

Abstract
Delivery of proteins and organelles to the growth cone during axon extension relies on anterograde transport by kinesin motors. Though critical for neural circuit development, the mechanisms of cargo-specific anterograde transport during axon extension are only starting to be explored. Cargos of particular importance for axon outgrowth are microtubule modifiers, such as SCG10 (Stathmin-2). SCG10 is expressed solely during axon extension, localized to growth cones, and essential for axon outgrowth; however, the mechanisms of SCG10 transport and activity were still debated. Using zebrafish mutants and in vivo imaging, we identified the Kif1B motor and its interactor Kif1 binding protein (KBP) as critical for SCG10 transport to axon growth cones and complete axon extension. Axon truncation in kbpst23 mutants can be suppressed by SCG10 overexpression, confirming the direct relationship between decreased SCG10 levels and failed axon outgrowth. Live imaging revealed that the reduced levels of SCG10 in kbpst23 mutant growth cones led to altered microtubule stability, defining the mechanistic basis of axon truncation. Thus, our data reveal a novel role for the Kif1B-KBP complex in the anterograde transport of SCG10, which is necessary for proper microtubule dynamics and subsequent axon extension.
SIGNIFICANCE STATEMENT Together, our data define the mechanistic underpinnings of failed axon outgrowth with loss of KBP or its associated motor, Kif1B. In addition, we provide conclusive evidence that this defect results from disruption of anterograde transport of SCG10. This is one of the first examples of a motor to be implicated in the essential transport of a discreet cargo necessary for axon extension. In addition, counter to previous in vitro and cell culture results, neither loss of the Kif1B motor nor KBP resulted in inhibition of mitochondrial transport. Altogether, our work links transport of SCG10 to the regulation of microtubule dynamics in the axon growth cone and enhances our understanding of this process during axon outgrowth.
Keywords: axon extension, KBP, KIF1B, SCG10, stathmin
Introduction
Precise regulation of microtubule dynamics is necessary for axon outgrowth. This process is modulated by a number of factors including the local concentration of free tubulin, microtubule severing proteins, and proteins that facilitate microtubule stabilization and destabilization. Disruption of microtubule dynamics by pharmacologically increasing stability or instability of the assembled structure results in failed axon outgrowth (Letourneau and Ressler, 1984; Tanaka et al., 1995). Microtubules are particularly dynamic in the growth cones of extending axons. During axon outgrowth, the half-life of distal microtubules is measured in minutes; in contrast, microtubules in mature axons are maintained for hours or days (Sabry et al., 1991; Hammond et al., 2010). Together, the high dynamicity of microtubules in axons during extension and the necessity of this process for axon elongation suggest that tight regulatory mechanisms must exist to modulate these microtubule dynamics during neural development.
One mechanism of microtubule dynamics regulation is the presence of microtubule destabilizing proteins, such as SCG10. SCG10 (superior cervical ganglion-10; also known as Stathmin-2) is a member of the stathmin family of proteins (Grenningloh et al., 2004; Morii et al., 2006). This protein binds tubulin dimers to titrate tubulin from growing microtubules, thereby decreasing stability (Fleury et al., 2000; Charbaut et al., 2001; Grenningloh et al., 2004; Morii et al., 2006). Consequently, increased levels of SCG10 can rescue axon truncation caused by microtubule stabilizing drugs (Riederer et al., 1997). SCG10 is carried to growth cones by anterograde transport after being packaged into vesicles in the Golgi (Lutjens et al., 2000; Shin et al., 2012). Work in cultured dorsal root ganglion neurons demonstrated that SCG10 is rapidly degraded once in axon growth cones (Shin et al., 2012). Consequently, the maintenance of SCG10 levels in growth cones requires active transport of this protein throughout axon outgrowth.
Kinesin-1 is the most well studied anterograde motor protein responsible for transporting cargo from the neuronal cell body toward axon terminals (Hirokawa et al., 2009, 2010). Although this motor transports a range of cargos in mature axons (Hirokawa et al., 2009), it initiates neurite outgrowth through facilitating microtubule sliding (Ferreira et al., 1992; Lu et al., 2013). Notably, in addition to Kinesin-1, there are 13 other kinesin families (encoded by 42 genes in humans), largely composed of plus-end directed motors. The function of other kinesin motors in axon elongation is largely unknown.
Recently, Kif1B, a Kinesin-3 family member, was shown to have a role in axon extension. Loss of Kif1B in zebrafish results in truncation of the long axons of the peripheral nervous system and central nervous system due to stalled outgrowth (Lyons et al., 2009). Kif1 binding protein (KBP) is a Kif1B interactor, which also functions in axon extension (Wozniak et al., 2005; Lyons et al., 2008). The function of the Kif1B-KBP complex in axon outgrowth was predicted to rely on either a function in mitochondrial transport or through its interaction with the microtubule modifier SCG10 (Alves et al., 2010). However, the mechanistic underpinnings of Kif1B-KBP function in axon outgrowth remained unexplored.
Using live imaging of cargo transport and microtubule dynamics, we defined the function of the KBP-Kif1B complex in axon extension. Counter to previous work (Nangaku et al., 1994; Wozniak et al., 2005), neither Kif1B nor KBP regulate mitochondrial transport during axon extension; rather, these proteins work together to transport SCG10 to axonal growth cones. Axon extension in kbpst23 mutants can be rescued by supplying exogenous SCG10, showing a causal link between the loss of SCG10 and axon truncation. Finally, we determined that loss of SCG10 increases microtubule stability in the growth cone, altering the microtubule dynamics necessary for axon outgrowth. Altogether, our data suggest a model in which KBP-Kif1B interaction is necessary for SCG10 transport to axon growth cones where it modulates microtubule dynamics to facilitate axon outgrowth.
Materials and Methods
Zebrafish husbandry.
Adult *AB zebrafish were maintained at 28.5°C and staged as described previously (Kimmel et al., 1995). Embryos were derived from natural matings or in vitro fertilization, raised in embryo media, and developmentally staged using a standard staging series (Westerfield, 2000). Strains used included the TgBAC(neurod:EGFP)nl1 transgenic (Obholzer et al., 2008), and kbpst23 (Lyons et al., 2008) and kif1bst43 (Lyons et al., 2009) mutants. All analyses were done in embryonic and larval zebrafish, which have indeterminate sex at this stage.
Genomic DNA isolation and genotyping.
Genomic DNA was isolated from larvae by incubating it overnight at 55°C in PCR extraction buffer (10 mm Tris, pH 8, 2 mm EDTA, 0.2% Triton X-100, 200 μg/ml proteinase K). Homozygous kbpst23 and kif1bst43 mutants were identified using established genotyping protocols (Lyons et al., 2008, 2009).
Generation of the SCG10-2b-mRFP BAC and kbpΔkif1b mRNA and quantification of axon length.
We modified a SCG10-2b-containing bacterial artificial chromosome (BAC) clone by Escherichia coli-based homologous recombination (Suster et al., 2011). BAC clone CH211-182a19 contains 52 kb of sequence upstream and 92 kb downstream of scg10-2b. After recombination, the modified BAC clone contained a monomeric red fluorescent protein (mRFP) gene positioned in frame at the 3′ end of the last scg10-2b exon, following SV40 polyadenylation signal and kanamycin resistance gene. The kanamycin resistance gene was flanked by FLP sites and was removed by FLP-mediated recombination (Suster et al., 2011). The accuracy of mRFP integration and FLP recombination were evaluated by PCR, sequencing, and by transient expression assays.
kbp and kbpΔkif1b mRNA were synthesized using the SP6 mMessage Machine kit (Applied Biosystems). The templates were produced by cloning the open reading from zebrafish KBP (NM001013462) into a the gateway compatible middle entry vector, pDONRp2Rp3, and subsequently recombining it into the sp6:egfp-kbp plasmid in the pDEST394 destination vector as described previously (Kwan et al., 2007). Deletion of the previously defined Kif1B binding domain was performed on kbp in pDONRp2p3 using the QuikChange II Site-Directed Mutagenesis kit (Agilent). Both kbp and kbpΔkif1b were sequence confirmed before recombination into the destination vector.
For overexpression essays, 40 pg of BAC plasmid was injected in embryos derived from kbpst23/+ crosses. The posterior lateral line (pLL) nerve developed normally in the injected wild-type siblings. Following injections, embryos were raised to 3 d postfertilization (dpf) and processed for immunofluorescence with an anti-DsRed antibody (Fisher, 632496) to better visualize SCG10-monomeric red fluorescent protein (mRFP) under low-magnification. For analysis, embryos in which a pioneer axon was expressing SCG10-mRFP were mounted on a slide and imaged using a Zeiss Axioimager under a 50 × magnification. Body and axon length were quantified using ImageJ. The ability of kbp and kbpΔkif1b mRNA injection to rescue pLL nerve length was quantified using the same protocol.
Immunohistochemistry and LysoTracker staining.
Whole-mount immunohistochemistry was performed following established protocols (Ungos et al., 2003). The following antibodies were used: anti-GFP (1:1000; Invitrogen, A11122), anti-GFP (1:1000; Aves, GFP-1020), anti-zebrafish postsynaptic density marker (1:500; NeuroMab, clone N286/74), and anti-SCG10 (1:100; ProteinTech, 10586-AP; primary incubation 3 d at 4°C). For LysoTracker red vital dye staining, 26–28 h postfertilization (hpf) embryos were incubated in LysoTracker red (10 μm in 0.5% DMSO containing embryo media; ThermoFisher, L-7528) for 10 min in embryo media, washed briefly, embedded in 1.2% low-melt agarose, and imaged immediately using the same acquisition parameters for all. All fluorescently labeled embryos were imaged using a FV1000 laser scanning confocal system (Olympus). Bright-field or Nomarski microscopy images were collected using a Zeiss Imager Z1 system. Images were processed using ImageJ software (Abramoff et al., 2004) and Imaris (Bitplane). When defining acidic vesicles contained within the nerve in Imaris, the surface parameters for neurod:egfp-positive nerve and LysoTracker-positive vesicles, were kept constant for all samples analyzed. Brightness and contrast were adjusted in Adobe Photoshop and figures were compiled in Adobe Illustrator.
Axon transport analysis.
Analysis of axonal transport was performed as described previously (Drerup and Nechiporuk, 2013, 2016). DNA constructs used to drive expression of cargos were produced using Gateway technology (Kwan et al., 2007). For transport analysis, zygotes were injected with plasmid DNA encoding fluorescently tagged cargos of interest under control of the 5kbneurod promoter (Mo and Nicolson, 2011). Between 26 and 30 hpf or at 4 dpf, embryos or larvae were sorted under epifluorescence to identify individuals with tagged cargo expression in a subset of cells of the pLL ganglion. For imaging, embryos were mounted in 1.2% low-melting-point agarose on a glass coverslip, submerged in embryo media containing 0.02% tricaine and a 30–100 μm region was imaged using a 60×/NA = 1.2 water objective on an upright FV1000 confocal microscope (Olympus). Scans were taken at the fastest possible speed (3–5 frames/s) for 600–750 frames. Embryos were subsequently released from agarose and processed for genotyping.
Transport parameters were analyzed using kymograph analysis in the MetaMorph software package (Molecular Devices). Kymographs were generated from each imaging session and used to determine distance moved in individual bouts of movement (uninterrupted straight lines) and velocity of movement (slope of uninterrupted straight lines). Typically, 10–50 traces were analyzed in each kymograph and these were averaged within individual embryos for statistical analysis. The number of particles moving in each direction was determined based on traces on the kymographs and then normalized to length of axonal segment and total imaging time.
Quantification of immunofluorescence.
For analysis of SCG10 intensity in growth cones, individuals were immunolabeled and confocal z-stacks (0.5 μm between planes) were taken of the area of interest using a 60×/NA = 1.3 oil objective with identical settings. Images were analyzed using ImageJ (Abramoff et al., 2004). For fluorescence intensity measurements, summed projections of the regions-of-interest (ROIs) were generated only through the growth cone (identified using neurod:EGFP signal) and converted to 8 bit in ImageJ. Before statistical comparison, the mean background fluorescence intensity, measured in a region adjacent to the growth cone, was subtracted.
For quantification of SCG10 fold-change with plasmid and BAC transient transgenesis, embryos were immunolabeled for SCG10 and a zebrafish axonal marker (NeuroMab, clone N286/74) and imaged as described above. Fold-changes were determined using the ratio of SCG10 immunofluorescence in expressing (mEos2 or mRFP-positive) to non-expressing (mEos2 or mRFP-negative) growth cones.
Analysis of microtubule growth and stability.
For analysis of microtubule growth dynamics, zygotes were injected with DNA constructs encoding EB3 fused to GFP (end-binding protein 3 tagged with green fluorescent protein), expressed by a neural promotor (5kbneurod:eb3-gfp). This construct was made using a template previously used to study microtubule dynamics in zebrafish neurons (Hendricks and Jesuthasan, 2009). Embryos expressing EB3-GFP in a single neuron were mounted as described above for analysis of axonal transport. Confocal z-stacks were acquired through the distal axon and growth cone every 5 s for 15 min. Stacks were projected into a single plane for every time-point and EB3 comets were analyzed using kymograph analysis in MetaMorph as described above for analyses of axonal transport.
For analysis of microtubule stability, we expressed α-tubulin tagged with monomeric Eos2 in neurons (5kbneurod:mEos2-αtubulin). Similar constructs have been used previously to analyze microtubule stability in cultured hippocampal neurons (Hammond et al., 2010). A region surrounding a single expressing pioneer axon growth cone was selected at 30 hpf and a z-series was set to encompass the region. After a pre-conversion stack was acquired, Eos2 was converted using 20 scans of the region (10 μs/pixel dwell time) with a 405 nm laser at 5% power at the center of the defined z-stack. Immediately after, a z-stack (0.5 μm between planes) was acquired every minute for 20 min to analyze decay of red fluorescence using a 60×/NA1.2 water objective. Red fluorescence intensity was quantified at each time-point using a summed projected stack as described above for quantification of immunofluorescence.
Analysis of SCG10 transport.
To analyze transport of SCG10 from the cell body to the growth cone, we transiently expressed an mEos2-SCG10-2b fusion in pLL neurons using the 5kbneurod promotor (neurod5kb:mEos2-SCG10). Five picograms of neurod5kb:mEos2-SCG10-2b and 2.5 pg of neurod5kb:EGFP (cell fill that helped to delineate growth cones of pioneer axons) were coinjected into one-cell stage embryos. Following injections, embryos were raised in the dark (to minimize mEos2 conversion under ambient light) until 25–27 hpf, sorted for EGFP expression in the pLL, and mounted in 1.2% low-melting-point agarose. Before photo-conversion, we acquired a z-stack of EGFP/mEos2-positive pLL cell bodies and pioneer axon growth cones to determine background levels of photoconverted mEos2. mEos2 was photoconverted using 25 scans of the cell body (defined as ROIs in a single z-plane) at 10 μs/pixel dwell time with 405 nm laser set at 25% power. Immediately after conversion, we recorded the level of photoconverted mEos2 in the cell body. Eighty minutes later, we recorded levels of photoconverted mEos2 in the pioneer axon growth cones. All images were acquired as confocal z-stacks (1.0 μm between planes) using a 60×/NA = 1.2 water objective. Throughout the course of these experiments, the acquisition parameters for photoconverted mEos2 were kept constant. Quantification was performed using mean fluorescence intensity measurement function in ImageJ: summed projections of the regions of interest were generated only through cell bodies and growth cones (identified using neurod:EGFP signal) and converted to 16 bit in ImageJ. Before statistical comparison, the mean background fluorescence intensity, measured in a region adjacent to either the cell body or growth cone, was subtracted. To quantify changes of photoconverted mEos2 in growth cones, we subtracted the mean fluorescent intensity before conversion from the mean fluorescent intensity 80 min after photoconversion and normalized to photoconverted mEos2 in the pLL ganglion cell bodies. One wild-type embryo was excluded from analysis as it was outside of 3 SD from the mean.
Statistical analysis.
Statistical analysis was performed using the JMP or Prism6 software package. Data suitable for parametric analysis were analyzed using ANOVA, with Tukey–Kramer highly significant difference post hoc contrasts for more than two variables. Data not suitable for parametric analysis were analyzed using Wilcoxon rank sum analysis. To compare expected to observed ratios for embryo truncation analyses, χ2 tests were used. For analyses of fluorescence decay, one-phase decay curves were analyzed using Prism6 to determine significance of half-life and tau.
Results
Mitochondrial anterograde transport is independent of Kif1B function
We investigated the function of the Kif1B motor and its interacting protein, KBP, in axon outgrowth and axonal transport using the zebrafish posterior lateral line (pLL) sensory axons. The primary pLL organ develops early and rapidly in the zebrafish, with initial organ patterning and axon extension completed by 48 hpf (Fig. 1A,B; Ghysen and Dambly-Chaudière, 2004). During extension, pLL pioneer axons, the first to extend from the pLL ganglion, are guided through the trunk by a migrating primordium. The primordium, a group of ∼100 cells, is a source of growth factors, some of which have been shown to facilitate pLL axon outgrowth (Schuster et al., 2010). Importantly, primordium migration is normal in both kif1bst43 and kbpst23 mutant strains and axon extension defects in these strains have previously been shown to be cell autonomous (Lyons et al., 2008, 2009).
Figure 1.
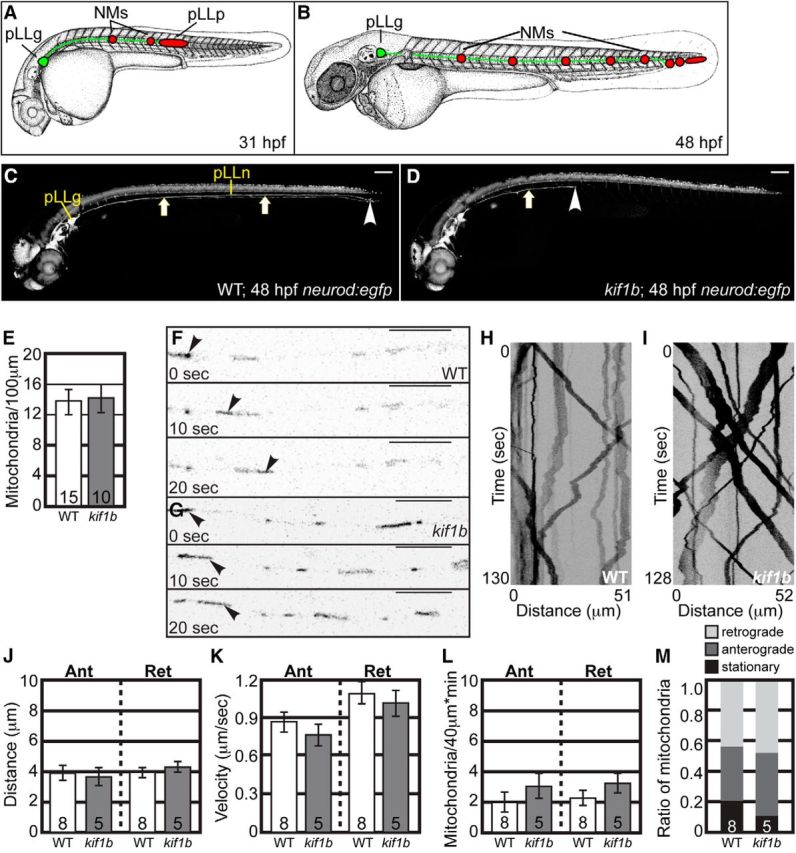
Mitochondrial transport is normal in kif1bst43 mutants. A, B, Schematic of pLL development. pLL pioneer axons (green) extend from neurons in the pLL ganglion (pLLg). The growth cones of pLL axons are embedded in the pLL primordium (red) which migrates along the trunk between 22 and 48 hpf. As it migrates, the pLL primordium deposits neuromasts (NMs; red) that will develop into the sensory organs of this system. Initial NM deposition and axon extension is complete by 48 hpf. C, D, Wild-type and kif1bst43 mutant neurod:egfp transgenic zebrafish embryos at 48 hpf. pLL axons (arrows) have fully extended by this time point in wild-types but fail to do so in kif1bst43 mutants. Scale bar, 100 μm. Arrowhead indicates the end of the nerve. E, Total mitochondria per 100 μm is unchanged with loss of Kif1B (ANOVA; p = 0.8776). F, G, Stills from Movies 1 and 2 showing mitochondrial transport in single pLL axons of wild-type (F) and kif1bst43 mutant (G) embryos between 24 and 28 hpf. Arrowheads point to a single mitochondria moving in the anterograde direction in each series. H, I, Kymographs of mitochondrial transport in wild-type (H) and kif1bst43 mutant (I) embryos. J, Distance of movement [ANOVA; Anterograde (Ant): p = 0.8426, Retrograde (Ret): p = 0.3128], (K) velocity of transport (ANOVA; Ant: p = 0.1664, Ret: p = 0.5649), (L) frequency of movement in either direction, and (M) mitochondrial population dynamics (ANOVA; Ant: p = 0.3520, Ret: p = 0.2734) are unchanged in kif1bst43 mutants. Numbers on graphs indicate sample size. Error bars represent standard error of the mean. Scale bars, 10 μm.
To visualize axon length and morphology in kif1bst43 zebrafish embryos, we crossed the mutation into the TgBAC(neurod:egfp)nl1 transgenic (hereafter referred to as neurod:egfp; Obholzer et al., 2008), which labels neurons with cytoplasmic enhanced green fluorescent protein (EGFP). The kif1bst43 mutation interrupts both α and β isoforms of this motor and has been shown to be a complete loss of function allele (Lyons et al., 2009). Mutant kif1bst43 embryos displayed truncated pLL nerves, with axons extending only 30–50% through the trunk (Fig. 1C,D; Lyons et al., 2009). To determine whether mitochondrial localization was affected by loss of Kif1B, we labeled mitochondria in single axons of kif1bst43 mutants and wild-type siblings by transient, plasmid-based expression of a mitochondrial targeting sequence tagged with the red fluorescent protein TagRFP (5kbneurod:mito-TagRFP; modified from Fang et al., 2012). Embryos were fixed at 30 hpf and the number of mitochondria per hundred micrometers was counted. This analysis revealed no change in total mitochondrial number in kif1bst43 pLL axons (Fig. 1E).
To determine whether Kif1B functions in mitochondrial movement, we performed in vivo imaging of mitochondrial transport before stalled axon outgrowth, ie, before the primordium has separated from the pioneer axon growth cones, ∼28 hpf in this strain. As growth factor signaling can modulate mitochondrial transport (Chada and Hollenbeck, 2004), we wanted to image the movement of this organelle in mutants before the growth cone detached from the primordium, a source of several growth factors during pLL axon extension (Nechiporuk and Raible, 2008; Schuster et al., 2010). To determine whether mitochondrial transport was perturbed with loss of Kif1B, mitochondria were labeled by expression of the 5kbneurod:mito-TagRFP construct and transport was imaged in individual pLL axons in real time between 24 and 28 hpf according to our established protocols (Fig. 1F,G; Movies 1, 2; Drerup and Nechiporuk, 2013, 2016). Kymograph analysis of mitochondrial transport revealed no change in the distance moved or velocity of movement (Fig. 1H–K). Additionally, we analyzed the frequency of mitochondrial movement in the anterograde and retrograde directions in our movies and found no change in the number moving in either direction (Fig. 1L). Finally, analyses of population dynamics, ie, the proportion of mitochondria moving in the anterograde or retrograde direction or stationary showed no change in this measure of mitochondrial dynamics in kif1bst43 mutants (Fig. 1M). Together, our data demonstrates no role for Kif1B in mitochondrial transport.
Mitochondrial transport in a pLL axon of a kif1bst43 wild-type sibling at ∼30 hpf. Mitochondria were tagged with TagRFP using a mitochondrial targeting sequence in neurons using the 5kbneurod:mito-TagRFP plasmid. Anterograde transport is from left to right. Scale bar, 10 μm. 
Mitochondrial transport in kif1bst43 mutant pLL axon at ∼30 hpf. Mitochondria were tagged with TagRFP using a mitochondrial targeting sequence and expressed in neurons using the 5kbneurod:mito-TagRFP plasmid. Anterograde transport is from left to right. Scale bar, 10 μm. 
As a positive control, we analyzed the transport of a known Kif1B-dependent anterograde cargo, synaptic vesicle precursors labeled by the protein Synaptophysin (Zhao et al., 2001; Niwa et al., 2008). For these experiments, Synaptophysin tagged with mRFP was expressed in pLL neurons using zygotic injection of a 5kbneurod:Synaptophysin-mRFP plasmid. Kymograph analysis demonstrated active Synaptophysin transport in wild-type embryos between 24 and 28 hpf and decreased movement in kif1bst43 mutants (Fig. 2A,B). Analysis of transport parameters demonstrated decreased anterograde distance and velocity as well as decreased numbers of Synaptophysin-labeled vesicles moving in the anterograde and retrograde directions in kif1bst43 mutants (Fig. 2C–E). The decrease in retrograde Synaptophysin-positive puncta is an expected consequence of the strong decrease in anterograde synaptic vesicle transport into the axon. Together, these experiments confirm that, unlike synaptic vesicle precursors, mitochondria do not depend upon Kif1B for anterograde transport.
Figure 2.

Transport of synaptic vesicle precursors is disrupted in kif1bst43 mutants. A, B, Wild-type and kif1bst43 mutant kymographs representing synaptic vesicle precursor (labeled by Synaptophysin-mRFP) transport in pLL axons. Schematic of wild-type and kif1bst43 mutant kymographs illustrating anterograde (black) and retrograde (gray) traces. C, D, Anterograde distance and velocity of Synaptophysin transport is decreased in kif1bst43 mutants (ANOVA; sample size indicated on graphs). E, Number of Synaptophysin labeled vesicles moving in the anterograde and retrograde directions is decreased in kif1bst43 mutants (ANOVA). Error bars represent standard error of the mean.
KBP, a Kif1B interactor, is not required for mitochondrial transport
KBP has been shown to interact with Kif1B and proposed to be a necessary factor for mitochondrial localization and transport through this interaction (Wozniak et al., 2005). To determine KBP's role in mitochondrial transport and axon extension, we performed similar analyses in kbpst23 mutants. In this strain, axon extension fails before ∼36 hpf when pioneer axons have extended ∼60–75% through the trunk as previously reported (Fig. 3A,B; Lyons et al., 2008). These axons, as in kif1bst43 mutants, fail to recover and never innervate the sensory organs of the distal trunk and tail.
Figure 3.

Mitochondrial transport is normal in kbpst23 mutant pLL axons between 24 and 32 hpf. A, B, Wild-type and kbpst23 mutant neurod:egfp transgenic zebrafish embryos at 48 hpf. pLL axons have fully extended by this time point in wild-types but fail to do so in kbpst23 mutants. Arrows point to the pLL nerve; arrowhead indicates the end of the pLL nerve. Scale bar, 100 μm. C, D, Stills from imaging sessions showing anterograde transport in individual axons. Arrowheads point to an individual mitochondria moving in a wild-type (C) and kbpst23 mutant (D) axon. E, F, Kymographs representing wild-type (E) and kbpst23 mutant (F) mitochondrial transport. E′, F′, Schematic of wild-type and kbpst23 mutant kymographs illustrating anterograde (black) and retrograde (gray) traces. G, Distance of transport [ANOVA; Anterograde (Ant): p = 0.9662, Retrograde (Ret): p = 0.9704], (H) velocity of movement (ANOVA; Ant: p = 0.2315, Ret: p = 0.5849), (I) mitochondrial transport frequency (ANOVA; Total: p = 0.7952, Ant: p = 0.2558, Ret: p = 0.2766), and (J) mitochondria population dynamics are similar between wild-type and kbpst23 mutants during axon extension. Numbers on graphs indicate sample size. Error bars represent standard error of the mean. Scale bars, 10 μm.
To determine whether KBP functions in mitochondrial movement, we performed in vivo imaging of mitochondrial transport between 24 and 32 hpf. These assays demonstrated active mitochondrial movement in both kbpst23 mutants and wild-type siblings between 24 and 32 hpf (Fig. 3C,D). Kymograph analysis revealed no change in distance or velocity of mitochondrial transport in kbpst23 mutants (Fig. 3E–H). To determine whether the frequency of mitochondria moving in kbpst23 mutant axons was altered, the number of mitochondria moving in the anterograde or retrograde direction was counted in a 40 μm axonal section in each movie. This revealed no change in the frequency of mitochondrial movement in either direction (Fig. 3I). Similarly, when population dynamics were assayed, there was no change in the proportion of stationary, anterograde or retrograde pools of mitochondria in kbpst23 mutants (Fig. 3J). Together, these experiments demonstrate that, similar to our work with Kif1B, mitochondrial transport in extending axons is independent of KBP function.
Lysosome localization and transport is unperturbed in kbpst23 mutants
To determine whether other cargo was also normally transported, we analyzed the localization and transport of lysosomes in kbpst23 mutant axons. First, acidified vesicles were labeled with LysoTracker red in intact kbpst23 mutants and wild-type siblings and their localization was assayed in pLL axons at 28 hpf. This analysis revealed no change in the presence of acidic vesicles in kbpst23 mutants (Fig. 4A–C). To confirm that the transport of lysosomes was unaffected, we marked late endosomes and lysosomes by expressing Lamp1 (Lysosome-associated membrane protein 1) tagged with mRFP using zygotic injection of the 5kbneurod:lamp1-mRFP plasmid (Drerup and Nechiporuk, 2013). Kymograph analysis demonstrated that, though there is a decrease in the number of lysosomes moving in either direction in these axons, the ability of motors to transport lysosomes, assayed by distance and velocity of movement, was unaffected by loss of KBP (Fig. 3D–H). These data supports the conclusion that axonal transport, particularly that of mitochondria, is largely intact in kbpst23 mutants before axon truncation.
Figure 4.

Lysosome transport is normal in kbpst23 mutants during axon extension. A, B, Representative images of neurod:egfp-positive wild-type (A) and kbpst23 mutant (B) embryos that were labeled with LysoTracker red and imaged live at 26–28 hpf. Top, neurod:egfp-positive pLL nerve, just proximal to the growth cone. Middle, LysoTracker red-positive labeling in the same area showing positive signal in all cells. Bottom, Imaris software was used to identity LysoTracker-positive vesicles (magenta) that were enclosed within the pLL nerve (green). Scale bar, 15 μm. C, Quantitation of LysoTracker-positive vesicles within the pLL nerve in wild-type and kbpst23 mutant embryos. y-Axes show total vesicle volume (μm3) normalized to the total nerve volume (μm3); these ratios are not significantly different between wild-type (n = 18) and kbpst23 mutants (n = 6; Student's t test). D, E, Kymographs representing lysosome transport in wild-type (D) and kbpst23 mutant (E) pLL axons. F, Quantification of moving lysosomes reveals a decrease in the number moving in the anterograde and retrograde directions (ANOVA). G, H, Distance and velocity of lysosome transport are normal in kbpst23 mutants (ANOVA). Numbers on graphs represent sample size. Error bars represent standard error of the mean.
Axonal transport of mitochondria and lysosomes is disrupted in kbpst23 mutant axons at later stages
In contrast to our work, a previous study demonstrated that KBP loss had an effect on mitochondrial localization (Wozniak et al., 2005). To reconcile these differences, we set out to determine whether there was a temporal effect on axonal cargo transport with loss of KBP. A temporal difference may indicate the reliance on KBP for transport of various cargos at distinct stages. Alternatively, loss of KBP may impact the development of microtubule cytoskeleton, disruption of which indirectly affects axonal transport at later stages. First, we analyzed mitochondrial transport at 4 dpf in kbpst23 mutants, well after axon extension has failed. Kymograph analysis of mitochondrial motility revealed decreased distance moved in the anterograde and retrograde directions in kbpst23 mutants (Fig. 5A–C). The limited mitochondrial movement that did occur was at a normal velocity (Fig. 5D). In addition, analysis of mitochondrial population dynamics revealed a shift toward primarily stationary mitochondria (Fig. 5E). These data show that mitochondrial motility is disrupted in kbpst23 mutants at later stages.
Figure 5.
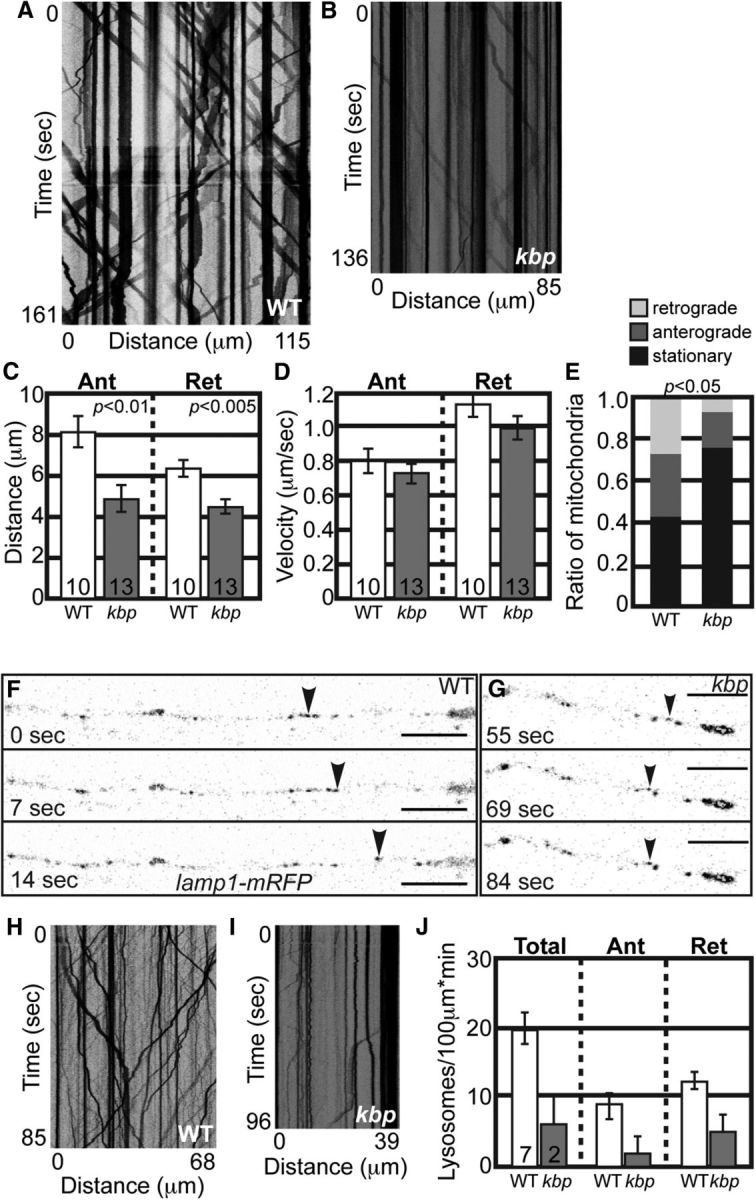
Mitochondrial and lysosomal transport is disrupted in kbpst23 mutants at 4 dpf. A, B, Kymographs representing mitochondrial transport in a wild-type (A) and kbpst23 mutant (B) at 4 dpf. C, Distance moved by mitochondria in both directions is decreased at 4 dpf (ANOVA). D, Velocity of transport is unchanged (ANOVA; Ant: p = 0.3971, Ret: p = 0.3275). E, Population dynamics show a shift to primarily stationary mitochondria at 4 dpf in kbpst23 mutants (ANOVA; p < 0.05). F, G, Stills from imaging sessions analyzing Lamp1-mRFP transport in a kbpst23 mutant (G) and a wild-type sibling (F) at 4 dpf. H, I, Kymographs representing lysosome transport. J, Total lysosome number and the number moving in either direction are decreased in kbpst23 mutants. There were too few moving lysosomes to calculate statistics of transport parameters, including distance and velocity of movement. Error bars represent standard error of the mean. Scale bars, 10 μm.
To determine whether the transport of other cargos was disrupted in kbpst23 at 4 dpf, we analyzed lysosomal transport at 4 dpf. Analysis of Lamp1-positive vesicles demonstrated active lysosome movement in wild-type siblings at 4 dpf, but almost no movement in kbpst23 mutants (Fig. 5F–J). Indeed, only 2 of 10 assayed kbpst23 larvae showed any lysosomal movement whatsoever, making statistical analysis of transport distance and velocity impossible. Together, these experiments suggest that KBP may not directly participate in mitochondrial transport in axons. Rather, the abnormal mitochondrial localization and movement in kbpst23 mutants in mature axons may be due to a more global disruption of axonal transport, though we cannot rule out developmental differences in the reliance of mitochondrial and lysosomal transport on KBP.
KBP and Kif1B function together to mediate SCG10 anterograde transport, which is necessary for axon extension
Abnormalities in the microtubule cytoskeleton, particularly the dynamic assembly and disassembly of this structure during axon extension, can dramatically impact axonal outgrowth and could cause cytoskeletal abnormalities that impact microtubule-based transport. One powerful modulator of microtubule dynamics is the stathmin-like protein SCG10 (Riederer et al., 1997; Grenningloh et al., 2004; Morii et al., 2006). As KBP has been shown to interact with SCG10 in a yeast two-hybrid assay (Alves et al., 2010), we asked whether SCG10 was properly localized in extending kbpst23 and kif1bst43 mutant pLL axons. Immunolabeling for SCG10 revealed a substantial loss of this protein from the growth cones of both mutants (Fig. 6A–D,F).
Figure 6.
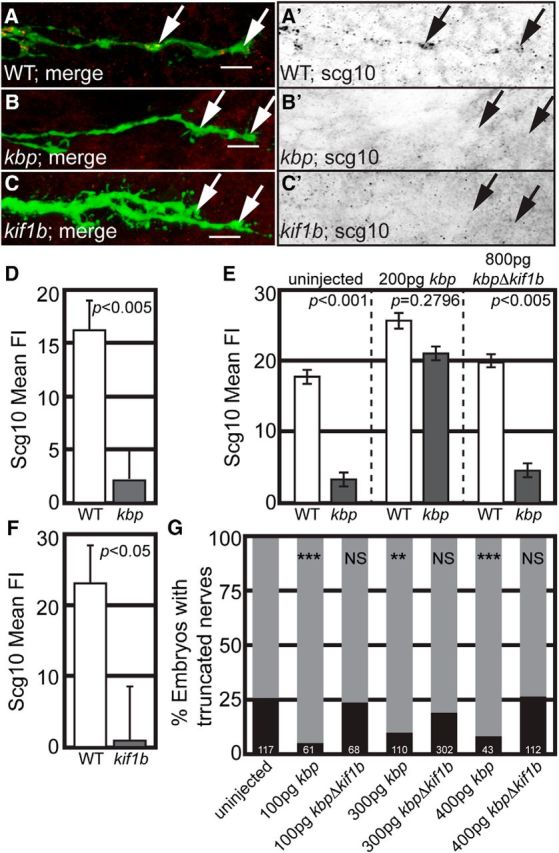
KBP interaction with Kif1B is essential for SCG10 growth cone localization and axon extension. A–C, SCG10 is absent in both kbpst23 and kif1bst43 mutant pLL axon growth cones during axon extension. A′–C′ show SCG10 immunofluorescence alone. Arrows point to growth cones. D, F, Quantification of SCG10 mean fluorescence intensity in kbpst23 (D) and kif1bst43 (F) mutants compared with wild-type siblings shows a dramatic reduction (background subtracted) in SCG10 protein levels in mutant growth cones (ANOVA). E, Whereas expression of full-length KBP in kbpst23 mutants rescues SCG10 mean fluorescence intensity, a version of KBP lacking the Kif1B binding domain (kbpΔkif1b) fails to do so (ANOVA). G, Expression of full-length KBP but not KBPΔKif1B rescues axon length in kbpst23 mutants. Zygotes were injected with mRNA encoding either KBP or KBPΔKif1B and larvae were scored at 3 dpf as either having truncated or full-length pLL nerves. The percentage of larvae with truncated (black bar) or full-length (gray bar) nerves in each experimental group is depicted. Twenty-five percent of larvae are expected to show axon truncation from a heterozygous cross, as observed in the uninjected controls (χ2; ***p < 0.0001, **p < 0.005, NS, not significant). Error bars represent standard error of the mean.
KBP and Kif1B could be working together or in parallel pathways to properly localize SCG10 to growth cones. To determine whether KBP-Kif1B interaction was necessary for proper localization of SCG10, we expressed wild-type KBP or a variant that lacks the previously defined Kif1B binding domain (KBPΔKif1B; Wozniak et al., 2005) in kbpst23 mutants and analyzed SCG10 localization in extending axons (30 hpf). Antibody labeling for SCG10 demonstrated that wild-type kbp mRNA rescued SCG10 immunofluorescence while kbpΔkif1b did not (Fig. 6E), supporting a role for KBP-Kif1B interaction in SCG10 localization to axonal growth cones. Next, we asked whether this interaction impacted axon outgrowth by analyzing the ability of this deletion construct to rescue axon length. Zygotes from a kbpst23/+ heterozygous cross were injected with mRNA encoding either KBP or KBPΔKif1B, raised to 3 dpf (well after axon extension complete), and larvae were scored for pLL axon extension phenotypes (truncated versus full-length). As expected, ∼25% of uninjected larvae from this cross-displayed axon truncation. Full-length kbp mRNA injection suppressed axon truncation in kbpst23 mutants (<25% display axon truncation), but kbpΔkif1b did not (∼25% display axon truncation; Fig. 6G). These results support a function for KBP-Kif1B interaction in both the proper localization of SCG10 to growth cones and axon elongation.
As Kif1B is an anterograde motor protein, we next asked whether the anterograde transport of SCG10 was altered with loss of the Kif1B interactor, KBP, using fluorescence accumulation after photoconversion. SCG10-2b tagged with the green to red monomeric photoconvertible protein mEos2 was expressed in wild-type and kbpst23 mutant pLL neurons using 5kbneurod:mEos2-SCG10-2b plasmid injection. This plasmid-based expression elevated levels of SCG10 in the growth cone ∼5-fold in both wild-type and mutants relative to non-expressing growth cones in the same animal (Fig. 7F). A 5kbneurod:egfp expression plasmid was coinjected to define the complete growth cone volume. pLL neuronal cell bodies were photoconverted with a 405 nm laser between 25 and 27 hpf and red fluorescence was measured in the associated growth cones 80 min later. This analysis revealed a strong decrease in SCG10 accumulation in kbpst23 mutant growth cones compared with wild-type siblings (Fig. 7A–E), supporting a function of KBP in SCG10 anterograde transport.
Figure 7.

Hindered SCG10 anterograde transport leads to pLL axon truncation. A–D, mEos2-SCG10-2b was transiently coexpressed with EGFP (cell fill that marks growth cones; green) in pLL neurons under control of the 5kbneurod promotor. mEos-SCG10-2b was converted from green to red in the pLL neuron cell bodies between 25 and 27 hpf. Following conversion, the levels of photoconverted mEos were captured in growth cones (80 min postconversion). Note a marked increase of photoconverted, red mEos2 in the wild-type growth cone compare with kbpst23 mutant 80 min after photoconversion. A–D and A′–D′ represent EGFP-labeled cell body and growth cones prior to and post photoconversion. A″–D″ and A″′–D″′ represent the red mEos2 fluorescence in these regions prior to and post photoconversion. E, Quantification of red mEos2-SCG10-2b fluorescence accumulation (background subtracted and normalized to cell body red mEos2) demonstrated loss of SCG10 transport to kbpst23 pLL growth cones (Wilcoxon/Kruskal–Wallis; p < 0.05). F, Fold-change in SCG10 levels in axonal growth cones due to plasmid-mediated mEos2-Scg10-2b or BAC-mediated SCG10-2b-mRFP expression. Ratio represents expressing to non-expressing growth cones from the same animals. G, Exogenous expression of SCG10-2b suppresses axon truncation in kbpst23 mutants (ANOVA with Tukey–Kramer HSD post hoc contrasts; p < 0.05). Error bars represent standard error of the mean. Scale bars, 10 μm.
As SCG10 promotes microtubule dynamics necessary for axon outgrowth, we then tested for a causal relationship between loss of SCG10 from growth cones and axon truncation. To address this, we overexpressed an SCG10-2b-mRFP fusion using zygotic injection of an SCG10-2b BAC. SCG10 levels could be augmented in kbpst23 mutant growth cones using BAC-based expression, though to a lesser degree than in wild-type siblings (Fig. 7F). The limited increase in the amount of SCG10 available in the growth cone with overexpression is likely due to the high cytosolic levels achieved with this exogenous expression. Overexpression of SCG10-2b in pioneer axons was able to partially suppress axon truncation in kbpst23 mutants and had no effect on wild-type siblings (Fig. 7G), supporting a direct relationship between decreased SCG10 levels and failed axon outgrowth in kbpst23 mutants. These data supports a model in which KBP and Kif1B function together to transport SCG10 to axon growth cones, which is necessary for axon extension.
Loss of SCG10 impacts growth cone morphology and microtubule stability
To understand how loss of SCG10 impacts the growth cone during axon outgrowth, we analyzed growth cone morphology in kbpst23 axons. Growth cones of pioneer axons were smaller and less elaborated in kbpst23 mutants than in wild-type siblings (Fig. 8A–D). To determine whether loss of SCG10 impacted microtubule outgrowth, which could affect growth cone morphology and axon extension, we analyzed EB3-GFP comets in individual pLL axons. Because EB3 marks the growing end of the microtubule, the movement of these comets can be used to determine microtubule growth parameters. EB3-GFP comets were readily apparent in extending wild-type and kbpst23 axons between 28 and 32 hpf (Fig. 8E). Kymograph analysis of these comets in the distal axon revealed no change in distance or velocity of microtubule growth bouts in kbpst23 mutants (Fig. 8F,G). Unfortunately, the high level of EB3-GFP in growth cones made analysis in this region impossible.
Figure 8.

Growth cone morphology and microtubule stability are altered in kbpst23 mutants. A, B, Growth cones of pLL axons in wild-type (A) and kbpst23 mutants (B) at 32 hpf. Yellow dotted line outlines the growth cone in each. C, D, Total number of processes extending from growth cones and the total growth cone area are reduced in kbpst23 mutants (ANOVA). E, Still of EB3-GFP transport from Movies 3. Asterisks indicate EB3-GFP-positive comets. F, G, Neither distance (ANOVA; p = 0.8287) nor velocity (ANOVA; p = 0.9367) of EB3 comets were altered in kbpst23 mutants. H, kbpst23 Mutants have increased microtubule stability in growth cones (p < 0.0001; F test; Tau: WT = 6.0, kbpst23 mutants = 18.7). Numbers on graphs indicate sample size. Error bars represent standard error of the mean. Scale bars, 10 μm.
GFP-EB3 imaging in a wild-type pLL axon at ∼30 hpf to analyze microtubule growth dynamics. EB3 was expressed in neurons using the 5kbneurod:GFP-EB3 expression plasmid. Growth cones are to the right. Scale bar, 10 μm. 
SCG10 has been implicated in microtubule stability in vitro, which could subsequently impact axon outgrowth (Riederer et al., 1997; Morii et al., 2006). To determine whether microtubule stability was altered in kbpst23 mutant growth cones, we analyzed the persistence of microtubules in this region using a photoconversion approach. α-Tubulin tagged with mEos2 was expressed in individual pLL axons using zygotic injection of the 5kbneurod:α-tubulin-mEos2 plasmid. The growth cone of a single pioneer axon was photoconverted with a 405 nm laser and a time series was immediately initiated to analyze the decay of red fluorescence. Similar analyses have been done previously in cultured hippocampal neurons to assay microtubule stability in growing and mature axons (Hammond et al., 2010). In the growth cone, red fluorescence persisted longer in kbpst23 mutants, indicating that the microtubule cytoskeleton in the central domain of the growth cone was significantly more stable (wild-type half-life: 4.16 min; kbpst23 half-life: 12.95 min; Fig. 8H). Together, our data support a role for the Kif1B-KBP interactions in the proper localization of SCG10 to growth cones, which in turn regulates microtubule stability and axon outgrowth.
Discussion
We have identified the Kif1B anterograde motor and its interaction with KBP as critical for SCG10 anterograde transport to axonal growth cones during axon extension. Our evidence supporting a specific role for Kif1B-KBP in this process includes: (1) SCG10 is lost from the growth cone in kbpst23 and kif1bst43 mutants; (2) KBP lacking the Kif1B binding domain cannot rescue SCG10 localization to the growth cone; (3) photoconversion experiments show very limited SCG10 movement from cell bodies to growth cones of kbpst23 mutants; and (4) transport of other cargos, specifically mitochondria and lysosomes, is unaffected before axon truncation. The proper transport and localization of SCG10 is in turn necessary for the regulation of microtubule stability and axon elongation. Together, our work identifies the specific mechanism of anterograde SCG10 transport and underscores the importance of proper localization of this microtubule regulatory protein during axon development.
The role of Kinesins in neural development
There are very few axonal cargos whose transport can be linked to a single kinesin motor. Forty-five genes exist in humans that code for motors in the kinesin superfamily (Hirokawa et al., 2009). Additional complexity is added by the several isoforms that exist for most of these motors in mammals (Hirokawa et al., 2010). The availability of this large cohort of anterograde motors for cargo transport oftentimes leads to functional redundancy in kinesin motor usage. Consequently, it is typically necessary to knockdown entire kinesin families to study the function of a particular class in neuronal development. One such study in mammalian cells focused on the function of Kif5 (Kinesin-1), of which there are three family members, and revealed a reliance on this motor for initial neurite outgrowth in cultured cells (Ferreira et al., 1992). The underlying mechanism of this reliance has been elegantly illustrated through experiments in Drosophila cells and larvae, which have a single Kinesin-1; Kinesin-1 slides microtubules into growing processes, facilitating neurite extension (Jolly et al., 2010; Lu et al., 2013). Interestingly, Kinesin-1 may have less of a role in the continued outgrowth of these axons, as loss of function of this motor at later stages has no impact on axon length (Lu et al., 2013). Thus, additional Kinesins and interacting proteins, such as Kif1B and KBP, must facilitate transport of cargo necessary for complete axon extension.
Because of the functional redundancy often observed between members of kinesin families, the dramatic effect of loss of a single Kinesin-3 motor (Kif1B) on SCG10 transport is striking. There are six members of the Kinesin-3 family. Kif1A and Kif1C are closely related to Kif1B, whereas Kif13A/B, Kif14, and Kif16A/B are more distantly related. The existence of these additional motors, some of which can even interact with KBP (Alves et al., 2010), increases the chance of functional redundancy; however, this is not the case for Kif1B-KBP in either the transport of SCG10 or the axon extension phenotype we observe. Loss of KBP and Kif1B result in similar loss of SCG10 immunofluorescence from growth cones and KBP-Kif1B interaction is necessary to rescue this phenotype as well as axon extension. Therefore, our experiments point to a lack of functional redundancy in kinesin usage for SCG10 transport.
The function of SCG10 in growth cones and the distal axon
Despite being studied intensively since its discovery in the 1980s (Mitchison and Kirschner, 1984), the role of microtubule instability in the growth cone during axon outgrowth is still a subject of intense investigation. It is clear that overstabilization or destabilization of the microtubule cytoskeleton impairs axon outgrowth (Letourneau and Ressler, 1984; Tanaka et al., 1995). Therefore, the proper localization of modulators of microtubule dynamics to areas of high microtubule restructuring activity, such as the growth cone, must be imperative to proper axon extension. SCG10 is a powerful regulator of microtubule dynamics (Riederer et al., 1997; Grenningloh et al., 2004). This protein binds to tubulin dimers and is thought to modulate microtubule dynamics by sequestering tubulin and limiting the available pool of building blocks for microtubule assembly. This is thought to increase catastrophe frequency, inherently destabilizing microtubules (Charbaut et al., 2001).
We offer several pieces of data supporting a destabilizing function of SCG10 in the growth cone. Similar to previous reports (Riederer et al., 1997; Lutjens et al., 2000; Morii et al., 2006), we observe the highest levels of SCG10 in the central domain of the growth cone, a region of highly dynamic microtubules (for review, see Dent and Gertler, 2003). We show that loss of SCG10 from this region correlates with failed axon outgrowth and increased microtubule stability as assayed by our photoconversion assays. These data align with biochemical studies indicating that SCG10 can bind free tubulin dimers (Charbaut et al., 2001), decrease microtubule polymerization in vitro (Grenningloh et al., 2004), and suppress defects caused by pharmacologically induced increases in microtubule stabilization (Riederer et al., 1997). Together, these data support the conclusion that SCG10 is a microtubule destabilizing factor, important for axon outgrowth in the growth cone.
Kif1B, KBP, and mitochondrial movement in axons
Previous work on the Kif1B motor has primarily focused on the differential regulation of transport by the two known isoforms, Kif1Bα and Kif1Bβ, of which Kif1Bβ has been the subject of the most investigation. These isoforms share a motor domain but have distinct cargo-binding tail domains. Kif1Bβ is responsible for anterograde transport of synaptic vesicle proteins, similar to the Kif1A Kinesin-3 motor (for review, see Hirokawa et al., 2009). In contrast, Kif1Bα has largely been considered to be the motor, in addition to Kinesin-1, to transport mitochondria toward microtubule plus ends in axons. This assertion is based on work performed on cell components reconstituted in vitro; in this system, loss of Kif1B impacts mitochondrial movement (Nangaku et al., 1994). This finding is somewhat controversial, as the major brain-specific Kif1B isoform, Kif1Bβ, lacks the putative mitochondrial binding domain and Kif1B zebrafish mutants (which lose both isoforms) do not show a substantial loss of mitochondria in axons (Conforti et al., 1999; Lyons et al., 2008; Campbell et al., 2014). To address this discrepancy, we analyzed mitochondrial transport in fully intact axons of zebrafish embryos in which Kif1Bα and β are lost (Lyons et al., 2009). Our data clearly show no reliance of mitochondria movement on Kif1B motor function in neurons. Because the kif1bst43 mutation results in loss of both the α and β isoforms, the isoform specificity of Kif1B for SCG10 transport is unknown. However, data from Lyons et al. (2009) indicate that the β isoform is likely the critical motor for SCG10 transport, because knockdown of Kif1Bβ but not Kif1Bα using antisense oligonucleotides phenocopied the mutant axon truncation.
KBP has previously been linked to mitochondrial transport through its interaction with Kif1Bα (Wozniak et al., 2005). Similar to the work implicating Kif1B in mitochondrial transport, this assertion relies on a select number of studies performed in cell culture. In one study, mitochondria in cultured fibroblasts were mislocalized in the absence of KBP or the presence of a KBP variant that does not interact with Kif1B; however, in the same study, Kif1B still colocalizes with mitochondria in the absence of KBP in cultured fibroblasts (Wozniak et al., 2005). This argues that KBP-Kif1B interaction is not directly involved in mitochondrial transport. Therefore, rather than serving as a mediator of mitochondrial motility, our data, in conjunction with this previous work, support an indirect function for KBP and Kif1B in the motility of mitochondria and other cargo through modulation of microtubule cytoskeleton development (Lyons et al., 2008). However, we cannot rule out other explanations, particularly developmental changes in the usage of KBP for cargo-specific transport regulation. This is especially intriguing in light of a recent report demonstrating that KBP can interact with the motor domain of several kinesin motors. In the case of Kif1A, this association regulates microtubule-motor interaction in vitro (Kevenaar et al., 2016). The potential developmental differences in KBP-Kif1B function for motor activity and cargo transport are a subject for future investigations.
Conclusions
Using the live imaging and genetic advantages of the zebrafish system, we have established that SCG10 transport to axon growth cones relies on the Kif1B-KBP complex. This is the first motor shown to be necessary for the transport of SCG10 and a novel example of a motor-cargo complex specifically necessary for axon extension. Loss of SCG10 from growth cones in kbpst23 or kif1bst43 mutants has dramatic effects on microtubule dynamics in the growth cone and on complete extension of long axons. Despite being implicated in mitochondrial transport previously, live imaging in these mutant lines did not reveal a direct role for either Kif1B or KBP in the movement of this organelle during axon extension. Therefore, our data reveal a novel mechanism for the intracellular localization of a microtubule modifying protein, which is essential for cytoskeletal development and subsequent axon formation and function. These data, in combination with the previous work on KBP function, offer insights into the basis of Goldberg–Shprintzen syndrome, a neurological disorder caused by mutations in KBP with clinical phenotypes indicative of abnormal neural circuit formation.
Footnotes
This work was supported by the NINDS/NIH (1K99NS086903-01A1) to C.M.D., the Collins Medical Trust, and the Tartar Trust. Funding to A.N. from the NICHD/NIH (R01HD072844-03). We thank the Nechiporuk laboratory for excellent feedback on this project and fish care, W. Talbot for the kbpst23 and kif1bst43 mutant strains, and Gary Banker and Marvin Bentley for their thoughtful comments on this work.
The authors declare no competing financial interests.
References
- Abramoff MD, Magalhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- Alves MM, Burzynski G, Delalande JM, Osinga J, van der Goot A, Dolga AM, de Graaff E, Brooks AS, Metzger M, Eisel UL, Shepherd I, Eggen BJ, Hofstra RM. KBP interacts with SCG10, linking Goldberg-Shprintzen syndrome to microtubule dynamics and neuronal differentiation. Hum Mol Genet. 2010;19:3642–3651. doi: 10.1093/hmg/ddq280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PD, Shen K, Sapio MR, Glenn TD, Talbot WS, Marlow FL. Unique function of Kinesin Kif5A in localization of mitochondria in axons. J Neurosci. 2014;34:14717–14732. doi: 10.1523/JNEUROSCI.2770-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr Biol. 2004;14:1272–1276. doi: 10.1016/j.cub.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Charbaut E, Curmi PA, Ozon S, Lachkar S, Redeker V, Sobel A. Stathmin family proteins display specific molecular and tubulin binding properties. J Biol Chem. 2001;276:16146–16154. doi: 10.1074/jbc.M010637200. [DOI] [PubMed] [Google Scholar]
- Conforti L, Buckmaster EA, Tarlton A, Brown MC, Lyon MF, Perry VH, Coleman MP. The major brain isoform of kif1b lacks the putative mitochondria-binding domain. Mamm Genome. 1999;10:617–622. doi: 10.1007/s003359901056. [DOI] [PubMed] [Google Scholar]
- Dent EW, Gertler FB. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron. 2003;40:209–227. doi: 10.1016/S0896-6273(03)00633-0. [DOI] [PubMed] [Google Scholar]
- Drerup CM, Nechiporuk AV. JNK-interacting protein 3 mediates the retrograde transport of activated c-Jun N-terminal kinase and lysosomes. PLoS Genet. 2013;9:e1003303. doi: 10.1371/journal.pgen.1003303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drerup CM, Nechiporuk AV. In vivo analysis of axonal transport in zebrafish. Methods Cell Biol. 2016;131:311–329. doi: 10.1016/bs.mcb.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C, Bourdette D, Banker G. Oxidative stress inhibits axonal transport: implications for neurodegenerative diseases. Mol Neurodegener. 2012;7:29. doi: 10.1186/1750-1326-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Niclas J, Vale RD, Banker G, Kosik KS. Suppression of kinesin expression in cultured hippocampal neurons using antisense oligonucleotides. J Cell Biol. 1992;117:595–606. doi: 10.1083/jcb.117.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleury D, Grenningloh G, Lafanechère L, Antonsson B, Job D, Cohen-Addad C. Preliminary crystallographic study of a complex formed between the alpha/beta-tubulin heterodimer and the neuronal growth-associated protein SCG10. J Struct Biol. 2000;131:156–158. doi: 10.1006/jsbi.2000.4283. [DOI] [PubMed] [Google Scholar]
- Ghysen A, Dambly-Chaudière C. Development of the zebrafish lateral line. Curr Opin Neurobiol. 2004;14:67–73. doi: 10.1016/j.conb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Grenningloh G, Soehrman S, Bondallaz P, Ruchti E, Cadas H. Role of the microtubule destabilizing proteins SCG10 and stathmin in neuronal growth. J Neurobiol. 2004;58:60–69. doi: 10.1002/neu.10279. [DOI] [PubMed] [Google Scholar]
- Hammond JW, Huang CF, Kaech S, Jacobson C, Banker G, Verhey KJ. Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol Biol Cell. 2010;21:572–583. doi: 10.1091/mbc.E09-01-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks M, Jesuthasan S. PHR regulates growth cone pausing at intermediate targets through microtubule disassembly. J Neurosci. 2009;29:6593–6598. doi: 10.1523/JNEUROSCI.1115-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- Jolly AL, Kim H, Srinivasan D, Lakonishok M, Larson AG, Gelfand VI. Kinesin-1 heavy chain mediates microtubule sliding to drive changes in cell shape. Proc Natl Acad Sci U S A. 2010;107:12151–12156. doi: 10.1073/pnas.1004736107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevenaar JT, Bianchi S, van Spronsen M, Olieric N, Lipka J, Frias CP, Mikhaylova M, Harterink M, Keijzer N, Wulf PS, Hilbert M, Kapitein LC, de Graaff E, Ahkmanova A, Steinmetz MO, Hoogenraad CC. Kinesin-binding protein controls microtubule dynamics and cargo trafficking by regulating kinesin motor activity. Curr Biol. 2016;26:849–861. doi: 10.1016/j.cub.2016.01.048. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien CB. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- Letourneau PC, Ressler AH. Inhibition of neurite initiation and growth by taxol. J Cell Biol. 1984;98:1355–1362. doi: 10.1083/jcb.98.4.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Fox P, Lakonishok M, Davidson MW, Gelfand VI. Initial neurite outgrowth in Drosophila neurons is driven by kinesin-powered microtubule sliding. Curr Biol. 2013;23:1018–1023. doi: 10.1016/j.cub.2013.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutjens R, Igarashi M, Pellier V, Blasey H, Di Paolo G, Ruchti E, Pfulg C, Staple JK, Catsicas S, Grenningloh G. Localization and targeting of SCG10 to the trans-Golgi apparatus and growth cone vesicles. Eur J Neurosci. 2000;12:2224–2234. doi: 10.1046/j.1460-9568.2000.00112.x. [DOI] [PubMed] [Google Scholar]
- Lyons DA, Naylor SG, Mercurio S, Dominguez C, Talbot WS. KBP is essential for axonal structure, outgrowth and maintenance in zebrafish, providing insight into the cellular basis of Goldberg-Shprintzen syndrome. Development. 2008;135:599–608. doi: 10.1242/dev.012377. [DOI] [PubMed] [Google Scholar]
- Lyons DA, Naylor SG, Scholze A, Talbot WS. Kif1b is essential for mRNA localization in oligodendrocytes and development of myelinated axons. Nat Genet. 2009;41:854–858. doi: 10.1038/ng.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison T, Kirschner M. Dynamic instability of microtubule growth. Nature. 1984;312:237–242. doi: 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- Mo W, Nicolson T. Both pre- and postsynaptic activity of nsf prevents degeneration of hair-cell synapses. PLoS One. 2011;6:e27146. doi: 10.1371/journal.pone.0027146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morii H, Shiraishi-Yamaguchi Y, Mori N. SCG10, a microtubule destabilizing factor, stimulates the neurite outgrowth by modulating microtubule dynamics in rat hippocampal primary cultured neurons. J Neurobiol. 2006;66:1101–1114. doi: 10.1002/neu.20295. [DOI] [PubMed] [Google Scholar]
- Nangaku M, Sato-Yoshitake R, Okada Y, Noda Y, Takemura R, Yamazaki H, Hirokawa N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell. 1994;79:1209–1220. doi: 10.1016/0092-8674(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Nechiporuk A, Raible DW. FGF-dependent mechanosensory organ patterning in zebrafish. Science. 2008;320:1774–1777. doi: 10.1126/science.1156547. [DOI] [PubMed] [Google Scholar]
- Niwa S, Tanaka Y, Hirokawa N. KIF1Bbeta- and KIF1A-mediated axonal transport of presynaptic regulator Rab3 occurs in a GTP-dependent manner through DENN/MADD. Nat Cell Biol. 2008;10:1269–1279. doi: 10.1038/ncb1785. [DOI] [PubMed] [Google Scholar]
- Obholzer N, Wolfson S, Trapani JG, Mo W, Nechiporuk A, Busch-Nentwich E, Seiler C, Sidi S, Söllner C, Duncan RN, Boehland A, Nicolson T. Vesicular glutamate transporter 3 is required for synaptic transmission in zebrafish hair cells. J Neurosci. 2008;28:2110–2118. doi: 10.1523/JNEUROSCI.5230-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer BM, Pellier V, Antonsson B, Di Paolo G, Stimpson SA, Lütjens R, Catsicas S, Grenningloh G. Regulation of microtubule dynamics by the neuronal growth-associated protein SCG10. Proc Natl Acad Sci U S A. 1997;94:741–745. doi: 10.1073/pnas.94.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabry JH, O'Connor TP, Evans L, Toroian-Raymond A, Kirschner M, Bentley D. Microtubule behavior during guidance of pioneer neuron growth cones in situ. J Cell Biol. 1991;115:381–395. doi: 10.1083/jcb.115.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster K, Dambly-Chaudière C, Ghysen A. Glial cell line-derived neurotrophic factor defines the path of developing and regenerating axons in the lateral line system of zebrafish. Proc Natl Acad Sci U S A. 2010;107:19531–19536. doi: 10.1073/pnas.1002171107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Miller BR, Babetto E, Cho Y, Sasaki Y, Qayum S, Russler EV, Cavalli V, Milbrandt J, DiAntonio A. SCG10 is a JNK target in the axonal degeneration pathway. Proc Natl Acad Sci U S A. 2012;109:E3696–E3705. doi: 10.1073/pnas.1216204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suster ML, Abe G, Schouw A, Kawakami K. Transposon-mediated BAC transgenesis in zebrafish. Nat Protoc. 2011;6:1998–2021. doi: 10.1038/nprot.2011.416. [DOI] [PubMed] [Google Scholar]
- Tanaka E, Ho T, Kirschner MW. The role of microtubule dynamics in growth cone motility and axonal growth. J Cell Biol. 1995;128:139–155. doi: 10.1083/jcb.128.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungos JM, Karlstrom RO, Raible DW. Hedgehog signaling is directly required for the development of zebrafish dorsal root ganglia neurons. Development. 2003;130:5351–5362. doi: 10.1242/dev.00722. [DOI] [PubMed] [Google Scholar]
- Westerfield M. The zebrafish book: a guide for the laboratory use of zebrafish (Danio rerio) Ed 4. Eugene, OR: University of Oregon; 2000. [Google Scholar]
- Wozniak MJ, Melzer M, Dorner C, Haring HU, Lammers R. The novel protein KBP regulates mitochondria localization by interaction with a kinesin-like protein. BMC Cell Biol. 2005;6:35. doi: 10.1186/1471-2121-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y, Hirokawa N. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bβ. Cell. 2001;105:587–597. doi: 10.1016/S0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mitochondrial transport in a pLL axon of a kif1bst43 wild-type sibling at ∼30 hpf. Mitochondria were tagged with TagRFP using a mitochondrial targeting sequence in neurons using the 5kbneurod:mito-TagRFP plasmid. Anterograde transport is from left to right. Scale bar, 10 μm.
Mitochondrial transport in kif1bst43 mutant pLL axon at ∼30 hpf. Mitochondria were tagged with TagRFP using a mitochondrial targeting sequence and expressed in neurons using the 5kbneurod:mito-TagRFP plasmid. Anterograde transport is from left to right. Scale bar, 10 μm.
GFP-EB3 imaging in a wild-type pLL axon at ∼30 hpf to analyze microtubule growth dynamics. EB3 was expressed in neurons using the 5kbneurod:GFP-EB3 expression plasmid. Growth cones are to the right. Scale bar, 10 μm.