode was positioned in the solution above the photoreceptors. The reference electrode was located in the chamber below the retina. A small aperture (1 mm) separated the two chambers, over which the retina was flat-mounted. The bottom chamber was filled with Ames solution that contained 2.36 g/L HEPES, pH 7.4, 40 μm dl-AP-4, and 100 μm BaCl2. Flash responses recorded from GNAT2−/− retinae contained exclusively the rod component of the ERG response, because the cones in this mouse strain are unresponsive.
Retinae were stimulated with 20 ms flashes of 500 nm light. The stimulus light source was calibrated to deliver (without attenuation) 1.26 × 107 photons μm−2 s−1, and its spot size was sufficient to illuminate the entire retina. We used calibrated neutral density filters to adjust stimulus light intensity. Flash responses were recorded using a Warner Instruments DP-311 differential amplifier (1000× gain) and low-pass filtered at 1000 Hz, before being digitized and recorded at 2 kHz using a Digidata 1322A acquisition board. Data analysis was performed using the pClamp 9 software system (Molecular Devices). DC offsets were removed during data analysis. Response families were obtained by averaging, seven trials for the dimmest flashes and decreasing to a single flash for the brightest intensities.
Single-cell suction electrode flash response measurements.
Single-cell suction electrode recordings allow for the measurement of changes in photocurrent attributable to flash stimuli in single rod cells. Procedures used were generally similar to those we have described previously (Nymark et al., 2012). Retinal recordings were made immediately after isolation from the eye cup or after a bleach and a period of incubation in darkness in the presence of 11-cis-retinal. The retinae were cut into smaller pieces and suspended in buffered Ames solution (HEPES, pH 7.4) and then injected into the recording chamber. The cells and retinal fragments were allowed sufficient time to adhere spontaneously to the bottom of the chamber before superfusion commenced. The superfusion system provided medium that contained heated (35–37°C) Ames medium that was buffered and gassed as described above. Single cells were drawn outer-segment first into a tight-fitting glass micropipette for recording.
The 500 nm flash stimulus had a duration of 20 ms. The maximum unattenuated light intensity was set to provide 4.18 × 107 photons μm−2 s−1. Calibrated neutral density filters were inserted into the stimulus beam to attenuate the light intensity. Data were low-pass filtered at 50 Hz, digitized at 2 kHz, and recorded as described above. Response families were obtained by averaging, seven trials for the dimmest flash responses and decreasing to a single flash for the brightest intensities.
SQRs were estimated by measuring the mean and time-dependent variance of current responses to dim flashes by methods described previously (Rieke, 2000; Ala-Laurila et al., 2007). First, we recorded response traces to 100 dim (3.56 × 102 photons μm−2 s−1) flashes. From these traces, we calculated the mean response trace, r(t), the mean square response trace, r2(t), and the total variance σtotal2(t) as a function of time. Second, we recorded 100 traces in darkness. From these, we calculated the dark variance σdark2(t). The light-induced variance σlight2(t) was calculated from the total variance and the dark variance by the following relation:
The number of photons that had been transduced per flash, R*, was calculated by scaling the mean square response to the light-induced variance:
Finally, SQR(t) was calculated by scaling the mean response r(t) by R*:
To obtain the activation constant, A, SQR(t)s of a given treatment were averaged, smoothed (Origin version 8.5.1), and normalized to the dark current (idark = 1, baseline = 0). The rising phases of these traces, y(t), were fitted with an activation model (Pugh and Lamb, 1993):
where t is the time, and teff is a temporal offset.
Microspectrophotometry.
Measurements of rhodopsin absorption spectra [optical density (OD) vs wavelength] from intact rod photoreceptors were made using a custom-built microspectrophotometer (MSP) that has been described previously (Frederiksen et al., 2012; Nymark et al., 2012). The retina was oriented photoreceptor-side up on a quartz coverslip window located in the bottom of a Plexiglas recording chamber. The retina was gently flattened with forceps and held in place at the bottom of the chamber with a slice anchor (Warner Instruments). The tissue was superfused at a rate of 4 ml/min with Ames solution that was buffered and gassed as described above. Temperature was maintained at 35–37°C.
Measurements of absorption spectra were made before and after bleaching and/or incubation in the presence of 11-cis-retinal. Spectra were determined from a region of the retina along its edge where isolated outer segments were visually identified. Generally, 10 sample scans and 10 baseline scans were each averaged over the wavelength range 350–700 nm in steps of 2 nm to increase the signal-to-noise ratio of the measured spectra. Absorbance spectra were calculated from Beers' law according to the following relation:
 |
where OD is the optical density or absorbance, Ii is the light transmitted through cell-free solution surrounding the outer segments, and It is the light transmitted through the tissue. All absorption spectra were measured with the polarization of light in the incident measuring beam parallel to the plane of the intracellular disks (T polarization). The relative concentration of rhodopsin was then determined by calculating the difference between that measured absorbance and the absorbance measured after a total (>99%) bleach.
Results
Our approach was designed to establish the relationship between rhodopsin dephosphorylation and dark adaptation. First, we used IEF in combination with Western blotting to measure the levels of rhodopsin phosphorylation in dark-adapted, bleached, and regenerating retinae. Concurrently, we correlated this (IEF) data with flash sensitivity recovery measured in isolated retinae by trans-retinal ERG or single-cell suction electrode recordings.
Measurement of phosphorylated rhodopsin species by IEF
Figure 2A illustrates IEF gels that we used to measure the extent of rhodopsin phosphorylation in isolated retinae from WT mice before bleaching (dark-adapted overnight) and at different times up to 180 min after exposure to a bright light that resulted in a 70% rhodopsin bleach. During the period before and after the bleach, each retina was incubated in the dark in bicarbonate-buffered, oxygenated Ames medium (in the absence of 11-cis-retinal) maintained at 35–37°C. The amount of rhodopsin phosphorylation is indicated in the IEF gels by the multiple bands representing differently phosphorylated species of opsin. Similar to human rhodopsin, mouse rhodopsin has three serine and three threonine residues (al-Ubaidi et al., 1990) that can be phosphorylated after bleaching light exposure (Kennedy et al., 2001; Lee et al., 2010). For instance, the band labeled 1P in Figure 2A represents singly phosphorylated species. Likewise, the band labeled 4P is when four of the six residues are phosphorylated. Finally, 6P represents the case in which all six sites were phosphorylated. It is evident from Figure 2A that, after 3 h of dark incubation, there is a persistent and substantial quantity of phosphorylated bleached photopigment. Although this method provides no information about the identity of the specific residues that are phosphorylated, the results suggest that, once each residue is phosphorylated after light exposure, its phosphorylation state is little changed for >3 h. A negative control for evaluating rhodopsin phosphorylation by IEF after bleaching light is illustrated by the gel displayed at the extreme right of Figure 2A. This gel was processed from the bleached GRK1−/− retina in which rhodopsin kinase was genetically deleted. In contrast, Figure 2B shows a similar experiment performed on live WT mice. Here the retinae were removed from mice either before or after exposure to a light measured to have bleached ∼90% of rhodopsin. Retinae were flash-frozen for IEF analysis and were processed in the same way as for gels in Figure 2A. After exposure to a ∼90% bleach, as shown in the lane marked 0 min in Figure 2B, we observed (after quantification of the data) that ∼90% of opsin was phosphorylated. These results are consistent with previous in vivo mouse studies (Ohguro et al., 1995; Mendez et al., 2000; Kennedy et al., 2001; Lee et al., 2010). However, our results and those cited above are inconsistent with observations of negligible rhodopsin phosphorylation made from amphibian retinae (Binder et al., 1996). The reason for this difference is likely attributable to lower bleach levels relative to our conditions. In our data presented in Figure 2B, we observed that rhodopsin dephosphorylation was nearly complete after 3 h of dark adaptation. These data clearly show that, by 120 min after bleach, no opsins had associated with them more than two phosphates. It is evident from comparison of gels in Figure 2, A and B, that dephosphorylation profiles are very different. In the living mouse, opsin dephosphorylation takes place over a time course of ∼3 h, as has been shown previously (Kennedy et al., 2001; Lee et al., 2010). However, in isolated retinae, the levels of phosphorylation at all residues had not measurably decreased from their initial post-bleach levels at any time measured.
Figure 2.

Opsin dephosphorylation measured by IEF gels before and after in vivo and ex vivo bleaching of mouse retinae. In vivo bleaching was performed by direct projection of 500 nm light into the eye of anesthetized mice. Ex vivo bleaching was performed by projection of light on retinae that had been isolated from the RPE and placed in Ames solution in a small plastic Petri dish. IEF gels show rhodopsin segregation based on the total number of attached phosphates as indicated in the right side of each series of gels. A, Gels from isolated retinae from WT mice. Retinae from dark-adapted mice were either kept in darkness or were bleached by 70% and incubated in darkness for the indicated duration without added 11-cis-retinal, before freezing and processing. The single lane at right was processed from a bleached Grk1−/− mouse retina. B, Gels from retinae of living animals processed as in A. Mice were either dark adapted or had their retinae subjected to a 90% bleach in vivo. After bleach, animals were left in darkness for various periods (as indicated in the panel). C, Gels from isolated retinae from GNAT2−/− mice. Retinae from dark-adapted mice were either kept in darkness or bleached by 50% and incubated with added 11-cis-retinal for the indicated times in darkness. D, Plots from IEF gels prepared from isolated retina preparations of GNAT2−/− mice in four different conditions (left to right): 50% bleached (n = 3), 50% bleached and incubated in darkness with 10 μm 11-cis-retinal for 100 min (n = 4), for 160 min (n = 7), and 220 min (n = 6). The band intensities from IEF gels were quantified and analyzed using NIH ImageJ. Fractional phosphorylation of opsin is plotted on the abscissa. Sites (residues) with zero or multiple phosphates are plotted on the ordinate. The error bars are ±SD. E, Plots as in D derived from retinae of living WT animals in five different conditions (left to right): bleached (90%) and then killed after 0 min in darkness (n = 4), bleached and killed after 30 min in darkness (n = 4), bleached and killed after 60 min in darkness (n = 4), bleached and killed after 120 min in darkness (n = 4), or bleached and killed after 180 min in darkness (n = 4). F, Fractional phosphorylation of opsin plotted versus time after bleaching in isolated retinae (black squares) or in retinae bleached in vivo (red triangles). The data points were calculated from D and E. For each lane, densities from bands 1P to 6P were summed and divided by the summed densities from all bands (0 to 6P) to obtain the fraction of phosphorylated rhodopsin in that sample. Error bars are ±SD. Red line (Ptot = −0.004t + 0.91) fitted to triangles by method of least squares. Black line is horizontal with a value of 0.5.
Because our studies were designed to compare post-bleach levels of rhodopsin phosphorylation with measurements of rod sensitivity recovery made by whole retina ERG, it was important to determine the rate of dephosphorylation in a mouse model in which only the rod component of the electrical response is present. The GNAT2−/− mouse model fills this requirement because the cone component of the ERG is absent and rod responses can be measured without complication of cone responses. Figure 2C shows that, similar to WT retinae, very little dephosphorylation of opsin was observed in isolated GNAT2−/− retinae during up to 4 h dark incubation subsequent to 50% bleach and, in addition, after total pigment regeneration with 11-cis-retinal. Indeed, as judged from the 0P band (unphosphorylated rhodopsin), the extent of unbleached and unphosphorylated pigment remains at 50% even after 220 min. This is consistent with our measurement of the amount of rhodopsin bleached. The plots in Figure 2, D and E, show averaged phosphorylation data from isolated GNAT2−/− retinae (as shown in Fig. 2C) and in retinae of WT mice bleached in vivo (as shown in Fig. 2B). These data illustrate graphically that the phosphorylation profiles observed in retinae of WT mice, whether intact or isolated, are also observed in the GNAT2−/− strain. Each graph in Figure 2, D and E, plots at different times the fraction of the total phosphorylated residues on opsin. Table 1 compares results similar to those in Figure 2 made at 20, 50, and 70% bleaches, showing that sustained phosphorylation is observed on residues over a wide range of bleach fractions. Here, it is also evident that the distribution of phosphorylation among 1P–6P seems to be independent of the fraction bleached. Figure 2F plots these data as fraction of phosphorylated species (summed signals from 1P to 6P divided by total rhodopsin population) as a function of time after bleach, for isolated retinae (black squares) and in the intact animal (red triangles). The solid red straight line that was fitted to the red triangles illustrates the time course of dephosphorylation measured in retinae of intact mice. The linear decline suggests that the dephosphorylation mechanism may be saturated. The black horizontal line plotted through the filled squares illustrates that the phosphorylation level remains virtually unchanged in isolated retinae after the bleach for up to 220 min at a level consistent with the fraction bleached. A linear regression analysis was performed on these data (data not shown). The linear regression line did not deviate significantly from that plotted (p = 0.2484). These data clearly show that there is no significant rhodopsin dephosphorylation taking place in isolated retina after bleaching, whereas efficient dephosphorylation takes place in vivo. Furthermore, the blockage of dephosphorylation in the absence (Fig. 2A) or presence (Fig. 2C) of 11-cis-retinal indicates that pigment regeneration has no effect on rhodopsin dephosphorylation in the isolated retina. Together, the data from the IEF experiments demonstrate that isolation of the retina from the living animal blocks virtually all opsin dephosphorylation compared with the dephosphorylation that normally occurs in vivo.
Table 1.
Rhodopsin phosphorylation in bleached isolated retinae
| Bound phosphates | 20% Bleach | 50% Bleach | 70% Bleach | Trend (100% bleach) |
|---|---|---|---|---|
| 6P-rhopsin | 2.7 ± 0.6 | 5.8 ± 0.7 | 8.2 ± 1.9 | 11.6 |
| 5P-rhopsin | 2.8 ± 0.3 | 6.3 ± 0.7 | 8.7 ± 1.3 | 12.3 |
| 4P-rhopsin | 2.0 ± 0.2 | 5.6 ± 0.6 | 8.5 ± 1.8 | 11.5 |
| 3P-rhopsin | 2.1 ± 0.1 | 6.1 ± 0.7 | 11.5 ± 1.3 | 14.5 |
| 2P-rhopsin | 3.7 ± 1.2 | 7.0 ± 0.8 | 16.9 ± 0.7 | 20.3 |
| 1P-rhopsin | 6.6 ± 0.5 | 15.7 ± 3.0 | 20.7 ± 2.8 | 29.7 |
| 0P-rhopsin | 80.1 (%)a | 53.1 ± 5.1 (%) | 25.2 ± 0.3 (%) | 0 (%) |
Retinae were bleached by 20% (n = 4), 50% (n = 6), or 70% (n = 3) and allowed to incubate in Ames medium for 3 h before IEF analysis. Errors are mean ± SD.
aOversaturated data point, value calculated.
This novel finding is robust. Similar results were obtained under a variety of experimental conditions commonly used in physiologic studies of isolated retinae, as shown in Figure 3. These included different superfusion/incubation buffers in which pH was regulated with HEPES or bicarbonate, over a pH range from 5.6 to 8.0, in the presence and absence of 11-cis-retinal (Fig. 3A), under different intensities of bleaching light (Fig. 3B), when the retina was intact or chopped (Fig. 3C), under conditions of continuous superfusion or static incubation, with or without attached RPE (Fig. 3D), with or without exogenous interphotoreceptor retinoid binding protein (data not shown), different extents of bleaching (from 20 to 70%; Table 1), and in WT and GNAT2−/− genetic models (Fig. 2).
Figure 3.

Opsin dephosphorylation in isolated retinae measured by IEF gels in different conditions. The gels show rhodopsin segregation based on the total number of attached phosphates as indicated in the right side of each series of gels. Except when noted, retinae were incubated in bicarbonate-buffered Ames medium under 95% O2/5% CO2. A, IEF gels measured after incubation in different conditions as indicated in the panel. The retinae were either dark adapted (left most lane) or bleached 70% and incubated in Ames medium in darkness for 180 min. The opsin band, representing the apoprotein attributable to incomplete regeneration with 11-cis-retinal, is sometimes observed in these gels. B, IEF gels of retinae that were bleached 70% at different bleaching rates (as indicated in the panel) and incubated in Ames medium in darkness for 180 min. C, IEF gels of retinae that were bleached 50 or 70%, chopped, and then incubated in Ames medium containing 11-cis-retinal in darkness for 180 min. D, Eyecups containing both the neural retina and RPE were bleached and incubated in darkness as in C. E, Retinae were incubated in Ames medium containing 4 mm Na l-lactate, bleached 70%, and immediately frozen (0 min) or frozen after incubated in darkness for 60 or 180 min. For each lane, signals from 1P to 6P were summed and divided by signals from all bands (0 to 6P) to obtain the fraction of phosphorylated rhodopsin in that sample. The value obtained for 0 min was 0.72 ± 0.04 (n = 8), corresponding well to the estimated 70% bleach, and 0.71 ± 0.07 (60 min, n = 4) and 0.44 ± 0.06 (180 min, n = 4). These values were subjected to one-way ANOVA. The p value for one-way ANOVA was 1.7 × 10−6, and the p value for Tukey's HSD was p < 0.002 for both 180 versus 0 min and 180 versus 60 min. F, Retinae were incubated with Ames medium (without added lactate) under 20% O2/5% CO2/75% N2 and subjected to bleach and dark incubation as described in E. The fractions of phosphorylated rhodopsin were 0.72 ± 0.04 (0 min, n = 8) and 0.73 ± 0.02 (n = 4) for 60 min and 0.64 ± 0.05 (n = 4) for 180 min. The one-way ANOVA showed a p value of 0.0049. This was followed by Tukey's HSD test, which showed a significant difference between 180 versus 0 min and 180 versus 60 min (p < 0.01 for both comparisons).
We found two conditions that promote rhodopsin dephosphorylation in the isolated retina. Both of these relate to the metabolic state of the retina. It is common practice to perform electrophysiological experiments on mammalian retina under conditions of high oxygen tension. Our experimental conditions have followed this norm, using 95% O2/5% CO2. However, a number of studies have established that O2 tension in the region of the photoreceptors is much lower in the intact mammalian retina at levels varying between 10 and 30% (Cringle et al., 2002; Yu and Cringle, 2006; Birol et al., 2007; Lau and Linsenmeier, 2012). In addition, photoreceptor cells of the mammalian retina use aerobic glycolysis because of their high metabolic demand (Adler and Southwick, 1992; Hurley et al., 2015). This results in high levels of lactate in the interphotoreceptor matrix. This lactate is taken up and used as fuel by Müller cells (Lindsay et al., 2014; Hurley et al., 2015). Therefore, some of our experiments were performed in the presence of 4.0 mm Na l-lactate added to the Ames medium (Fig. 3E). Other experiments were performed under a level of O2 similar to that suggested as physiological by the work referenced above (20% O2/5% CO2/75% N2; Fig. 3F). These experiments were designed to test whether dephosphorylation may be enhanced under these more physiological conditions. There was a pronounced effect on rhodopsin dephosphorylation when 4 mm l-lactate was included in the medium (Fig. 3E). Interestingly, we found a modest, but statistically significant, enhancement of rhodopsin dephosphorylation when retinae were exposed to 20% O2 (Fig. 3F). Results from these experiments suggest the involvement of metabolic processes in rhodopsin dephosphorylation. The molecular mechanism(s) for inhibition of dephosphorylation that we have observed in isolated retina were not further investigated but left for subsequent study. However, we found that the blockage of rhodopsin dephosphorylation in isolated retina is a useful tool for determining mechanisms of dark adaptation.
Microspectrophotometric measurement of phosphorylated rhodopsin
To determine whether phosphorylated opsin affects pigment regeneration, we performed MSP recordings of the visual pigment content of mouse rod outer segments in intact retina isolated and treated under the same conditions used for IEF analysis. Figure 4A plots absorption spectra taken from a cluster of rod outer segments located along the cut edge of a piece of isolated retina that had been bleached by an amount calculated to be 50%, incubated in darkness for 3 h in the incubation chamber, and then transferred to the MSP stage. Spectrum 1 was measured after superfusion in darkness and transfer to the MSP stage. After this point, the retina was superfused with solution containing 10 μm 11-cis-retinal. Spectrum 2 shows that, during this intervening period, substantial rhodopsin was regenerated. Spectrum 3 was taken after exposure to bright light calculated to bleach >99% of the visual pigment. The peak absorbance of spectrum 1 corresponds to a 50% bleach (see Materials and Methods). Spectrum 3 is consistent with having bleached >99% of the rhodopsin at the end of the experiment. The observation that spectrum 2 has twice the OD of spectrum 1 confirms that the visual pigment was completely regenerated. The experiment illustrated in Figure 4B was performed to demonstrate that incubation for long periods up to 3 h in the light-tight incubation container did not compromise the physiological state of the isolated retina so as to impair rhodopsin regeneration. Spectrum 4 was measured 3 h after a retina had been bleached by 50% and then placed in the light-tight incubation container in Ames solution containing 11-cis-retinal. The peak absorbance in this trace, whose level is similar to that measured in dark-adapted retinae, suggests that most of the rhodopsin that had been bleached had been regenerated during this 3 h period of incubation. To confirm this, the retina was treated with additional 11-cis-retinal on the stage of the MSP yielding the black trace (Figure 4B, 5), which shows no additional increase in absorbance. The total amount of rhodopsin in the outer segments was then determined by calculation of the difference in peak absorbance between this measurement and that illustrated by spectrum 6 made after a >99% bleach. Thus, the experiments in Figure 4 illustrate that we were able to bleach retinae with high precision and then fully regenerate the visual pigment by incubating the retina in a solution containing 11-cis-retinal, whether this was performed in the light-tight incubation container or on the stage of the MSP. Furthermore, this spectrophotometric data, together with the IEF data illustrated in Figure 2, demonstrate that dephosphorylation is not required for pigment regeneration. This observation is consistent with a recent report using carp photoreceptors (Yamaoka et al., 2015). It is noteworthy that the spectrum of phosphorylated rhodopsin is very similar to that of native rhodopsin. This is indicated by comparisons of the MSP traces in Figure 4, A and B, with the cyan solid curve calculated for an ideal solution of rhodopsin (Govardovskii et al., 2000).
Figure 4.
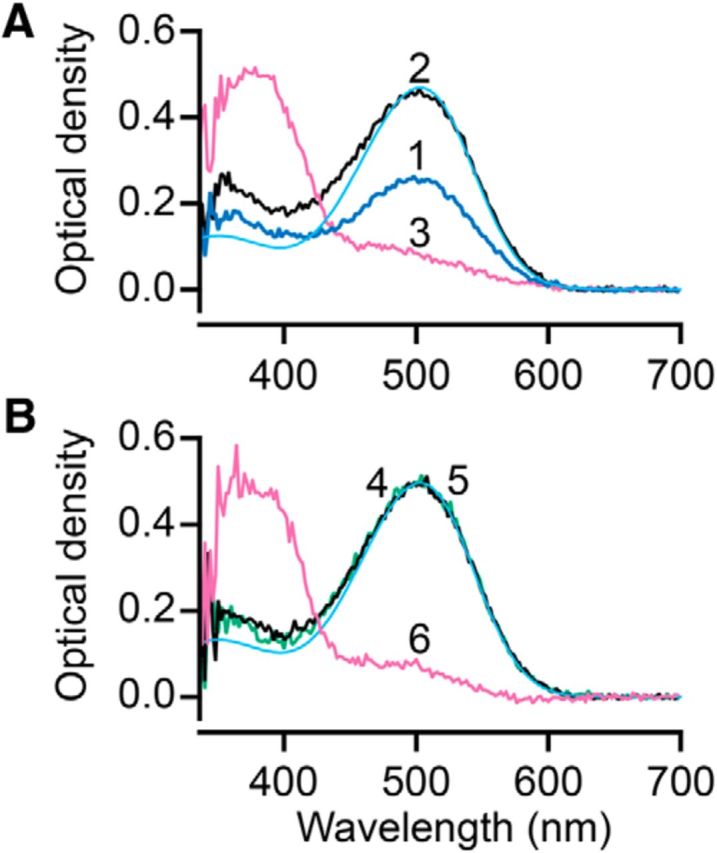
MSP absorbance spectra of bleached and regenerated WT (C57BL/6) retinae. A, Absorbance spectrum of a retina that had been bleached 50% (trace 1, blue), then treated with 11-cis-retinal on the stage of the MSP for 40 min (trace 2, black), and finally after exhaustive bleach (trace 3, pink). The cyan trace is a normalized rhodopsin template (Govardovskii et al., 2000) calculated at λmax = 503 nm, ODα = 0.47. B, Trace 4 (green) shows an absorbance spectrum of a retina that had been bleached 50% and incubated for 3 h with 10 μm 11-cis-retinal in darkness in a light-tight incubation container. Trace 5 (black) shows an absorbance spectrum of the same preparation after 40 min additional exposure to solution containing 10 μm 11-cis-retinal on the stage of the MSP. Trace 6 (pink) shows absorbance spectrum after an exhaustive bleach (>99%). The cyan trace is a fitted rhodopsin template (λmax = 503 nm, ODα = 0.50).
Figure 5.

Trans-retinal ERGs demonstrating the recovery of flash sensitivity in GNAT2−/− mouse rods having persistent rhodopsin phosphorylation. A, Response family of isolated a-waves from a dark-adapted retina (left), from a retina that was bleached 50% and incubated in darkness for 60 min (middle), and from a retina that was bleached (50%) and incubated in darkness for 180 min in 10 μm 11-cis-retinal (right). B, Intensity–response relations that plot maximum a-wave amplitude versus time under the above conditions. Each relation is fitted with exponential saturation function: V = Vmax(1 − exp(−I/τ)), where V is the voltage amplitude, Vmax is the voltage amplitude of the saturated response, I is light intensity, and τ is a sensitivity fitting parameter. Data were recorded from dark-adapted retinae (black circles, n = 8, Vmax = 324 μV, τ = 94 hν μm−2), 50% bleached retinas (pink triangles, n = 6, Vmax = 144 μV, τ = 1860 hν μm−2), and retinae that had been bleached 50% and incubated for 3 h (blue squares, n = 7, Vmax = 297 μV, τ = 102 hν μm−2). The recordings were made at 35–37°C. Error bars are ±SEM. The inset shows normalized dim flash responses from a dark-adapted retina (black trace), a 50% bleached retina (pink trace), and a retina that had been bleached 50% and incubated with 10 μm 11-cis-retinal in darkness for 3 h (blue trace). C, Western blots showing transducin translocation. Retinae were either dark adapted or bleached 90% and then incubated in Ames medium containing 11-cis-retinal in darkness as indicated above the lanes. Retinae were frozen, and the ROSs were isolated from the remaining retina. Antibodies were used to visualize the amount of rod transducin (Gtα) in the outer segment. Gβ5L was used as a loading control. Gβ5S serves as a quality control for contaminants. A sample of whole-retinal homogenate was carried as control. Signal intensities were quantified using NIH ImageJ software, and the values for Gtα were normalized against that of Gβ5L. The normalized values for Gtα, when compared with the dark-adapted value (set to 1.0) for 30 min was 0.6 ± 0.1 and for 240 min was 0.9 ± 0.2 (mean ± SD, n = 3).
In addition to spectral measurements of phosphorylated rhodopsin, we also compared the molecular photosensitivity of rhodopsin in rods outer segments containing 100% unphosphorylated rhodopsin and 90% P-rhodopsin with MSP. This was done by exposing the retina to a light intensity, I, for different times, t. The fractional bleaching of pigment, F, after each exposure was then measured microspectrophotometrically. The photosensitivity, P, was calculated from Equation 1. We measured photosensitivities of (6.0 ± 1.3) × 10−9 μm2 for dark-adapted retinas containing 100% unphosphorylated rhodopsin and (6.7 ± 1.2) × 10−9 μm2 for retinas containing 90% P-rhodopsin. These values were statistically indistinguishable from one another (t test, t = 0.73, p = 0.49) and very similar to the photosensitivity that has been reported previously for rhodopsin of 5.7 × 10−9 μm2 (Woodruff et al., 2004; Nymark et al., 2012).
Whole-retina ERG measurement of flash sensitivity and response kinetics during dark adaptation
We then determined how dark adaptation is affected when rhodopsin dephosphorylation is inhibited. Here, we used whole-retina ERG recording of GNAT2−/− retinae. These recordings were obtained from isolated retinae in the presence of AP-4 to block synaptic transmission from rod to rod bipolar cell as well as Ba2+ to block Müller cell activity (see Materials and Methods). Thus, the GNAT2 strain allows flash sensitivity of the whole population of rods to be evaluated under conditions that were identical to those under which IEF and MSP experiments were performed. Figure 5A (left to right) shows response families from trans-retinal ERGs measured under dark-adapted conditions (Fig. 5A, left), 60 min after a 50% bleach and incubation in Ames solution (Fig. 5A, middle) and after 3 h incubation in Ames solution containing 11-cis-retinal (Fig. 5A, right). These measurements were made from a subset of retinae that were processed subsequently for IEF analysis, the results of which are illustrated in Figure 2C. The responses in the family illustrated in the middle panel are accelerated compared with dark-adapted responses (Fig. 5B, inset) and show smaller peak amplitudes, consistent with persistent opsin adaptation (Cornwall et al., 1990; Cornwall and Fain, 1994). After incubation for 3 h with 11-cis-retinal in darkness, families displayed a virtually complete return to dark-adapted kinetics, although overall response amplitude was somewhat smaller. Average response/intensity relationships measured in retinae treated in this way are illustrated in Figure 5B. Here we have plotted average response amplitude as a function of flash intensity for retinae that are dark adapted (circles), bleached 50% (triangles), bleached 50% and incubated in 11-cis-retinal for 3 h (squares). Exponential saturation functions are fitted to the data (for details, see figure legend). On average, after bleaching, we observed a shift of the response/intensity function to higher intensity (∼1.3 log units) compared with pre-bleach conditions, and response amplitude was reduced approximately by half. Over the 3 h of incubation in 11-cis-retinal solution, response amplitude and sensitivity steadily increased until, at 3 h, response amplitude and sensitivity were almost completely recovered and response kinetics were indistinguishably different from dark adapted. This is also illustrated in the inset of Figure 5B, in which average dark-adapted dim flash responses (black trace, 158 ms time-to-peak) are compared with those measured ∼60 min after a 50% bleach (pink trace, 72 ms time-to-peak) and the following 3 h of incubation in 11-cis-retinal solution (blue trace, 166 ms time-to-peak). This recovery of sensitivity and return to dark-adapted response kinetics in bleached and regenerated isolated retinae suggests that, aside from blockage of rhodopsin dephosphorylation, the physiology of the rods is not significantly altered by our experimental conditions.
Light stimulates rhodopsin phosphorylation and transducin translocation from the outer segment to the inner compartments (Whelan and McGinnis, 1988; Sokolov et al., 2002). In addition to rhodopsin phosphorylation, local depletion of transducin concentration is also known to reduce the activity of rhodopsin (Sokolov et al., 2002; Mao et al., 2013). However, a greater than twofold reduction is required to observe a change in sensitivity because little change was observed in mice heterozygous for rod transductin, GNAT1+/− (Calvert et al., 2000). In contrast, Arr1 moves in the opposite direction in response to light (Whelan and McGinnis, 1988; Nair et al., 2005; Strissel et al., 2006), although an increase in Arr1 concentration does not appear to affect transduction gain (Song et al., 2011). To determine the amount of transducin in the outer segment under our experimental conditions, we isolated outer segments from retinae in the dark or after 90% bleach, followed by reconstitution with 11-cis-retinal. Determinations were made after 30, 180, or 240 min of dark incubation by Western blots to visualize the amount of rod transducin α (Gtα) in the outer segment. Gβ5L (Keresztes et al., 2004), a component of the transducin GAP complex, is anchored to the outer segment compartment by R9AP (Cao et al., 2010) and was used as a loading control (Moaven et al., 2013). Conversely, Gβ5S is an isoform produced from alternative splicing of the same gene. It is present in the inner segment compartments and inner retinal neurons and serves as a quality control for contaminants (Watson et al., 1996). Under our experimental conditions, light exposure caused the level of α-subunit of GNAT1 (Gtα) to fall and Arr1 to rise in the outer segment (Fig. 5C), similar to previous reports. Quantification of the signals from Gtα normalized to that of Gβ5L shows that the amount of Gtα has returned to the outer segment similar to the dark-adapted level by 180 min (0.9 ± 0.2, mean ± SD, n = 4; in which the dark-adapted value is 1.0) and is maintained at that level thereafter. We conclude that sensitivity differences detected under the steady-state experimental conditions that we used in whole-retinal ERG experiments described above and single-cell measurements of sensitivity (see below) can be attributed to rhodopsin phosphorylation but not transducin concentration in the outer segment.
Single-cell measurements of sensitivity and flash response kinetics
From the ERG data presented in Figure 5, it appears that retinae that have undergone a 50% bleach and subsequent rhodopsin regeneration contain half their total rhodopsin in the phosphorylated state. To know more of the state of sensitivity and response kinetics under conditions of persistent phosphorylation, we conducted single-cell suction electrode experiments. Figure 6A shows response families from such current recordings of rod flash responses in different cells under four different conditions. From left to right, these include the following: (1) dark adapted; (2) bleached by 50% and incubated >90 min in darkness; (3) bleached by 50% and incubated in darkness with exogenous 11-cis-retinal for 4 h; and (4) bleached by 90% and incubated in darkness with 11-cis-retinal for 4 h. We were unable to observe any significant differences in the time course of dark-adapted flash responses measured before bleach compared with those measured after bleach by either 50 or 90%, followed by incubation with 11-cis-retinal (Fig. 7). Figure 6B plots average response/intensity functions measured under the different conditions in Figure 6A. The response/intensity relation of dark-adapted rods (0% P-rhodopsin) are represented by black circles, that from bleach-adapted rods in the steady state after the 50% bleach (no 11-cis-retinal treatment) are represented by pink triangles, that from rods that had been bleached 50% and incubated for 4 h with 11-cis-retinal (50% P-rhodopsin) are represented by blue squares, and that from rods that had been bleached 90% and incubated for 4 h with 11-cis-retinal (90% P-rhodopsin) are represented by orange diamonds. In each case, the data were fitted by exponential saturation functions as in Figure 5 (for details, see legend). It is apparent that the magnitude of the dark current is approximately the same after bleaching and regeneration in all three states, independent of the extent of rhodopsin phosphorylation. Thus, there is no evidence of steady-state activation of transducin by phosphorylated rhodopsin, as is the case when excess free opsin is present under bleach-adapted conditions (Cornwall and Fain, 1994). Moreover, although the intensity response curve and flash sensitivities of the 50% P-rhodopsin state (S50% = 0.33 ± 0.04 pA/hv) are indistinguishable from the 0% P-rhodopsin dark-adapted state (SDA = 0.33 ± 0.03 pA/hv), a small but significant shift (less sensitive) can be detected in the 90% P-rhodopsin rods (S90% = 0.24 ± 0.05 pA/hv). This shift represents an average desensitization of ∼33%.
Figure 6.

Suction electrode recordings demonstrating the flash sensitivity recovery in bleached mouse rods that have persistent rhodopsin phosphorylation. A, Response families from a dark-adapted rod (left), from a 50% bleached rod that was incubated in darkness for 90 min (middle left), from a rod that was bleached 50% and incubated with 10 μm 11-cis-retinal in darkness for 3 h (middle right), and from a rod that was bleached 90% and incubated with 10 μm 11-cis-retinal in darkness for 3 h (right). B, Average response–intensity relations recorded from dark-adapted rods (black circles, n = 8), rods that had been bleached 50% and incubated in darkness for 90 min (pink triangles, n = 4), rods that had been bleached 50% and incubated with 10 μm 11-cis-retinal in darkness for 3 h (50% P-rhodopsin, blue squares, n = 10), and rods that had been bleached 90% and incubated with 10 μm 11-cis-retinal in darkness for 3 h (90% P-rhodopsin, orange diamonds, n = 10). Exponential saturation functions: i = imax(1 − exp(−I/τ)) are fitted to data, where i is the peak photocurrent, imax is the peak photocurrent of a saturating flash response, I is intensity, and τ is a sensitivity fitting parameter. Dark-adapted, black line, imax = 12.9 ± 0.7 pA, τ = 73.8 ± 12.6 hν μm−2; 50% P-rhodopsin, blue line, imax = 13.8 ± 0.7 pA, τ = 81.5 ± 13.1 hν μm−2; 90% P-rhodopsin, orange line, imax = 12.8 ± 0.8 pA, τ = 111.0 ± 22.7 hν μm−2. The recordings were made at 35–37°C. Error bars are ±SEM.
Figure 7.
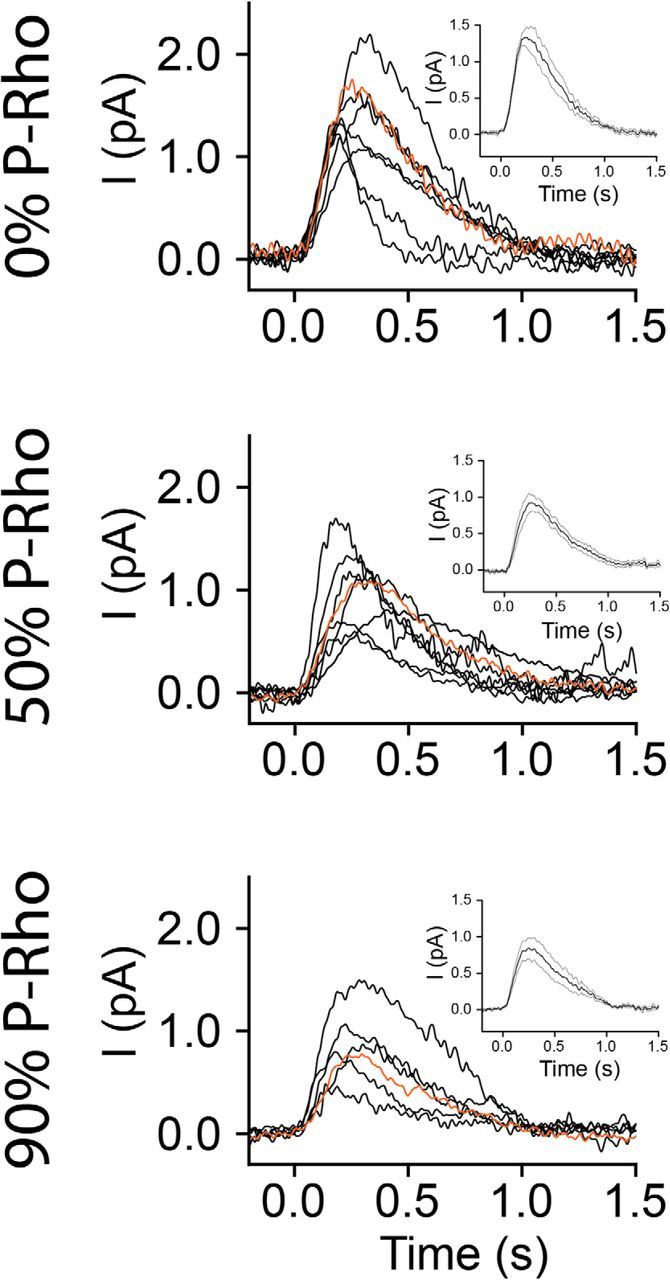
SQRs recorded from WT rods containing rhodopsin (top), 50% phosphorylated rhodopsin (middle), and 90% phosphorylated rhodopsin (bottom). The red traces show representative SQRs that are shown in Figure 8. Insets, Mean waveform of all SQRs presented in each panel ± SEM. All recordings were made at 35–37°C.
SQRs resulting from light activation of phosphorylated rhodopsin
To explore further the mechanism of the desensitization caused by persistent phosphorylation of rhodopsin after high bleaches, we recorded responses to a large number of dim flashes and calculated the SQR (see Materials and Methods). Plotted in Figure 7 are superimposed SQRs recorded from dark-adapted rods (top row), rods containing 50% P-rhodopsin (middle row), and rods containing 90% P-rhodopsin (bottom row). The insets in the panels show the average ± SEM waveforms of the recorded SQRs. It is evident from the traces in Figure 7 and the average data presented in Table 2 that the SQR amplitudes in rods containing P-rhodopsin are smaller than those of the dark-adapted rods containing exclusively unphosphorylated rhodopsin. Moreover, the averaged data presented in Table 2 demonstrate that the response kinetics of SQRs in all three conditions are similar. These data show that, although there is a significant reduction of SQR amplitude caused by the phosphorylation of rhodopsin, the fractions of the incident photons that lead to a response in each case are statistically indistinguishable. The same is true for the response kinetics. The mean time-to-peak, as well as the time constant of deactivation of the response, τrec, are statistically indistinguishable in cells containing 0, 50, or 90% P-rhodopsin. Moreover, we calculated the activation constant, A, of the averaged SQRs, by fitting Equation 5 (see Materials and Methods), a phototransduction activation model (Pugh and Lamb, 1993). We obtained activation values for the different treatments of A0% P-rhodopsin = 4.1 s−2, A50% P-rhodopsin = 3.3 s−2, and A90% P-rhodopsin = 2.9 s−2 (Fig. 8A, inset). This calculation demonstrates a lower level of activation of phototransduction by photoactivated phosphorylated rhodopsin compared with unphosphorylated rhodopsin.
Table 2.
SQR properties from suction cell recordings of single mouse rods
| Amplitude (pA) | R*a | Time-to-peak (s) | τrec (s)b | |
|---|---|---|---|---|
| Rhodopsin | 1.50 ± 0.11 | 1.07 ± 0.14 | 0.26 ± 0.02 | 0.37 ± 0.05 |
| 50% P-rhodopsin | 1.02 ± 0.14 | 1.27 ± 0.19 | 0.27 ± 0.03 | 0.35 ± 0.05 |
| 90% P-rhodopsin | 0.81 ± 0.09 | 0.87 ± 0.09 | 0.25 ± 0.03 | 0.40 ± 0.06 |
| ANOVAc | F = 7.48* | F = 0.82 (NS) | F = 0.18 (NS) | F = 0.19 (NS) |
SQRs were recorded in dark-adapted retinae containing rhodopsin (n = 8), 50% phosphorhodopsin (n = 9), and 90% phosphorhodopsin (n = 6). Values are mean ± SEM.
aR*was calculated according to Equation 3. The incident light was 3.56 × 102 photons μm−2 s−1 in all treatments.
bτrec was calculated by fitting an exponential decay function to the falling phase of the response.
cOne-way ANOVA with a Tukey's test for means comparison.
* Indicates significance at the 5% level between rhoopsin and 50% P-rhopsin, as well as between rhodopsin and 90% P-rhodopsin. NS, Not significant.
Figure 8.
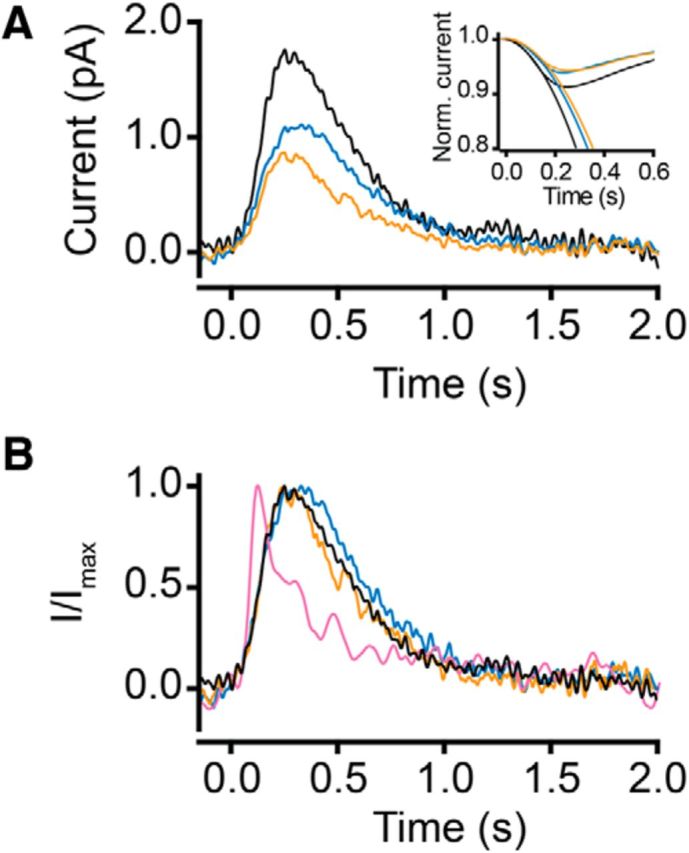
Average SQR recorded from WT rods containing rhodopsin and phosphorylated rhodopsin. A, Representative SQRs from a rod containing rhodopsin (black), 50% phosphorylated rhodopsin (blue), and 90% phosphorylated rhodopsin (orange). Inset, Averaged, smoothed, and normalized SQRs fitted with an activation model (Pugh and Lamb, 1993). For details, see text. B, Normalized SQRs from A. Also shown is a normalized average dim flash response from a 50% bleached rod (pink). The recordings were made at 35–37°C.
To facilitate comparison of the waveforms under the three conditions, we have selected one representative recording in each condition for additional analysis. These recordings are indicated by the red traces in respective rows in Figure 7. The differences in SQR amplitudes are shown in Figure 8A, with the representative SQR from each condition plotted together. Here, it is clear that the SQR amplitude decreases when rhodopsin is phosphorylated before activation. These traces are normalized in Figure 8B. Also included in Figure 8B is a normalized average dim flash response from a rod bleached by 50% (pink) in which the rhodopsin was not regenerated. As expected, this trace has a significantly more rapid time course because of opsin adaptation (Cornwall and Fain, 1994; Nymark et al., 2012).
The data presented in Figures 7 and 8 and the data in Table 2 indicate that, whereas SQR arising from phosphorylated rhodopsin has a decreased amplitude, the response time course appeared similar between SQR generated from unphosphorylated rhodopsin (0% P-rhodopsin) and phosphorylated rhodopsins (50% P-rhodopsin and 90% P-rhodopsin; Fig. 7). An analysis of response variability failed to show differences between the three groups of responses. Notably, Doan et al. (2006) have shown previously that each phosphorylated site provides an independent step in rhodopsin deactivation that collectively confer reproducibility of the SQR of the rod by genetically eliminating phosphorylatable sites on rhodopsin (Doan et al., 2006; Azevedo et al., 2015). Our experiment is opposite to their approach; we eliminated no phosphorylatable sites but rather prephosphorylated them, and our analysis did not reveal this variation in SQR reproducibility. However, we must exercise caution in the interpretation of these data. Proper estimations of response variability (coefficient of variation) require recording with very low drift, cellular noise exceeding instrumental noise by at least twofold, and the ability to resolve all single-photon responses, noting that much of the variance arises from responses at the tails of the amplitude distribution. Although the issues noted above restricted an in-depth noise analysis, we observed that the averaged SQR was able to accurately reflect the kinetics and amplitudes of individual SQRs under different conditions of rhodopsin phosphorylation (Fig. 8).
Discussion
Effects of P-rhodopsin on visual sensitivity
The central findings of our work are that mouse rods exposed to bright light in the presence of 11-cis-retinal contain a significant fraction of their rhodopsin in a phosphorylated form that is spectrally identical to ground state rhodopsin, and its light activation results in physiological responses with reduced amplitudes but with kinetics similar to the dark-adapted state. A model that incorporates these results on the phototransduction cascade is shown in Figure 9. The box in Figure 9 shows that unphosphorylated rhodopsin (R) and phosphorylated rhodopsin (R-P) coexist and give rise to different response amplitudes. After light activation, unphosphorylated rhodopsin forms R* [metarhodopsin II (MII)], which maximally activates the G-protein transducin. The extent of transducin activation (G) is indicated by the bold arrows. MII is rapidly phosphorylated within tens of milliseconds to form MII-P, which triggers high-affinity Arr1 binding to form MII-P · Arr1 in which the catalytic activity of rhodopsin is quenched fully. All-trans retinal is then gradually released from MII-P · Arr1, leaving phosphorylated opsin, which has diminished affinity for Arr1. Because large amounts of MII-P · Arr1 accumulate under bright light exposure and because the dephosphorylation reaction for P-opsin is slow in vivo, high levels of P-rhodopsin are formed upon reconstitution of P-opsin with 11-cis-retinal (Lee et al., 2010). Furthermore, light-activated P-rhodopsin has diminished ability to activate transducin (one bold arrow).
Figure 9.

A model illustrating the effects of R-P on the phototransduction cascade. When stimulated by light, activated rhodopsin (R* or MII) interacts with the G-protein transducin (G) to initiate phototransduction (indicated by 2 bold arrows). Rhodopsin kinase (GRK1) quickly phosphorylates R* to form MII-P in a process that reduces its capacity to activate transducin (as indicated with only 1 bold arrow). Shortly thereafter, the catalytic activity of the MII-P is terminated through binding with Arr1 to form MII-P · Arr1. After this step, phospho-opsin (Op-P · Arr1) is hydrolyzed from all-trans retinal, which is recycled in the RPE (data not shown). The dashed arrow indicates that opsin itself has very low transduction activity. Pigment regeneration occurs when phospho-opsin (Op-P) recombines with 11-cis-retinal (supplied from the RPE) to form phospho-rhodopsin (R–P). Phospho-rhodopsin can either be slowly dephosphorylated by rhodopsin phosphatase or reactivated by light. The latter outcome results in diminished activation of transducin (G).
An important manifestation of the ability of P-rhodopsin to activate transducin is a diminished amplitude of the SQR. Previous investigations of the nature and kinetics of SQRs were conducted on fully dark-adapted rods and therefore reflect the activity of R* produced from rhodopsin in its unphosphorylated form. To our knowledge, our study is the first to characterize SQRs arising from P-rhodopsin regenerated during exposure to exogenous 11-cis-retinal. Our comparison of SQRs from rhodopsin and P-rhodopsin show that the phototransduction gain decreased as the concentration of P-rhodopsin increased. We extrapolate from our data that the gain reduction would be ∼50% for P-rhodopsin, a value similar to that observed in the Arr1−/− rods in which the catalytic activity of P-rhodopsin was also ∼50% of the peak value (Xu et al., 1997). This result is consistent with biochemical studies showing a decrease in the efficiency of transducin activation by photolyzed P-rhodopsin (Miller et al., 1986; Wilden et al., 1986; Bennett and Sitaramayya, 1988; Wilden, 1995), a point further supported by our calculations of the amplification constants. Although the IEF data show that the P-rhodopsin is a heterogeneous population comprising rhodopsin that has one to six phosphates attached, it has been shown previously that one or two attached phosphates is sufficient to reduce phototransduction gain by half (Mendez et al., 2000). Although this reduction in sensitivity may seem to be relatively minor compared with the sensitivity loss produced by bleaching, a more serious consequence is expected for the final phases of dark adaptation toward absolute visual threshold. Here the size of the SQR has important implications. The detection of SQRs by rod bipolar cells, the primary synaptic output of rod photoreceptors in the mammalian retina (Dacheux and Raviola, 1986), requires passage of responses through a nonlinear synaptic threshold at the rod-to-rod bipolar synapse. (Field and Rieke, 2002). Under fully dark-adapted conditions, synaptic saturation renders the rod bipolar current insensitive to small changes in transmitter release from the rod. This mechanism serves to improve visual sensitivity by rejecting the noise arising from rods that did not absorb photons. Thus, for SQRs to be transmitted faithfully to rod bipolar cells, their size must be sufficiently large to relieve saturation at rod-to-rod bipolar cell synapses (Sampath and Rieke, 2004). This nonlinearity in signal transfer is likely to filter out small SQRs arising from P-R* and thereby reduce sensitivity at visual threshold during dark adaptation. This is important because, in the lowest four orders of magnitude in the visual range (from one photon capture per 10,000 rods to one photon capture per rod), scotopic vision depends exclusively on the detection of individual photons.
Effects of P-rhodopsin on Arr1 binding
In rods, stepwise rhodopsin deactivation occurs as follows: (1) GRK1 binding to MII; (2) Arr1 competition with GRK1 binding; (3) GRK1 catalyzed phosphate attachment to form MII-P; (4) Arr1 activation; and (5) Arr1 binding to MII-P, which fully quenches the catalytic activity of rhodopsin (Fig. 9). All steps contribute to shape the kinetics and variability of the SQR (Doan et al., 2006, 2009). Arr1 exists in a basal state that transitions into a binding-competent conformation by encounters with MII-P (Gurevich et al., 2011). Arr1 in the basal state competes with GRK1 and slows MII deactivation (Doan et al., 2009). As GRK1 sequentially adds phosphates onto MII, interaction of MII-P with Arr1 may be favored over GRK1. By comparing SQRs generated by rhodopsin and P-rhodopsin, we have the unique opportunity to investigate whether GRK1 catalyzed MII phosphorylation, or Arr1 activation and binding to MII-P, is rate limiting in MII deactivation. If MII-P has a significantly higher affinity to Arr1, then SQRs generated by P-rhodopsin should deactivate faster when compared with SQRs generated from R*, which still requires additional steps of phosphate attachment by GRK1. Conversely, if Arr1 activation is rate limiting, then the rate of recovery should remain the same. We observed that SQRs generated from rhodopsin and P-rhodopsin display similar response kinetics. This similarity suggests that the affinity of Arr1 to MII is insensitive to phosphate attachment and that Arr1 activation is a slow step in quenching the catalytic activity of MII-P.
A recent study of Arr1 interaction with different functional forms of rhodopsin using solution nuclear magnetic resonance spectroscopy shows that the affinity of Arr1 for P-rhodopsin was sufficiently high such that active monomeric Arr1 in the outer segment is predicted to bind P-rhodopsin in the intact rod (Zhuang et al., 2013). However, our observation that the efficiency of SQR generation by P-rhodopsin is virtually identical to that of ground-state SQRs indicates that Arr1 is not bound to P-rhodopsin in a manner that prevents photolyzed P-rhodopsin from generating a light response.
Phosphorylated rhodopsin, the visual cycle, and dark adaptation
The visual cycle is defined as the ensemble of biochemical reactions that reside in the neural retina and the RPE whereby visual pigment, once bleached, is returned to its fully functional ground state containing 11-cis-retinal. Because rhodopsin cannot be defined as being in its ground state until it has been fully dephosphorylated, we argue that the dephosphorylation of rhodopsin is a critical step in the visual cycle (Lee et al., 2010).
Vertebrate rods have the ability to undergo huge changes in their sensitivity in response to differences in background and bleaching light intensity. This endows them with the ability to respond over a range of three to four orders of magnitude of light intensities. The cellular mechanisms that have been identified within rods that are responsible for these changes include the following: (1) the transduction-driven decrease of cytosolic calcium concentration that, in turn, modulates the activities of cGMP phosphodiesterase, guanylyl cyclase, and recoverin within rod outer segments; (2) the prolonged lifetime of photoproducts of rhodopsin bleaching that result in persistent activation of the transduction cascade; (3) bleaching of rhodopsin that by virtue of its decreased concentration within the outer segment reduces quantum catch; and (4) the translocation of transducin from the outer to the inner segment of rods leading to decreased transducin activation by R*. Our results, which show that high levels of P-rhodopsin can persists within rod outer segments for many minutes after bright light exposure and that its photoactivation activates transducin with diminished efficiency suggest that this mechanism of adaptation be added to the above list. Except at the highest levels of rhodopsin phosphorylation, this sensitivity reduction is modest, but it is predicted to have its most significant effects near visual threshold.
Why is rhodopsin dephosphorylation blunted in isolated mammalian retina?
Our observation that isolation of the retina from the mouse eye results in substantial inhibition of rhodopsin dephosphorylation is novel and surprising. Importantly, biochemical and electrophysiological measurements of isolated retina and solitary photoreceptor cells under the same conditions we describe here have been routine for >40 years. Thus, our documentation emphasizes that isolation of the retina from the intact animal may have important implications for these studies. Furthermore, we demonstrate that low oxygen tension in the superfusion medium and/or the presence of lactate both promote dephosphorylation. These latter results clearly suggest the involvement of metabolic mechanisms in the regulation of rhodopsin dephosphorylation but provide no insights into specific mechanisms.
Footnotes
This work was supported by National Institutes of Health Grants EY01157 (C.C.) and EY12155 (J.C.) and Academy of Finland Grant 260375. We express appreciation to Drs. Gordon Fain and A. P. Sampath for their comments on a previous version of this manuscript. We thank Paul Hargrave for providing the rhodopsin N-terminal monoclonal antibody, R2-12N. We thank Howard Cohen for construction of electronic components in the microspectrophotometer.
The authors declare no competing financial interests.
References
- Adamus G, Arendt A, Hargrave PA, Heyduk T, Palczewski K. The kinetics of multiphosphorylation of rhodopsin. Arch Biochem Biophys. 1993;304:443–447. doi: 10.1006/abbi.1993.1373. [DOI] [PubMed] [Google Scholar]
- Adler AJ, Southwick RE. Distribution of glucose and lactate in the interphotoreceptor matrix. Ophthalmic Res. 1992;24:243–252. doi: 10.1159/000267174. [DOI] [PubMed] [Google Scholar]
- Ala-Laurila P, Donner K, Crouch RK, Cornwall MC. Chromophore switch from 11-cis-dehydroretinal (A2) to 11-cis-retinal (A1) decreases dark noise in salamander red rods. J Physiol. 2007;585:57–74. doi: 10.1113/jphysiol.2007.142935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- al-Ubaidi MR, Pittler SJ, Champagne MS, Triantafyllos JT, McGinnis JF, Baehr W. Mouse opsin. Gene structure and molecular basis of multiple transcripts. J Biol Chem. 1990;265:20563–20569. [PubMed] [Google Scholar]
- Ames A, 3rd, Nesbett FB. In vitro retina as an experimental model of the central nervous system. J Neurochem. 1981;37:867–877. doi: 10.1111/j.1471-4159.1981.tb04473.x. [DOI] [PubMed] [Google Scholar]
- Azevedo AW, Doan T, Moaven H, Sokal I, Baameur F, Vishnivetskiy SA, Homan KT, Tesmer JJ, Gurevich VV, Chen J, Rieke F. C-terminal threonines and serines play distinct roles in the desensitization of rhodopsin, a G protein-coupled receptor. Elife. 2015:4. doi: 10.7554/eLife.05981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett N, Sitaramayya A. Inactivation of photoexcited rhodopsin in retinal rods: the roles of rhodopsin kinase and 48-kDa protein (arrestin) Biochemistry. 1988;27:1710–1715. doi: 10.1021/bi00405a049. [DOI] [PubMed] [Google Scholar]
- Binder BM, O'Connor TM, Bownds MD, Arshavsky VY. Phosphorylation of non-bleached rhodopsin in intact retinas and living frogs. J Biol Chem. 1996;271:19826–19830. doi: 10.1074/jbc.271.33.19826. [DOI] [PubMed] [Google Scholar]
- Birol G, Wang S, Budzynski E, Wangsa-Wirawan ND, Linsenmeier RA. Oxygen distribution and consumption in the macaque retina. Am J Physiol Heart Circ Physiol. 2007;293:H1696–H1704. doi: 10.1152/ajpheart.00221.2007. [DOI] [PubMed] [Google Scholar]
- Calvert PD, Krasnoperova NV, Lyubarsky AL, Isayama T, Nicoló M, Kosaras B, Wong G, Gannon KS, Margolskee RF, Sidman RL, Pugh EN, Jr, Makino CL, Lem J. Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha-subunit. Proc Natl Acad Sci U S A. 2000;97:13913–13918. doi: 10.1073/pnas.250478897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Kolesnikov AV, Masuho I, Kefalov VJ, Martemyanov KA. Membrane anchoring subunits specify selective regulation of RGS9.Gbeta5 GAP complex in photoreceptor neurons. J Neurosci. 2010;30:13784–13793. doi: 10.1523/JNEUROSCI.1191-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV, Benovic JL. G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol. 1998;8:335–344. doi: 10.1016/S0959-4388(98)80058-5. [DOI] [PubMed] [Google Scholar]
- Chang B, Dacey MS, Hawes NL, Hitchcock PF, Milam AH, Atmaca-Sonmez P, Nusinowitz S, Heckenlively JR. Cone photoreceptor function loss-3, a novel mouse model of achromatopsia due to a mutation in Gnat2. Invest Ophthalmol Vis Sci. 2006;47:5017–5021. doi: 10.1167/iovs.05-1468. [DOI] [PubMed] [Google Scholar]
- Chen J, Makino CL, Peachey NS, Baylor DA, Simon MI. Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH- terminal truncation mutant. Science. 1995;267:374–377. doi: 10.1126/science.7824934. [DOI] [PubMed] [Google Scholar]
- Chen J, Shi G, Concepcion FA, Xie G, Oprian D. Stable rhodopsin/arrestin complex leads to retinal degeneration in a transgenic mouse model of autosomal dominant retinitis pigmentosa. J Neurosci. 2006;26:11929–11937. doi: 10.1523/JNEUROSCI.3212-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concepcion F, Chen J. Q344ter mutation causes mislocalization of rhodopsin molecules that are catalytically active: a mouse model of Q344ter-induced retinal degeneration. PLoS One. 2010;5:e10904. doi: 10.1371/journal.pone.0010904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornwall MC, Fain GL. Bleached pigment activates transduction in isolated rods of the salamander retina. J Physiol. 1994;480:261–279. doi: 10.1113/jphysiol.1994.sp020358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornwall MC, Fein A, MacNichol EF., Jr Cellular mechanisms that underlie bleaching and background adaptation. J Gen Physiol. 1990;96:345–372. doi: 10.1085/jgp.96.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cringle SJ, Yu DY, Yu PK, Su EN. Intraretinal oxygen consumption in the rat in vivo. Invest Ophthalmol Vis Sci. 2002;43:1922–1927. [PubMed] [Google Scholar]
- Dacheux RF, Raviola E. The rod pathway in the rabbit retina: a depolarizing bipolar and amacrine cell. J Neurosci. 1986;6:331–345. doi: 10.1523/JNEUROSCI.06-02-00331.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan T, Mendez A, Detwiler PB, Chen J, Rieke F. Multiple phosphorylation sites confer reproducibility of the rod's single-photon responses. Science. 2006;313:530–533. doi: 10.1126/science.1126612. [DOI] [PubMed] [Google Scholar]
- Doan T, Azevedo AW, Hurley JB, Rieke F. Arrestin competition influences the kinetics and variability of the single-photon responses of mammalian rod photoreceptors. J Neurosci. 2009;29:11867–11879. doi: 10.1523/JNEUROSCI.0819-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field GD, Rieke F. Nonlinear signal transfer from mouse rods to bipolar cells and implications for visual sensitivity. Neuron. 2002;34:773–785. doi: 10.1016/S0896-6273(02)00700-6. [DOI] [PubMed] [Google Scholar]
- Frederiksen R, Boyer NP, Nickle B, Chakrabarti KS, Koutalos Y, Crouch RK, Oprian D, Cornwall MC. Low aqueous solubility of 11-cis-retinal limits the rate of pigment formation and dark adaptation in salamander rods. J Gen Physiol. 2012;139:493–505. doi: 10.1085/jgp.201110685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govardovskii VI, Fyhrquist N, Reuter T, Kuzmin DG, Donner K. In search of the visual pigment template. Vis Neurosci. 2000;17:509–528. doi: 10.1017/S0952523800174036. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Hanson SM, Song X, Vishnivetskiy SA, Gurevich EV. The functional cycle of visual arrestins in photoreceptor cells. Prog Retin Eye Res. 2011;30:405–430. doi: 10.1016/j.preteyeres.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen H, Vinberg F, Pitkänen M, Kommonen B, Koskelainen A. Flash responses of mouse rod photoreceptors in the isolated retina and corneal electroretinogram: comparison of gain and kinetics. Invest Ophthalmol Vis Sci. 2012;53:5653–5664. doi: 10.1167/iovs.12-9678. [DOI] [PubMed] [Google Scholar]
- Hurley JB, Spencer M, Niemi GA. Rhodopsin phosphorylation and its role in photoreceptor function. Vision Res. 1998;38:1341–1352. doi: 10.1016/S0042-6989(97)00459-8. [DOI] [PubMed] [Google Scholar]
- Hurley JB, Lindsay KJ, Du J. Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J Neurosci Res. 2015;93:1079–1092. doi: 10.1002/jnr.23583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Lee KA, Niemi GA, Craven KB, Garwin GG, Saari JC, Hurley JB. Multiple phosphorylation of rhodopsin and the in vivo chemistry underlying rod photoreceptor dark adaptation. Neuron. 2001;31:87–101. doi: 10.1016/S0896-6273(01)00340-3. [DOI] [PubMed] [Google Scholar]
- Keresztes G, Martemyanov KA, Krispel CM, Mutai H, Yoo PJ, Maison SF, Burns ME, Arshavsky VY, Heller S. Absence of the RGS9.Gbeta5 GTPase-activating complex in photoreceptors of the R9AP knockout mouse. J Biol Chem. 2004;279:1581–1584. doi: 10.1074/jbc.C300456200. [DOI] [PubMed] [Google Scholar]
- Kühn H. Light-dependent phosphorylation of rhodopsin in living frogs. Nature. 1974;250:588–590. doi: 10.1038/250588a0. [DOI] [PubMed] [Google Scholar]
- Kühn H, McDowell JH. Isoelectric focusing of phosphorylated cattle rhodopsin. Biophys Struct Mech. 1977;3:199–203. doi: 10.1007/BF00535820. [DOI] [PubMed] [Google Scholar]
- Lau JC, Linsenmeier RA. Oxygen consumption and distribution in the Long-Evans rat retina. Exp Eye Res. 2012;102:50–58. doi: 10.1016/j.exer.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KA, Nawrot M, Garwin GG, Saari JC, Hurley JB. Relationships among visual cycle retinoids, rhodopsin phosphorylation, and phototransduction in mouse eyes during light and dark adaptation. Biochemistry. 2010;49:2454–2463. doi: 10.1021/bi1001085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ. Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol Sci. 2004;25:413–422. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Lindsay KJ, Du J, Sloat SR, Contreras L, Linton JD, Turner SJ, Sadilek M, Satrústegui J, Hurley JB. Pyruvate kinase and aspartate-glutamate carrier distributions reveal key metabolic links between neurons and glia in retina. Proc Natl Acad Sci U S A. 2014;111:15579–15584. doi: 10.1073/pnas.1412441111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao W, Miyagishima KJ, Yao Y, Soreghan B, Sampath AP, Chen J. Functional comparison of rod and cone Galpha(t) on the regulation of light sensitivity. J Biol Chem. 2013;288:5257–5267. doi: 10.1074/jbc.M112.430058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Roca A, Lem J, Wu LW, Simon MI, Baylor DA, Chen J. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 2000;28:153–164. doi: 10.1016/S0896-6273(00)00093-3. [DOI] [PubMed] [Google Scholar]
- Miller JA, Paulsen R, Bownds MD. Control of light-activated phosphorylation in frog photoreceptor membranes. Biochemistry. 1977;16:2633–2639. doi: 10.1021/bi00631a009. [DOI] [PubMed] [Google Scholar]
- Miller JL, Fox DA, Litman BJ. Amplification of phosphodiesterase activation is greatly reduced by rhodopsin phosphorylation. Biochemistry. 1986;25:4983–4988. doi: 10.1021/bi00366a002. [DOI] [PubMed] [Google Scholar]
- Moaven H, Koike Y, Jao CC, Gurevich VV, Langen R, Chen J. Visual arrestin interaction with clathrin adaptor AP-2 regulates photoreceptor survival in the vertebrate retina. Proc Natl Acad Sci U S A. 2013;110:9463–9468. doi: 10.1073/pnas.1301126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nymark S, Heikkinen H, Haldin C, Donner K, Koskelainen A. Light responses and light adaptation in rat retinal rods at different temperatures. J Physiol. 2005;567:923–938. doi: 10.1113/jphysiol.2005.090662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nymark S, Frederiksen R, Woodruff ML, Cornwall MC, Fain GL. Bleaching of mouse rods: microspectrophotometry and suction-electrode recording. J Physiol. 2012;590:2353–2364. doi: 10.1113/jphysiol.2012.228627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohguro H, Van Hooser JP, Milam AH, Palczewski K. Rhodopsin phosphorylation and dephosphorylation in vivo. J Biol Chem. 1995;270:14259–14262. doi: 10.1074/jbc.270.24.14259. [DOI] [PubMed] [Google Scholar]
- Pugh EN, Jr, Lamb TD. Amplification and kinetics of the activation steps in phototransduction. Biochim Biophys Acta. 1993;1141:111–149. doi: 10.1016/0005-2728(93)90038-H. [DOI] [PubMed] [Google Scholar]
- Ramulu P, Kennedy M, Xiong WH, Williams J, Cowan M, Blesh D, Yau KW, Hurley JB, Nathans J. Normal light response, photoreceptor integrity, and rhodopsin dephosphorylation in mice lacking both protein phosphatases with EF hands (PPEF-1 and PPEF-2) Mol Cell Biol. 2001;21:8605–8614. doi: 10.1128/MCB.21.24.8605-8614.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieke F. Mechanisms of single-photon detection in rod photoreceptors. Methods Enzymol. 2000;316:186–202. doi: 10.1016/S0076-6879(00)16724-2. [DOI] [PubMed] [Google Scholar]
- Sampath AP, Rieke F. Selective transmission of single photon responses by saturation at the rod-to-rod bipolar synapse. Neuron. 2004;41:431–443. doi: 10.1016/S0896-6273(04)00005-4. [DOI] [PubMed] [Google Scholar]
- Sokolov M, Lyubarsky AL, Strissel KJ, Savchenko AB, Govardovskii VI, Pugh EN, Jr, Arshavsky VY. Massive light-driven translocation of transducin between the two major compartments of rod cells: a novel mechanism of light adaptation. Neuron. 2002;34:95–106. doi: 10.1016/S0896-6273(02)00636-0. [DOI] [PubMed] [Google Scholar]
- Song X, Vishnivetskiy SA, Seo J, Chen J, Gurevich EV, Gurevich VV. Arrestin-1 expression level in rods: balancing functional performance and photoreceptor health. Neuroscience. 2011;174:37–49. doi: 10.1016/j.neuroscience.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang SH, Burns ME, Calvert PD, Gouras P, Baylor DA, Goff SP, Arshavsky VY. Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science. 1998;282:117–121. doi: 10.1126/science.282.5386.117. [DOI] [PubMed] [Google Scholar]
- Watson AJ, Aragay AM, Slepak VZ, Simon MI. A novel form of the G protein beta subunit Gbeta5 is specifically expressed in the vertebrate retina. J Biol Chem. 1996;271:28154–28160. doi: 10.1074/jbc.271.45.28154. [DOI] [PubMed] [Google Scholar]
- Whelan JP, McGinnis JF. Light-dependent subcellular movement of photoreceptor proteins. J Neurosci Res. 1988;20:263–270. doi: 10.1002/jnr.490200216. [DOI] [PubMed] [Google Scholar]
- Wilden U. Duration and amplitude of the light-induced cGMP hydrolysis in vertebrate photoreceptors are regulated by multiple phosphorylation of rhodopsin and by arrestin binding. Biochemistry. 1995;34:1446–1454. doi: 10.1021/bi00004a040. [DOI] [PubMed] [Google Scholar]
- Wilden U, Hall SW, Kühn H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci U S A. 1986;83:1174–1178. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff ML, Lem J, Fain GL. Early receptor current of wild-type and transducin knockout mice: photosensitivity and light-induced Ca2+ release. J Physiol. 2004;557:821–828. doi: 10.1113/jphysiol.2004.064014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Dodd RL, Makino CL, Simon MI, Baylor DA, Chen J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997;389:505–509. doi: 10.1038/39068. [DOI] [PubMed] [Google Scholar]
- Yamaoka H, Tachibanaki S, Kawamura S. Dephosphorylation during bleach and regeneration of visual pigment in carp rod and cone membranes. J Biol Chem. 2015;290:24381–24390. doi: 10.1074/jbc.M115.674101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DY, Cringle SJ. Oxygen distribution in the mouse retina. Invest Ophthalmol Vis Sci. 2006;47:1109–1112. doi: 10.1167/iovs.05-1118. [DOI] [PubMed] [Google Scholar]
- Zhuang T, Chen Q, Cho MK, Vishnivetskiy SA, Iverson TM, Gurevich VV, Sanders CR. Involvement of distinct arrestin-1 elements in binding to different functional forms of rhodopsin. Proc Natl Acad Sci U S A. 2013;110:942–947. doi: 10.1073/pnas.1215176110. [DOI] [PMC free article] [PubMed] [Google Scholar]