

Abstract
The formation and refinement of thalamocortical axons (TCAs) is an activity-dependent process (Katz and Shatz, 1996), but its mechanism and nature of activity are elusive. We studied the role of spike timing-dependent plasticity (STDP) in TCA formation and refinement in mice. At birth (postnatal day 0, P0), TCAs invade the cortical plate, from which layers 4 (L4) and L2/3 differentiate at P3-P4. A portion of TCAs transiently reach toward the pia surface around P2-P4 (Senft and Woolsey, 1991; Rebsam et al., 2002) but are eventually confined below the border between L2/3 and L4. We previously showed that L4-L2/3 synapses exhibit STDP with only potentiation (timing-dependent long-term potentiation [t-LTP]) during synapse formation, then switch to a Hebbian form of STDP. Here we show that TCA-cortical plate synapses exhibit robust t-LTP in neonates, whose magnitude decreased gradually after P4-P5. After L2/3 is differentiated, TCA-L2/3 gradually switched to STDP with only depression (t-LTD) after P7-P8, whereas TCA-L4 lost STDP. t-LTP was dependent on NMDA receptor and PKA, whereas t-LTD was mediated by Type 1 cannabinoid receptors (CB1Rs) probably located at TCA terminals, revealed by global and cortical excitatory cell-specific knock-out of CB1R. Moreover, we found that administration of CB1R agonists, including Δ9-tetrahydrocannabinol, caused substantial retraction of TCAs. Consistent with this, individual thalamocortical axons exuberantly innervated L2/3 at P12 in CB1R knock-outs, indicating that endogenous cannabinoid signaling shapes TCA projection. These results suggest that the developmental switch in STDP and associated appearance of CB1R play important roles in the formation and refinement of TCAs.
SIGNIFICANCE STATEMENT It has been shown that neural activity is required for initial synapse formation of thalamocortical axons with cortical cells, but precisely what sort of activities in presynaptic and postsynaptic cells are required is not yet clear. In addition, how activity is further translated into structural changes is unclear. We show here that the period during which spike timing-dependent long-term potentiation and depression (t-LTP, t-LTD) can be induced closely matches the time course of synapse formation and retraction, respectively, at the thalamocortical synapse. Moreover, administration of cannabinoid agonists, which mimic t-LTD, caused TCA retraction, suggesting that cannabinoids translate physiological changes into morphological consequences.
Keywords: CB1R, cortical plate, DiI, mouse, somatosensory, STDP
Introduction
Despite evidence that coordinated patterns of neuronal activity influence the organization of circuits throughout the brain, how activity is translated into structural changes remains unclear (Katz and Shatz, 1996; Catalano and Shatz, 1998). Long-term potentiation (LTP) and long-term depression (LTD) have been explored as candidate mechanisms (Kimura et al., 1989; Katz and Shatz, 1996; Foeller and Feldman, 2004; Malenka and Bear, 2004); however, causal links between LTP/LTD and anatomical rearrangements remain tentative. We have previously shown that, during synapse formation, L4-L2/3 synapses exhibit spike timing-dependent plasticity (STDP) with only LTP (timing-dependent long-term potentiation [t-LTP]) for 1 week; this is switched to standard Hebbian STDP with LTP and LTD at P13-P15 (Itami and Kimura, 2012). During synapse formation, the spiking order between presynaptic and postsynaptic cells is unstable or even random, due to weakness or absence of synaptic connections. t-LTP rule would be suitable for strengthening weak synaptic connections under such unregulated presynaptic and postsynaptic activity because only close temporal correlation, regardless of spiking order, is required for strengthening synapses under t-LTP. One week before L4-L2/3 synapse formation, thalamocortical axons (TCAs) start to form synapses with the cortical plate (CP), which later differentiates into L4 and L2/3. TCA projections to future L2/3 appeared to be largely transient (Senft and Woolsey, 1991; Rebsam et al., 2002), and the innervations are eventually confined to L4, the main target layer of TCAs. In the present study, we first asked whether TCA-CP synapses exhibit t-LTP during synapse formation. If so, how about TCAs to L2/3 while possible retraction is going on, and TCAs to L4? The results indicated that TCA synapsing to CP cells showed t-LTP, and TCA to differentiated L2/3 cells changed into t-LTD, whereas TCA to differentiated L4 lost STDP. We also identified that t-LTD was mediated by cannabinoid receptors (CB1R). We then attempted to mimic these morphological changes by exogenous administration of agonists or antagonist of CB1R, which produced physiological effects. Finally, we found that endogenous CB1R signaling also played an important role in shaping the TCA axons during development by examining TCAs in CB1R−/−. Our results indicate that CB1R may provide a causal link between synaptic and morphological consequences.
Materials and Methods
Experiments were conducted according to the guidelines of the animal welfare committees of the Saitama Medical University, Baylor College of Medicine, Hokkaido University, and Osaka University Graduate School of Medicine.
Animals.
Global CB1R-KO (CB1R−/−) mice and CB1R-floxed (CB1Rfl/fl) mice on a C57BL/6 background were generated as described previously (Marsicano et al., 2002, 2003). NEX-Cre mice were generated by inserting the Cre gene into the NEX locus (Goebbels et al., 2006). NEX-Cre/+ mice were backcrossed onto C57BL/6 background for five generations. CB1R+/− males were mated with CB1R+/− females to generate CB1R−/−, CB1R+/−, and CB1R+/+ mice. NEX-Cre/+; CB1Rfl/fl males were mated with CB1Rfl/fl females to produce NEX-Cre/+; CB1Rfl/fl (Cx-CB1R-KO) and +/+; CB1Rfl/fl; (control) mice.
To generate a TCA-green fluorescent protein (TCA-GFP) reporter line, we generated a double-transgenic retinoid-related orphan receptor (ROR)αCre-TaumGFP line (designated TCAmGFP) by crossing RORαCre mice with TaumGFP mice. RORαCre mice were generated by inserting an IRES-cre expression cassette into the 3′ noncoding region of the RORα gene. TaumGFP is a Cre-reporter line containing a floxed “stop transcription” sequence in front of membrane anchored GFP (mGFP) and an IRES-NLS-lacZ gene in the Tau locus (Hippenmeyer et al., 2005). This reporter line allows the detection of neurons with Cre-mediated recombination events by the presence of nuclear β-galactosidase (β-gal) and membrane-anchored GFP. Mouse colonies were maintained in a pathogen-free environment with a 14/10 h light/dark cycle (lights on at 06:00) with access to food and water ad libitum. All experiments and data analyses were performed by experimenters who were blinded to the genotype information. Animals were treated in compliance with guidelines set by the United States Department of Health and Human Services.
Genotyping.
Tail lysates were prepared by immersing the tail pieces in tail digestion buffer (50 mm KCl, 10 mm Tris-HCl, Triton X-100, 0.1 mg/ml proteinase K, pH 9.0) and vortexing gently for 3 h at 60°C to lyse the tail. Tail lysates were heated to 94°C for 10 min to denature by the proteinase K, and then centrifuged at 16,000 ×g for 15 min. The supernatants were used as DNA templates for PCRs. For the CB1R allele, PCRs were conducted with a mixture of primers CB50: 5′-GCTGTCTCTGGTCCTCTTAAA-3′ and CB51: 5′-GGT GTCACCTCTGAAAACAGA-3′ to detect the CB1R-WT allele (400 bp); or CB50 and CB54: 5′-CCTACCCGGTAGAATTAGCTT-3′ to detect the CB1R-KO allele (300 bp).
For the CB1R-floxed allele, PCRs were conducted with a mixture of primers CB50 and CB51 to detect the CB1R-WT allele (400 bp) and flox-inserted allele (500 bp). For Nex-Cre, PCRs were conducted using primers that have been described previously (Ballester-Rosado et al., 2010).
Slice preparation.
Mice were deeply anesthetized with Ethrane (Abbott Laboratories) and decapitated. The brain was rapidly transferred to ice-cold slicing ACSF consisting of the following (in mm): 124 NaCl, 3 KCl, 1.2 NaH2PO4, 1.3 MgSO4, 2 CaCl2, 10 glucose, and 26 NaHCO3, bubbled with 95% O2-5% CO2, and balanced at pH 7.4 (295–305 mOsm). Thalamocortical slices (350–400 μm) were prepared from P1-P19 from C57BL/6J mice and mutants using a rotor slicer or vibratome, as described previously (Agmon and Connors, 1991; Itami et al., 2001). Then, slices were immediately transferred to a holding chamber where they remained submerged in oxygenated ACSF for at least 1 h until being transferred to a recording chamber that was placed on the stage of an upright microscope.
Electrophysiology.
Whole-cell patch pipettes (5–7 mΩ) were used to record the membrane voltage from visually identified cells in cortical plate, L4, and L2/3 in response to ventrobasal nucleus of thalamus (VB) or L4 stimulation under differential interference optics with upright stage-fixed microscopes (BX51WI, Olympus, or Axioskop 2 FS plus, Zeiss), as described previously (Kimura et al., 2010; Itami and Kimura, 2012). The temperature was maintained at 27°C–30°C. Micropipettes were pulled from borosilicate, thick-walled glass capillary tubing (Sutter Instruments). The pipette solution contained the following: 130 mm K-methane sulfonate, 10 mm KCl, 10 mm HEPES, 0.5 mm K-EGTA, 5 mm Mg-adenosine triphosphate, 1 mm Na-guanosine-5′-triphosphate, 10 mm Na-phosphocreatinine, pH 7.3 (295 mOsm). Responses were recorded using a MultiClamp 700B or an Axoclamp 2B amplifier (Molecular Devices) in current-clamp mode. Signals were low-pass filtered at 3–5 kHz, digitally sampled at 10–20 kHz, and monitored with the pCLAMP software (Molecular Devices). Upon inserting the electrode in the bath, the stray pipette capacitance was compensated, as was the bridge balance, through a built-in circuit of the amplifier. The bridge balance was checked repeatedly and readjusted when necessary. A concentric bipolar stimulating electrode (Frederick Haer) was placed in L4 or in the VB area of the thalamus, through which electrical stimuli consisting of square pulses for 100 μs, up to 0.5 mA, were applied every 7500 ms (0.133 Hz), unless otherwise specified. Upon establishing whole-cell recording of synaptic responses in response to afferent stimulation, monosynaptic nature was examined by short (typically <4 ms from VB, depending on age) (for details, see Kimura et al., 2010) constant latency, following 1 Hz afferent stimulations without failure or latency jittering. To avoid responses resulting from antidromic activation of corticothalamic axons, only EPSPs with paired pulse depression that displayed no supernormality were adopted (Beierlein and Connors, 2002; Kimura et al., 2010).
Induction protocol for STDP.
Single postsynaptic action potentials (APs) were evoked by somatic current injection using the smallest current possible (typically <1.5 nA for 5–10 ms). The stimulus intensity to presynaptic cells, either in L4 or in the thalamus, was also adjusted to evoke EPSPs with a single component, without notches in the rising or decaying phases. The pairing interval was defined from the onset of EPSPs to the peak of each AP. A total of 90 pairings was applied to presynaptic and postsynaptic cells with fixed delays, except in random stimulation experiments (see Fig. 6). Stimulation patterns were constructed using a custom program written by LabVIEW (National Instruments) running on a Windows computer and delivered through an interface (USBX-I16P, Technowave) by the stimulator. In Gaussian random-delay timing stimulation, the computer generated 90 random numbers with a Gaussian distribution, with a mean of 0 and 99.7% (3 SD) of the numbers occurring within ±50. These numbers were then taken as the value of AP-EPSP delay in milliseconds.
Figure 6.
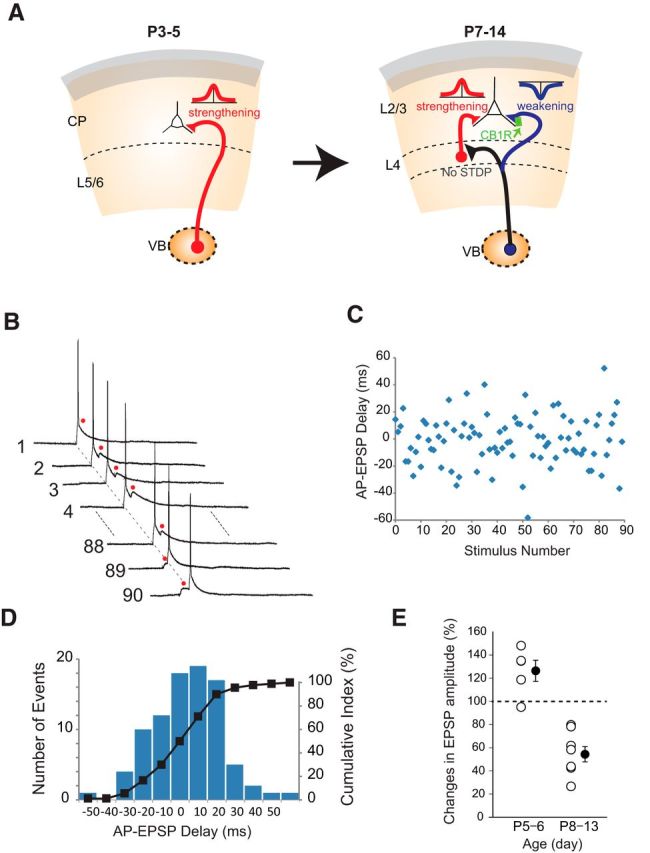
Developmental switch of STDP and associated transition of the effects of random-delay timing stimulation of thalamus and cortical cells. A, Schematic illustration of a developmental switch of STDP at thalamocortical synapses during the first 2 postnatal weeks. B–E, An example of random-delay timing stimulation (B–D) and developmental transition of its effect (E). B, APs were elicited by postsynaptic current injection at the fixed timing, whereas EPSPs (marked by red dots) were elicited by presynaptic stimulation such that the delay between AP and EPSP (AP-EPSP delay) was randomly assigned along a Gaussian distribution (for details, see Materials and Methods). C, A graph showing the distribution of individual AP-EPSP delays (±50 ms Gaussian) plotted against each of 90 stimulations. D, Histogram of AP-EPSP delay showing that the distribution is Gaussian with mean value of 0 ms and 99.7% (3 SD) of each value occurring within ±50 ms of the mean. E, Effect of random-delay stimulation. Random-delay stimulation led to LTP during the first postnatal week but led to LTD during the second postnatal week.
Drugs and drug injection.
WIN 55, 212–2 (WIN) mesylate, AM251, and AM281 were obtained from Tocris Bioscience. Ifenprodil was obtained from Sigma-Aldrich. WIN (1 or 0.1 mg/kg) was suspended in vehicle solution (10% or 1% DMSO, respectively) and vortexed immediately before intraperitoneal injection. AM281 (5, 10, or 15 mg/kg) was suspended in vehicle solution (4%, 8%, or 10% DMSO, respectively) and vortexed immediately before intraperitoneal injection. Δ9-Tetrahydrocannabinol (Δ9-THC) was obtained from the National Institute on Drug Abuse (Rockville, MD), dissolved in 100% ethanol (100 mg/5 ml) and diluted to final concentration (1 or 0.1 mg/kg), and then injected (i.p.). All stock solutions were kept at −80°C.
Triple immunofluorescence for CB1R and neurochemical markers.
Under pentobarbital anesthesia (100 mg/kg of body weight, i.p.), C57BL/6, CB1R−/− and TCA-reporter (ROR-GFP) mice were perfused transcardially with 4% PFA in 0.1 m sodium phosphate buffer, pH 7.2. After additional immersion in the same fixative for 4 h, the sections (50-μm-thick) were prepared with a microslicer (VT1000S, Leica Microsystems). All incubations were done at room temperature, and PBS containing 0.1% Triton X-100 was used as an antibody diluent and washing solution. After blocking with 10% normal donkey serum for 20 min, the sections from CB1R+/+ and CB1R−/− were subjected to triple immunofluorescence for guinea pig anti-CB1R (Fukudome et al., 2004), goat or rabbit anti-vesicular inhibitory amino acid transporter (VIAAT), anti-vesicular glutamate transporter 1 (VGluT1), and anti-VGluT2 (Miyazaki et al., 2003). The sections from ROR-GFP mice were subjected to triple immunofluorescence staining using guinea-pig anti-CB1R, goat anti-GFP (Takasaki et al., 2010), and rabbit anti-VGluT2. Sections from different genotypes were treated simultaneously, incubated overnight in a mixture of primary antibodies raised in different species. After several washes, the sections were incubated for 2 h in a mixture of 1:200 diluted species-specific secondary antibodies linked to Alexa-488 (Invitrogen), Cy3, or Cy5 (Jackson ImmunoResearch).
Image acquisition and analysis of CB1R immunoreactivity.
Images of single optical sections were acquired using a confocal laser-scanning microscope (FV1000, Olympus). All images were obtained by restricting the width of the emission wavelength using spectral slits, and by using sequential laser scanning to minimize fluorescence cross talk. Confocal images from sections of different genotypes stained with the same antibody combinations were obtained using the same acquisition settings. Intensity of CB1R immunoreactivity was measured using MetaMorph software (Molecular Devices). In brief, images were separated into individual channels and converted to grayscale. Then, VGluT1, VGluT2, or VIAAT signals from each channel that were above the threshold were automatically identified as objects. Subsequently, corresponding regions were created using the Create Regions Around Objects tool. CB1R intensity in individual puncta was measured using the Region Measurements tool for each region. In each mouse, 500–1000 puncta for each class of terminals were analyzed and averaged. Statistical significance among multiple groups was assessed using a one-way ANOVA with post hoc Bonferroni's test.
Immunohistochemistry for VGluT2 staining in thalamocortical slices.
Under isoflurane anesthesia (2%–4% for induction), C57BL/6 mice were perfused transcardially with 4% PFA in PBS, pH 7.4. Brains were postfixed with the same fixative overnight at 4°C; then, the thalamocortical sections (100 μm thick) were prepared with a microslicer (VT-1000S, Leica Microsystems). Six floating sections (per animal) were used for all of the subsequent steps. Sections were washed with PBS with 0.01% Triton X-100 (PBST) and permeabilized with 0.3% Triton X-100 in PBS for 20 min and then washed again with PBST. Nonspecific reactions were blocked with 3% normal goat serum in PBST for 1 h. Sections were incubated with anti-VGluT2 antibodies (Millipore Bioscience Research Reagents; 1:5000) in PBST containing 2 mg/ml BSA and 1% normal goat serum at 4°C overnight. Sections were washed with PBST and then incubated with AlexaFluor-647-conjugated anti-(guinea-pig immunoglobulin G) goat antibody (Molecular Probes; 1:1000) in PBST. DAPI (Invitrogen) staining (1:10,000) was also performed for 1 h to identify L4 areas and barrel borders. All processes were performed at room temperature, unless otherwise described.
Fluorescent images were obtained using a Zeiss Axioplan2 Imaging MOT with 2.5×/0.12 and 5×/0.25 Zeiss objectives (magnification/NA), and were analyzed with AxioVision software.
Immunohistochemical enhancement of GFP for TCA-reporter mice (ROR-GFP).
To confirm the expression pattern of the reporter gene in the ROR-GFP mice, double immunohistochemical staining of the GFP fluorescence and β-gal was performed. Thalamocortical sections were prepared like those for VGluT2 immunohistochemistry. A chicken anti-GFP antibody was used to enhance the GFP fluorescence (Sigma; 1:1000), together with the rabbit anti-β-gal antibody (Sigma; 1:1000). For blocking nonspecific responses, sections were first incubated with 3% normal goat serum with 0.01% Triton X-100 in PBS for 1 h, and then transferred to a solution containing rabbit anti-GFP antibody overnight at 4°C. Sections were washed with PBS 3 times for 10 min each, and then incubated with a solution containing Alexa-488-conjugated goat anti-chicken antibody (Molecular Probes; 1:1000) and Alexa-647-conjugated goat anti-rabbit antibody (Molecular Probes; 1:1000) for 1 h. After being washed with PBS, sections were mounted on slides and imaged with a Zeiss AxioImager M1 system with 2.5×/0.8 and 5×/0.16 objectives, with the same exposure time for all sections.
Analysis of fluorescence intensity.
In the thalamocortical slices from the right hemisphere, four or five barrels representing the left side of whiskers were selected. For each barrel, fluorescence intensity along a vertical line through the center of the barrel was analyzed from the pia surface to the white matter by using densitometry in the ImageJ software. For comparison across slices, differential values at each point from internal standard points at white matter (or in some cases, just below the pia surface) were obtained. Mean ± SEM values from each slice were calculated and plotted as a function of the distance from the pia surface.
Staining thalamocortical afferents with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) crystals.
For morphological characterization of the thalamocortical afferents, living thalamocortical slices (350–400 μm) were prepared from P12 CB1R+/+, CB1R+/−, and CB1R−/− mice in exactly the same way as the slices prepared for electrophysiology. Slices were kept in a holding chamber in oxygenated ACSF. One or two tiny crystals of DiI (Molecular Probes) were chosen and inserted into the VB region of the thalamus with a 30 gauge needle under a dissection microscope. The slices containing DiI crystals were fixed by immersion into 4% PFA containing 4% sucrose in PBS at 4°C for 1–2 d (overnight). Slices were moved to PBS with 0.01% NaN2 and kept in the dark for 4–8 weeks at room temperature for dye diffusion.
To identify the L4 barrel pattern, DAPI staining was also performed as described above. Stained axons were carefully checked, and only those running deep from and in parallel with the section surface were selected to avoid incomplete representation of axonal arbors by slicing processes. The arborization patterns of identifiable single DiI-labeled axons were acquired in stacks of 1-μm-interval images using either Zeiss LSM 710 confocal microscope with 20×/0.8/0.55 (Plan-Apochromat) and 10×/0.3/5.2 (EC Plan-Neofluar) objectives (magnification/NA/working distance) or Leica TCS SP8 confocal microscope with 10×/0.3/11 (HC PL FLUOTAR) and 25×/0.95/0.17 water-immersion (HCX IRAPO L) objectives. Single thalamocortical axons were identified and reconstructed above the layer V/VI boundary, and all the arbors found within layers I-V were traced using the NeuronJ software. For quantitative analysis of axonal arbor density, a region of interest covering all parts of the axonal arbor was first determined by connecting the most external axonal tips or axonal parts to a bifurcating point occurring just below layer 4. This was done to ensure that the region of interest area is as low as possible. The density of the axonal arbor was defined by length/area of the region of interest (μm/μm2).
Statistical analysis.
All values in the text and figures are presented as mean ± SEM, unless otherwise specified. Student's paired and unpaired t tests were performed. For comparison of multiple groups, ANOVA was first performed, and significance was determined by a post hoc test when necessary. A p value of <0.05 was considered statistically significant.
Results
Target-dependent developmental switch in STDP at thalamocortical synapses
In the barrel cortex, L5 and L6 are already differentiated from the CP at birth (P0), whereas the rest of the layers (L2–4) are not (Rice and Van der Loos, 1977). At this stage, TCAs can be found in L5–6, and some have reached the CP (Rebsam et al., 2002). Thereafter, TCAs continue to invade the CP, and transient extension of TCAs reaches near the pial surface around P2-P4 (Senft and Woolsey, 1991; Rebsam et al., 2002). These largely retract later, although portions of axons remain in L2/3 at P5 (Agmon et al., 1993). To examine whether the plastic property of synapses at TCAs to cortical cells changes during extension and retraction of axons, whole-cell recordings were made from cells above the L4-L3 border. Because L4 is differentiated from CP at P3-P4, the L4-L3 border becomes clearer at this age, whereas L2/3 is not well differentiated until around P4 (Rice and Van der Loos, 1977; Senft and Woolsey, 1991); cells above L4-L3 border were designated as CP cells until P4. We showed previously that L4-L2/3 synapses exhibit t-LTP during axon extension and synapse formation (Itami and Kimura, 2012); here we tested whether TCA-CP synapses exhibit similar properties. Consistent with previous work, stimulation of the VB elicited EPSPs with a large decay constant, which was sensitive to d-AP5, a selective antagonist against the NMDA-type glutamate receptor (Agmon and O'Dowd, 1992; Crair and Malenka, 1995; Isaac et al., 1997; Itami et al., 2003; Yanagisawa et al., 2004). Pairing stimulation with pre-before-post timing (75 ms, positive timing delay; + timing) induced clear long-term potentiation (t-LTP) (Fig. 1A). By testing various timing delays, including positive and negative (150 to −200 ms) values, we found that only t-LTP was induced, but the magnitude of t-LTP changed in a timing-dependent manner (Fig. 1B), similar to L4-L2/3 synapses at P7-P13 (Itami and Kimura, 2012). t-LTP at L4-L2/3 synapse during the second postnatal week requires NMDA receptors and activation of PKA (Itami and Kimura, 2012). We found that pairing-induced t-LTP at thalamocortical synapses during the first postnatal week was also dependent on NMDA receptors because d-AP5 (50 μm) successfully blocked the induction of t-LTP (Fig. 1C,E; p < 0.01, n = 8, 25 ms timing delay, one-way ANOVA). Similarly, involvement of PKA in the t-LTP induction was confirmed by experiments with Rp-cAMP-S, an inhibitor of cAMP cascade, which blocked t-LTP induction when applied to postsynaptic cells directly from recording pipettes (50 μm, Fig. 1D,E; p < 0.05, n = 16, 25 ms timing delay, one-way ANOVA). We then examined whether properties of t-LTP changed with ages as L2/3 differentiated from CP. Recordings were made from L2/3 cells on and after P5 and STDP of TCA to L2/3 cells was tested by applying negative timing stimulation (−10 ms, post-before-pre). We found that the magnitude of t-LTP decreased gradually after P5, at which point depression began to be induced. After P7-P8, only depression was induced (Fig. 1F).
Figure 1.

Target-dependent developmental switch in the STDP at thalamocortical synapses. A, An example of t-LTP induced by pairing stimulation with pre-before-post timing (75 ms) at P3. B, Both positive and negative (150 ms to −200 ms) timing stimulation produced t-LTP in a timing-dependent manner before P6. C, D, d-AP5 (50 μm) (C) and Rp-cAMP (50 μm) (D) blocked the induction of t-LTD, indicating that t-LTP requires NMDA receptor and activation of PKA, respectively. E, Summary of the effect of Rp-cAMP and d-AP5. F, Developmental changes in the magnitude of t-LTP at TCA-CP (until P4) and TCA-L2/3 (P5–P19) synapses. G, Experimental scheme showing VB stimulation and whole-cell recording from L4. H, TCA-L4 synapse did not exhibit STDP by negative timing (−25 ms) stimulation after P9. I, None of the timing stimulations (150 ms to −200 ms) produced t-LTP or t-LTD at TCA-L4 synapse at P8–P13.
Next, we tested whether TCA-L4 synapses were capable of inducing STDP of any type because the canonical target layer of TCAs is L4. One previous study demonstrated that simple pairing of afferent stimulation and postsynaptic depolarization (from −10 to 0 mV) induced LTP at the VB-L4 synapse, but only until P7 (Crair and Malenka, 1995). We tested the spike timing stimulation (−25 ms) at VB-L4 synapses and found that t-LTP was unable to be induced after P8 (Fig. 1G,H). Indeed, none of the timing stimulations ranging from −200 to 150 ms caused potentiation or depression at VB-L4 synapses after P8 (Fig. 1I; n = 33). These results demonstrate that the properties of STDP at thalamocortical synapses changes during development, depending on the target layers and age.
Thalamus to L2/3 projections possess CB1Rs at the terminal during the second postnatal week
We next investigated what caused t-LTD at TCA-L2/3 after P7-P8. Because the LTD component of Hebbian STDP with LTP/LTD at the L4-L2/3 synapse after P13-P15 is mediated by CB1R (Sjöström et al., 2003; Bender et al., 2006; Nevian and Sakmann, 2006; Itami and Kimura, 2012), we examined whether TCA-L2/3 synapses possess functional CB1Rs. Application of WIN, a CB1R agonist, suppressed VB-evoked EPSPs (5 μm; Fig. 2B), as summarized in Figure 2F (CB1R+/+, VB → L2/3, 69.3 ± 4.1% of control, n = 8, p < 0.01, t test, second from right). However, because CB1Rs are reportedly localized at L4 terminals to L2/3 cells at least after P16 (Sjöström et al., 2003; Bender et al., 2006; Nevian and Sakmann, 2006; Itami and Kimura, 2012), it is possible that WIN suppression was disynaptically induced, and TCA terminals may not possess functional CB1Rs. To rule out this possibility, we used both global CB1R-KOs (CB1R−/−) and cortical glutamatergic-specific CB1R-KO mice (NEX-Cre+/−; CB1Rfl/fl) to confirm the localization of CB1Rs responsible for the suppression of EPSPs at the thalamic terminals. Using global KOs, we confirmed that WIN suppression was mediated by CB1Rs because WIN application did not suppress VB-evoked EPSPs in L2/3 cells (Fig. 2C,F; CB1R−/− VB → L2/3, 99.7 ± 1.5% of control, n = 10, p = 0.85, t test). Then, we found that WIN suppressed VB-evoked EPSPs in NEX-Cre+/−;CB1Rfl/fl, in which CB1Rs were knocked out only in cortical excitatory cells (Fig. 2D,F; NEX-Cre+/−;CB1Rfl/fl, VB → L2/3, 80.6 ± 2.9% of control, n = 15, p < 0.0001, t test). This was recapitulated in control flox/flox mice (Fig. 2E,F; CB1Rfl/fl, VB → L2/3, 77.7 ± 6.5% of control, n = 10, p < 0.01, t test) to a similar extent (p = 0.66, unpaired t test). These results suggest that the CB1Rs responsible for WIN suppression were located directly on thalamic terminals.
Figure 2.
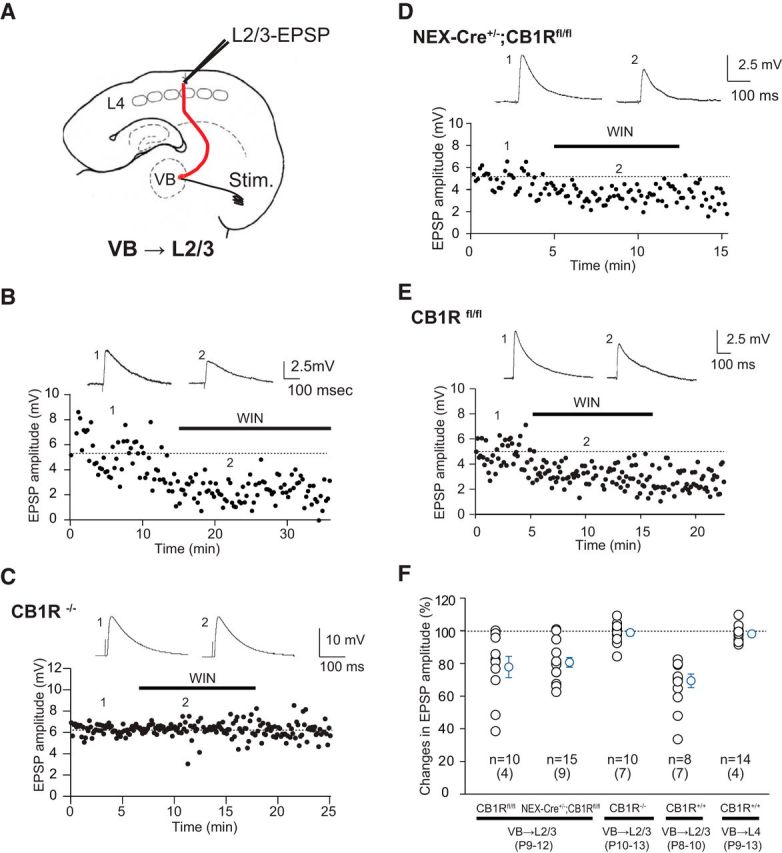
Thalamocortical terminals to L2/3 possess functional CB1Rs during the second postnatal week, examined by using cortical glutamatergic cell-specific knock-out of CB1R (NEX-Cre+/−;CB1Rfl/fl). A, Schematic depiction of the experiments. Recordings were made from L2/3 pyramidal cells while stimulation was applied to VB. B, WIN application (5 μm) suppressed VB-evoked EPSPs. C, WIN application failed to suppress EPSPs in CB1R−/−, as summarized in F (middle, CB1R−/− VB → L2/3, 99.7 ± 1.5% of control, n = 10, p = 0.85), indicating that the suppression was mediated by CB1Rs. D, In NEX-Cre+/−;CB1Rfl/fl, VB-evoked EPSPs were suppressed by WIN application, as summarized in F (NEX-Cre+/−;CB1Rfl/fl VB → L2/3, 80.6 ± 2.9% of control, n = 15, p < 0.0001). E, WIN application also suppressed VB-evoked EPSPs in control flox/flox mice, as summarized in F (CB1Rfl/fl VB → L2/3, 77.7 ± 6.5% of control, n = 10, p < 0.01). There was no significant difference in WIN suppression between NEX-Cre+/−;CB1Rfl/fl and CB1Rfl/fl (p = 0.66, unpaired t test). F, Summary of WIN suppression. The genotypes, connections, and ages of the animals in each experiment are indicated below. Results from the WIN sensitivity test at VB → L2/3 and VB → L4 synapses are shown at the right (CB1R+/+ VB → L2/3, P8–P10, 69.3 ± 4.1% of control, n = 8, p < 0.01, t test, CB1R+/+ VB → L4, P9–P13, 98.0 ± 1.2% of control, n = 14, p = 0.13, t test). Numbers in parentheses indicate the number of animals.
We showed that negative, as well as positive timing delay stimulation at the VB-L4 synapse did not exhibit t-LTD during the second postnatal week (Fig. 1H,I). This suggests that thalamic terminals to L4 cells do not possess CB1R during the second postnatal week. To address this, we tested WIN sensitivity and found that VB-L4 synapses were not sensitive to applied WIN (5 μm) (Fig. 2F; CB1R+/+, VB → L4), which is consistent with other morphological studies indicating the lack of CB1R localization at L4 (Bodor et al., 2005; Yoneda et al., 2013). These results suggest that differential expression of functional CB1R, depending on the target layers, underlies the expression of t-LTD.
Immunohistochemical localization of CB1R at thalamocortical terminals during the second postnatal week
Next, we conducted morphological tests to determine whether thalamocortical terminals possess CB1Rs. We investigated the expression pattern of CB1Rs in L2/3 using triple-labeling immunofluorescence with several neuronal markers. Consistent with previous reports, CB1R labeling demonstrated fibrous structures (Bodor et al., 2005; Kawamura et al., 2006) (Fig. 3A–C). At a higher magnification, intense CB1R labeling was frequently associated with VIAAT-positive terminals (Fig. 3B, arrowheads). Low- to moderate-intensity CB1R staining overlapped with VGluT1-positive terminals (Fig. 3C, arrowheads) and VGluT2-positive terminals (Fig. 3B,C, arrows), the latter of which correspond to thalamocortical terminals (Fremeau et al., 2001; Fujiyama et al., 2001). In contrast, we observed practically no staining in CB1R−/− mice (Fig. 3D), confirming the specificity of the antibody. We quantified the fluorescence intensity of CB1R staining and found that it was significantly higher in VGluT1- or VGluT2-positive terminals in L2/3 in CB1R+/+ mice than in CB1R−/− mice (Fig. 3B–D,H), although it was much lower than that found in VIAAT-positive structures (Fig. 3B,H). We further confirmed CB1R immunoreactivity in thalamocortical terminals using TCA-reporter mice (ROR-GFP), which express GFP in thalamic axons (Fig. 3E) (Nakagawa and O'Leary, 2003). We found weak but significant CB1R immunoreactivity in VGluT2- and GFP-positive terminals (Fig. 3F,G, arrows), which was also significantly higher than that observed in CB1R-KO mice (Fig. 3H). These results indicate that low, but significant, CB1R expression occurs in thalamic terminals in L2/3 of the barrel cortex at P10. Because some thalamic terminals express both VGluT1 and VGluT2 at this age (Nakamura et al., 2007), we could not determine whether L4 terminals possessed CB1Rs.
Figure 3.
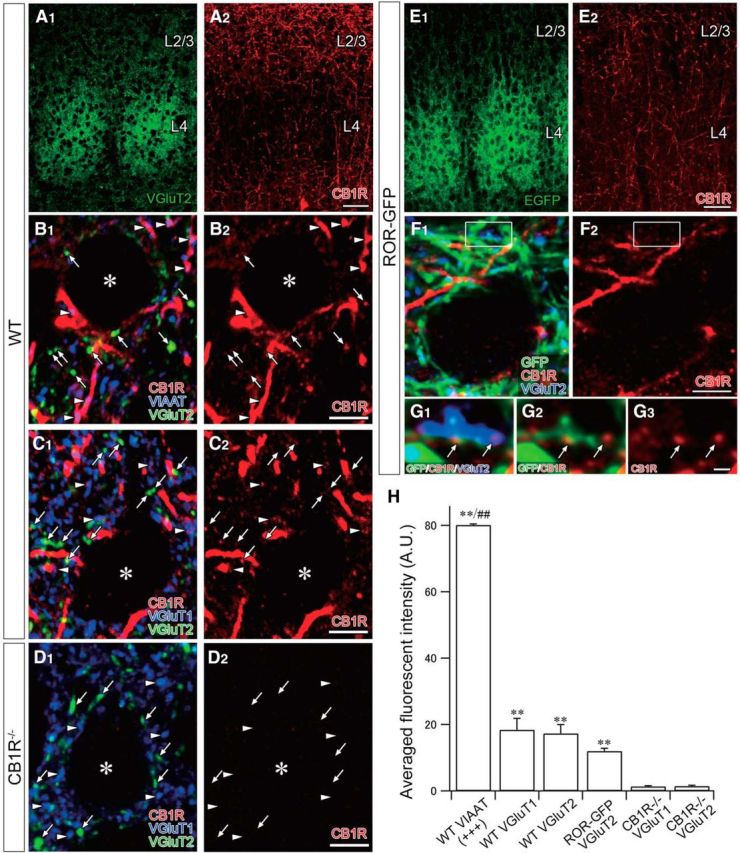
Fluorescence immunohistochemistry for the localization of CB1Rs at thalamocortical terminals at P10. A, Low-magnification images of the L4–L2/3 showing double immunofluorescence staining for VGluT2 (A1) and CB1R (A2) in wild-type mice. B–D, Triple immunofluorescence for CB1R, VGluT1, VGluT2, and VIAAT. B, Arrows and arrowheads indicate VGluT2- and VIAAT-positive terminals in L2/3, respectively. C, Arrows and arrowheads indicate VGluT2- and VGluT1-positive terminals in L2/3, respectively. D, Almost blank labeling in CB1R-KO mice. Arrows and arrowheads indicate VGluT2- and VGluT1-positive terminals in L2/3, respectively. E, Low-magnification images of the L4–L2/3 showing double immunofluorescence for GFP (E1) and CB1R (E2) in ROR-GFP mice. F, Triple immunofluorescence for CB1R, GFP, and VGluT2. The boxed region is enlarged in G. G, Arrows indicate CB1R-positive puncta in VGluT2- and GFP-positive terminals in L2/3. H, Comparison of the fluorescence intensity of CB1R staining in each terminal from CB1R+/+, CB1R−/−, and ROR-GFP mice. Although much weaker than VIAAT-positive terminals, VGluT2-positive terminals show significant immunoreactivity for CB1R. VIAAT-positive terminals showed either strong (+++) or null immunoreactivity for CB1R, and data were corrected only from those displaying strong immunoreactivity. **p < 0.01, comparison between WT or ROR-GFP and CB1R−/− mice at each VGluT1- and VGluT2-positive terminal. ##p < 0.01, comparison between VIAAT and VGluT1- or VGluT2-positive terminals in wild-type mice. Data were obtained from three mice for each genotype. Error bars indicate SEM. Asterisks indicate cell somas. Scale bars: A, E, 50 μm; B–D, F, 5 μm; G, 1 μm.
t-LTD at thalamus-L2/3 synapses is mediated by CB1R, and induced in both positive and negative timing delays during the second postnatal week
We then examined whether CB1R was involved in the induction of t-LTD at VB-L2/3 synapses. Post-before-pre stimulation (−25 ms) at VB-L2/3 synapse consistently induced t-LTD (Fig. 4A), as summarized in Figure 4D (VB → L2/3, post-before-pre, 55.6 ± 6.3% of control, n = 18, p < 0.05, t test). AM251 (5 μm), a CB1R inverse agonist, successfully blocked the induction of t-LTD (Fig. 4B,D; VB → L2/3, AM251 + post-before-pre, 96.7 ± 3.4% of pre-pairing, n = 16, p = 0.22, t test), indicating that the t-LTD was mediated by CB1Rs. This was also confirmed by occlusion experiments using WIN (Fig. 4C,D; VB → L2/3 WIN + post-before-pre, 101.4 ± 3.1% of pre-pairing after WIN treatment, n = 9, p = 0.64, t test). In contrast, we confirmed that, at L4-L2/3 synapses, post-before-pre stimulation resulted in modest LTP (114.4 ± 6.4% of control, range 57.0%–175.7%) during the second postnatal week, as shown in Figure 4D (left column) and consistent with a previous study (Itami and Kimura, 2012).
Figure 4.

VB-L2/3 synapses exhibited t-LTD in both positive and negative timing delays in a cannabinoid-dependent manner. A, Example of t-LTD by post-before-pre stimulation (25 ms delay: −25 ms). B, t-LTD by −25 ms stimulation was blocked by the CB1R antagonist AM251 (5 μm). C, t-LTD by −25 ms timing stimulation was occluded by prior application of the CB1R agonist WIN (5 μm). D, Summary of t-LTD (−25 ms delay) and the effects of WIN and AM251 on t-LTD at VB-L2/3 synapses, together with the effect of post-before-pre stimulation at L4–L2/3 synapses during the second postnatal week for comparison. E, VB-L2/3 synapses show t-LTD by both positive and negative timing stimulation in a timing-dependent manner.
We then examined the timing dependence of t-LTD, i.e., whether the magnitude of the depression changes depending on the spike timing of presynaptic and postsynaptic cells. When tested with various pairing stimulation timing, we found that pre-before-post, as well as post-before-pre, timing led to a variable magnitude of depression in a timing-dependent manner: that is, the shorter the timing difference, the larger the depression (Fig. 4E). This STDP profile is opposite to that observed in L4-L2/3 during the second postnatal week (Itami and Kimura, 2012), which exhibits only potentiation in both pre-before-post and post-before-pre timing, with the peak of potentiation at the time of zero.
Localization of CB1Rs responsible for t-LTD using global and cortical excitatory cell-specific CB1R-KOs
The requirement of CB1Rs in t-LTD at VB-L2/3 synapses during P9-P13 was confirmed by the lack of t-LTD in global CB1R-KOs (Fig. 5A,C; CB1R−/−, 97.9 ± 1.4%, n = 10), in contrast to the modest and robust t-LTD formation in CB1R heterozygotes (Fig. 5C; CB1R+/−, 85.4 ± 2.7%, n = 23, p = 0.008, t test) and in CB1R wild-type mice (Fig. 5A,C; CB1R+/+, 77.2 ± 4.5%, n = 13, p < 0.001, t test). However, t-LTD at VB-L2/3 was induced in cortical glutamatergic-specific CB1R-KOs (Fig. 5B,C; NEX-Cre+/−;CB1Rfl/fl, 78.9 ± 6.5%, n = 12, p < 0.01, t test) to the same extent as in their littermate control flox/flox mice (Fig. 5B,C; CB1Rfl/fl, 77.7 ± 6.6% of baseline, n = 10, p < 0.01, t test). These results indicate that t-LTD at VB-L2/3 synapses was mediated by CB1Rs that were most likely localized at thalamic terminals, but not at cortical cells. On the contrary, we confirmed that t-LTD at L4-L2/3 after P15 was mediated by CB1Rs located at cortical cells, consistent with the previous studies (Bender et al., 2006; Itami and Kimura, 2012). During P15-P19, global KOs failed to exhibit t-LTD at L4-L2/3 synapses (Fig. 5D,F; CB1R−/−, 103.6 ± 4.2%, n = 10, p = 0.41, t test), as did the cortical glutamatergic cell-specific KOs (Fig. 5E,F; NEX-Cre+/−;CB1Rfl/fl, 101.1 ± 3.4%, n = 14, p = 0.62, t test). t-LTD was induced in control flox/flox mice (Fig. 5E,F; CB1Rfl/fl, 77.9 ± 5.1%, n = 14, p < 0.001, t test). These results indicate that t-LTD at L4-L2/3 synapses is mediated by CB1Rs located at cortical glutamatergic neurons, most likely at L4 cell terminals to L2/3 cells after P15.
Figure 5.

Conditional KOs for CB1R account for pathway- and age-dependent t-LTD. A–C, During P9–P13, t-LTD at VB-L2/3 synapses was mediated by CB1R, localized most likely at thalamic terminals, but not at cortical cells. t-LTD at VB-L2/3 was induced in cortical glutamatergic cell-specific CB1R KOs (B,C; NEX-Cre+/−;CB1Rfl/fl) to the same extent as in their control flox/flox littermates (B,C; CB1Rfl/fl). The requirement of CB1R in t-LTD at VB-L2/3 synapse was confirmed by the lack of t-LTD in global CB1R KOs (A,C; CB1R−/−), in contrast to modest and robust t-LTD formation in CB1R heterozygotes and CB1R wild-type (A,C; CB1R+/−, CB1R+/+). C, F, Numbers in parentheses indicate the number of animals. D–F, During P15–P19, t-LTD at L4–L2/3 synapses was mediated by CB1R located at cortical cells because global KOs failed to exhibit t-LTD (D,F; CB1R−/−), as did the cortical glutamatergic cell-specific KOs (E,F; NEX-Cre+/−;CB1Rfl/fl), whereas it was induced in control flox/flox mice (E,F; CB1Rfl/fl).
Together, our results demonstrate that thalamic terminals to L2/3 pyramidal cells possess functional CB1Rs and that these receptors most likely mediate t-LTD during the second postnatal week.
Developmental transition of the effect of random activation of presynaptic and postsynaptic cells
We have thus far observed that thalamocortical synapses alter properties of STDP in a manner that depends on synapse target and age. Before CP is differentiated to L4, L2/3, TCA-CP exhibited t-LTP in both pre-before-post and post-before-pre timing. After L4 and L2/3 are differentiated, TCA-L2/3 synapses exhibited t-LTD in both pre-before-post and post-before-pre timing, whereas TCA-L4 lost STDP (Fig. 6A). Thus, thalamocortical STDP switched its direction from strengthening to weakening with the emergence of L4, L2/3. We previously demonstrated that, under t-LTP mechanism, random firing of presynaptic and postsynaptic cells within a short time window (e.g., −50 to 50 ms) strengthened the synaptic connection (Itami and Kimura, 2012). A developmental switch of STDP from t-LTP (Fig. 1B) to t-LTD (Fig. 4E) predicts that closely associated firing of presynaptic and postsynaptic cells would result in a transition from strengthening (LTP) to weakening (LTD) of synaptic connections during development. We tested whether such a transition actually occurs at thalamocortical synapses. Computer-generated random-delay timing stimulation (Fig. 6B–D) was applied to presynaptic and postsynaptic cells. We found that this random-delay stimulation led to potentiation (126.4 ± 9.1% of control, n = 5) in P5-P6 animals, whereas depression (54.4 ± 6.8% of control, n = 8) was induced in P8-P13 animals (Fig. 6E). This result indicates that correlated presynaptic and postsynaptic activity leads to a stronger connection during the first postnatal week, but a weaker connection during the second postnatal week, regardless of spiking order. A similar transition was demonstrated at L4-L2/3, when STDP was shown to switch from t-LTP to Hebbian STDP at P13-P15 (Itami and Kimura, 2012).
Exogenous administration of cannabinoid agonist leads to morphological attenuation of thalamocortical projections
Next, we asked whether continuous physiological changes due to an activation of CB1R could lead to any morphological changes in thalamocortical projections. To test this, we observed morphology following chronic administration of CB1R agonists or antagonist. In this experiment, we again used TCA-reporter mice (ROR-GFP), which express GFP in thalamic axons (Nakagawa and O'Leary, 2003). Our finding that the magnitude of t-LTP gradually decreased from P4-P5 and switched to t-LTD after P7-P8 (Fig. 1F) raises the possibility that t-LTD begins around P4-P5. In support of this, WIN administration (1 mg/kg, P2-P3 to P7, i.p. injection, daily) strongly suppressed thalamic innervation in the cortex at P7 (Fig. 7A, bottom). In contrast, AM281 administration (15 mg/kg, P5 to P12, i.p. injection, daily) slightly enhanced thalamic innervations (Fig. 7C, bottom). Quantitative analyses of the WIN and AM281 effects were performed with VGluT2 staining, which confirmed that both effects were dose-dependent (Fig. 8A–D). We further tested Δ9-THC, another CB1R agonist and an active ingredient of marijuana, on thalamic axons. Δ9-THC injection (10 mg/kg, P2-P7, i.p., daily) strongly suppressed normal thalamic innervation, as visualized by GFP (Fig. 7E,F). This suppression was also dose-dependent, as confirmed by VGluT2 staining (Fig. 8E,F). From these results, we concluded that exogenous administration of CB1R agonists attenuated thalamocortical innervation. Therefore, physiological changes due to CB1R activation could be linked to morphological alterations.
Figure 7.
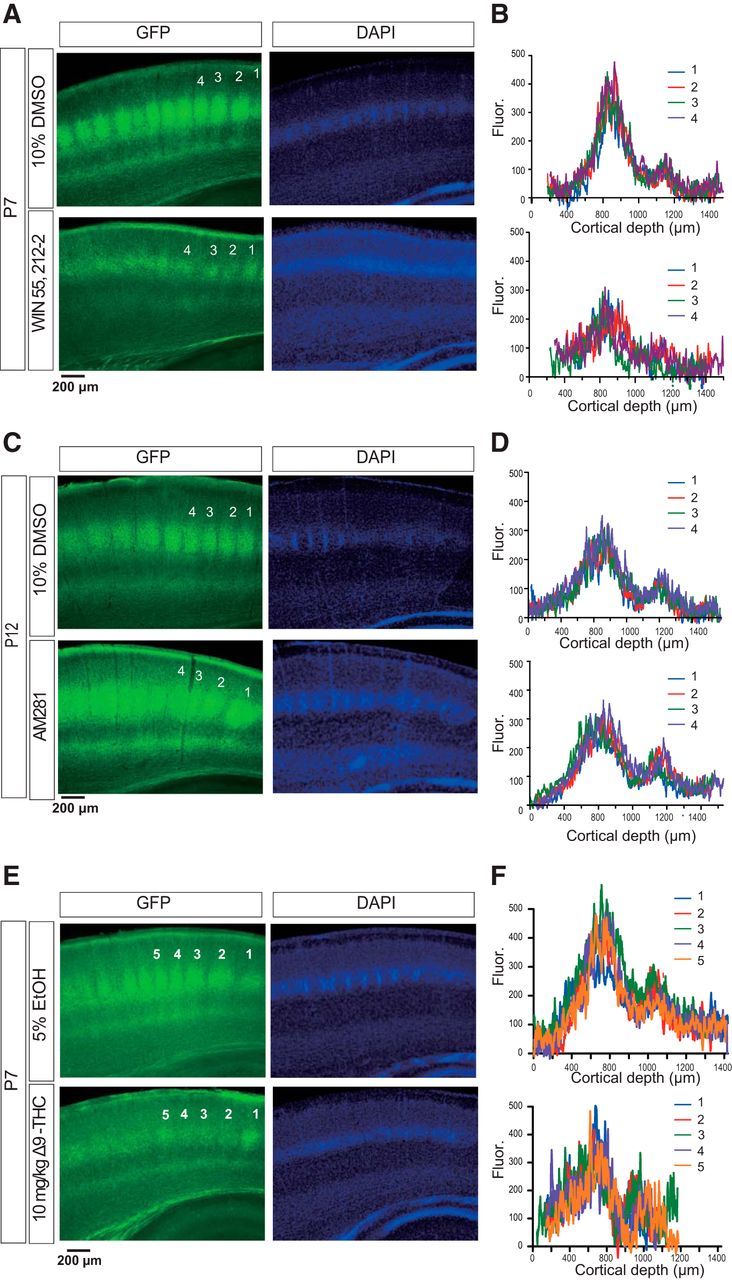
Exogenous administration of CB1R agonists disrupted TCA projections, whereas that of CB1R antagonist enhanced TCA projections. A, WIN injection (bottom, 1 mg/kg, P2–P7, i.p., daily) strongly suppressed TCA projections compared with vehicle injection (top) revealed by TCA reporter mice, which express GFP in TCA. B, Plots of fluorescence density from the pia surface (as depth = 0) to the white matter crossing the barrel centers at representative four barrels as indicated by numbers in the pictures. C, AM251 injection (bottom, 1 mg/kg, P5–P12, i.p. daily) slightly expanded the TCA projection, as revealed by the plots of fluorescence density (D) compared with vehicle injection (top). E, Δ9-THC injection (10 mg/kg, P2–P7, i.p., daily) also had a suppressive effect on TCA projections. F, Fluorescence density in the five columns is shown, following vehicle (top) or Δ9-THC (bottom) injection.
Figure 8.
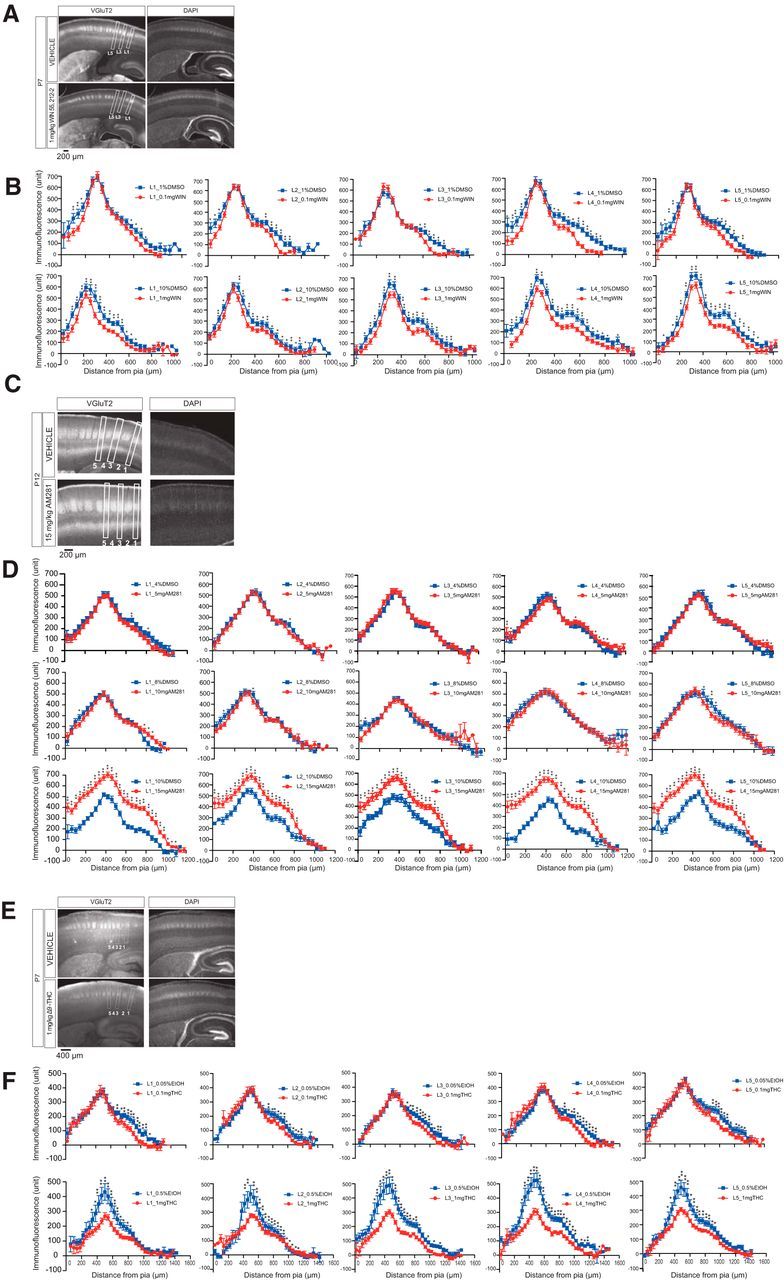
Chronic administration of WIN or Δ9-THC suppresses TCA projections, whereas AM281 administration enhances these projections. A, Photomicrograph of VGluT2 immunofluorescence (left) and DAPI staining (right) after administering WIN (1 mg/kg, i.p. injection P2–P3 to 7, daily) (bottom) and vehicle (top). B, Fluorescence density plotted at the indicated distance from the pia surface. WIN suppressed the thalamic projections in a dose-dependent manner. At 0.1 mg/kg, suppression was often observed only in the upper (L2/3) and lower (L5/6) layers (top row). However, a higher dose of WIN (1 mg/kg) suppressed the projections throughout the layers (bottom row). Data were collected from 38 slices (7 pups) for WIN 1 mg/kg, 28 slices (7 pups) for vehicle (DMSO), 32 slices (8 pups) for WIN 0.1 mg/kg, and 26 slices (6 pups) for vehicle. Asterisks indicate significant differences between the vehicle and WIN. C, D, Photomicrograph (C) and plots of fluorescence density (D) from AM281-treated mice, as in A and B. AM281 at the highest dose (15 mg/kg, i.p. injection P5-P12, daily) increased thalamocortical innervation throughout the layers. Data were collected from 43 slices (8 pups) for 5 mg AM281, 40 slices (8 pups) for vehicle (DMSO), 46 slices (9 pups) for 10 mg AM281, 34 slices (8 pups) for vehicle, 42 slices (11 pups) for 15 mg AM281, and 27 slices (6 pups) for vehicle. Asterisks indicate significant differences between the vehicle and AM281. E, F, Photomicrograph (E) and plots of fluorescence density (F) from Δ9-THC-treated mice, as in A and B, and C and D, respectively. Δ9-THC (1 mg/kg, i.p. injection P2-P7, daily) greatly suppressed thalamic projections (F). Data were collected from 34 slices (5 pups) for 0.1 mg Δ9-THC, 15 slices (2 pups) for vehicle (ethanol), 23 slices (3 pups) for 1 mg Δ9-THC, and 37 slices (5 pups) for vehicle. Asterisks indicate significant differences between the vehicle and Δ9-THC. B, D, F, Mean ± SEM values are plotted. Statistical significance was determined using two-way ANOVAs and post hoc tests; *p < 0.05. **p < 0.01.
Endogenous cannabinoid signaling regulates thalamocortical projections
The next question we addressed was whether endogenous cannabinoid signaling participates in the regulation of thalamocortical projections. To explore this, we stained individual TCAs with DiI in thalamocortical slices from CB1R−/−, CB1R+/−, and CB1R+/+ mice at P12, and compared morphologies between the genotypes. TCAs are known to be confined under the L4-L2/3 border and within a single barrel in L4 in matured cortex (Bernardo and Woolsey, 1987). In support of this, individual TCAs labeled with DiI terminated within each barrel at P12 (Fig. 9A,C) in CB1R+/+ animals. In contrast, in CB1R+/− and CB1R−/− animals, a number of TCAs persisting in L2/3 could be observed, and they seemed to ignore the barrel borders (Fig. 9B,D,E). Quantitatively, the total axon length and the number of branches in L4-L2/3 areas were not significantly different between the genotypes (Fig. 10A,B; p = 0.102 and 0.936, respectively, one-way ANOVA). However, the proportion of the axon length found in L2/3 compared with the total length found in L4-L2/3 was significantly larger in CB1R+/− and CB1R−/− (Fig. 10C; p < 0.001, one-way ANOVA). In addition, we measured the density of the axonal arbor within an arborization area to quantify the diffuseness of the branching. We found that the arborization of axons in CB1R+/− and CB1R−/− mice was significantly less dense than that of CB1R+/+ mice (Fig. 10D; p < 0.001, one-way ANOVA). These results are consistent with the idea that CB1R signaling regulated thalamic axons in L2/3 to be confined within L4. This implies that endogenous CB1R signaling participate in the regulation of thalamocortical projections.
Figure 9.

Endogenous cannabinoid signaling regulates TCA projections, as revealed by DiI staining of individual TCAs in CB1R−/−. A–E, Analyses of individual thalamocortical axons stained using DiI crystals placed in the thalamus at P12 indicate that CB1Rs play a major role in confining axons below the L4-L2/3 border. A, B, Photomicrographs of low (left) and high (middle) magnification and a reconstructed illustration (right) of a thalamocortical axon from CB1R+/+ (A) and CB1R−/− (B) mice. Left, Red arrowheads indicate septa between barrels. Right, Hatched squares represent barrels. Scale bars: left, 500 μm; middle, right, 100 μm. C–E, Other examples of axon illustrations from CB1R+/+ (C), CB1R−/− (D), and CB1R+/− (E). The pia surface is delineated for reference. Scale bars, 100 μm.
Figure 10.

Quantification of axon length, branches, and axon density in CB1R−/−, revealing regulation of TCA axons by endocannabinoid. Neither total axon length within L4 and L2/3 (A) nor total branch numbers (B) differed significantly across genotypes (one-way ANOVA). C, The proportion of axon length in the L2/3 area to the total axon length in L4, L2/3 area was significantly larger in CB1R+/− and CB1R−/− compared with CB1R+/+ (p < 0.001, one-way ANOVA, Bonferroni post hoc test). D, Axonal arbor density in each arborization area was also significantly lower in CB1R+/− and CB1R−/− compared with CB1R+/+ (p < 0.001, one-way ANOVA, Bonferroni post hoc test). **Significant difference, p < 0.001.
Discussion
Differential expression of CB1Rs at thalamocortical terminals to L4 and L2/3 cells
We found that thalamic projections to L2/3 express CB1Rs and show t-LTD, while thalamic projections to L4 do not express functional CB1Rs, nor do they show STDP during the second postnatal week. These findings seem to indicate that the thalamic neurons projecting to L4 are distinct from those projecting to L2/3, as it is likely that different terminals of the same neuron express the same presynaptic receptors (Kimura and Baughman, 1997). However, the opposite case can also occur, in which the expression of presynaptic receptors (Shigemoto et al., 1996; Sun and Dobrunz, 2006), or other presynaptic properties, such as release probabilities (Reyes et al., 1998; Rozov et al., 2001), is distinct across terminals of the same neuron and determined by the postsynaptic identity. Thus, we cannot rule out the possibility that distinct branches of the same thalamic axons project to L4 and L2/3 cells, whereas only those terminating onto L2/3 cells express CB1Rs during the second postnatal week.
Site of action of cannabinoids
We used both a NEX-Cre animal and a global knock-out of CB1R and demonstrated that t-LTD at L4-L2/3 synapses is not induced in either animals. We thus concluded that the sites of action of cannabinoids responsible for t-LTD at L4-L2/3 synapses are most likely the cortical synapses of L4 cell terminals onto L2/3 cells. This is important, as a previous study claims that CB1Rs in astrocytes are responsible for cortical t-LTD (Min and Nevian, 2012). We also demonstrated that t-LTD at VB-L2/3 synapses was induced, even in NEX-Cre+/−;CB1Rfl/fl mice, suggesting that the CB1Rs responsible for this t-LTD are likely to be at TCA terminals. Strictly speaking, however, we could not completely rule out the possibility of the involvement of astrocytes or inhibitory interneurons, which express abundant CB1Rs (Bodor et al., 2005; Min and Nevian, 2012). Similarly, we demonstrated morphological reorganizations following systemic administration of WIN, AM281, and Δ9-THC. Although not a very likely possibility, we again could not exclude the possibility that these substances acted on CB1Rs at inhibitory cells, astrocytes, or further trigeminal cells. One possible experiment to address this issue would be to use animals in which CB1Rs are knocked out in the thalamus. Experiments using such animals are expected to further elucidate the precise site of action of cannabinoids for producing the physiological and morphological effects observed here.
Molecular mechanisms leading to afferent retraction: cannabinoids versus semaphorins
A recent study identified semaphorins (Semas) as retrograde signals that are involved in regulating the elimination of early formed redundant fibers in the cerebellum (Uesaka et al., 2014). Sema7A facilitates this process and Sema3A suppresses the elimination of multiple immature climbing fibers, thus shaping the network into the adult type with only single innervations. Interestingly, Sema7A is expressed in the barrel cortex, although not in L2/3, but rather in L4 spiny stellate cells, which are the main target neurons of thalamocortical afferents. Therefore, Sema7A is unlikely to induce the elimination of afferents in the barrel cortex (Roland et al., 2014). In support of this, a previous study reported that Sema7A facilitates branching and synapse formation in thalamocortical axons (Fukunishi et al., 2011). It is not uncommon for the same molecules to play different roles in different areas of the brain. For example, Sema3A suppresses elimination in the cerebellum (Uesaka et al., 2014) but works as a repellent molecule and inhibits axon branching in the cortex (Dent et al., 2004).
Through what molecular mechanism does CB1R activation cause axon disruption? The exact role of cannabinoid signaling in the alteration of neuronal morphology is controversial, with some studies reporting neurite retraction (Berghuis et al., 2007; Argaw et al., 2011) and others indicating neurite outgrowth (Mulder et al., 2008; Vitalis et al., 2008; Watson et al., 2008). However, a recent study identified one molecular pathway that leads to rapid neural remodeling by the retraction of neurites and growth cones. They found that CB1R activation coupled to G12/13 proteins triggers rapid nonmuscle myosin II-dependent contraction of the actomyosin cytoskeleton through a Rho-GTPase and Rho-associated kinase (Roland et al., 2014), which may underlie the current observations. This is also interesting because t-LTD is most likely mediated by the Gi/o protein (Castillo et al., 2012). If thalamocortical neurons exhibit both t-LTD and retraction through CB1R in the same cell, as suggested from the present study, CB1R may work as a molecule that links physiological and morphological changes by using distinct G-proteins. Further study will be necessary to determine whether Gi/o, G12/13, and CB1R colocalize at the same thalamic neurons.
t-LTP and t-LTD may underlie network formation and reshaping
It has been shown that neural activity is required for initial synapse formation of thalamocortical axons on cortical cells (Katz and Shatz, 1996), but the precise activities of the presynaptic and postsynaptic cells required were unknown. In addition, the mechanism by which activity is further translated into structural changes has been unclear. We have shown here that robust t-LTP at the TCA-CP synapse can be induced until P4-P5, but its magnitude decreases gradually, likely resulting from an addition of t-LTD and reduction of t-LTP. This then switches to t-LTD after P7-P8 at the noncanonical target layer of L2/3, and STDP is lost at the canonical target of L4. Thus, the period during which t-LTP and t-LTD can be induced matches closely the time of synapse formation and retraction, respectively, at the thalamocortical synapse. We also showed that, under t-LTP and t-LTD rule, closely related activity of presynaptic and postsynaptic cells caused strengthening and weakening of synaptic connections regardless of spike order (Fig. 6E). These findings indicate that STDP rule and spontaneous activity may underlie network formation and reshaping at thalamocortical projections at least during the first two postnatal weeks. t-LTP has also been suggested to underlie synapse formation at L4-L2/3 synapse during the second postnatal week (Itami and Kimura, 2012). Interestingly, in both cases, t-LTP is switched to a distinct form of STDP in about a week; t-LTP is switched to t-LTD at TCA synapses for shaping aberrant innervations to L2/3. Similarly, t-LTP is switched to Hebbian STDP with LTP/LTD at L4-L2/3 synapses, which is highly relevant to the initiation of critical period of map plasticity, as we have discussed (Itami and Kimura, 2012). This is because the developmental switch of STDP at L4-L2/3 synapses coincides with the developmental establishment of sequential “L4 followed by L2/3” spiking activity by sensory inputs, due to the fast activation of parvalbumin-GABA cells in L4 by thalamic inputs (Kimura et al., 2010; Itami and Kimura, 2012). Thus, STDP and its alteration are closely related to the formation and/or function of neuronal circuits.
What causes the developmental switch of STDP at thalamocortical synapses?
We demonstrated that CB1R was expressed during the second postnatal week when t-LTD could be induced. It is most likely that the expression of CB1R caused the developmental switch of STDP. Future immunohistochemical studies identifying developmental changes in the CB1R expression at TCA terminals or studies using quantitative PCR to assess CB1R mRNA levels at the VB nucleus are needed. The production of endocannabinoids may well be developmentally regulated. However, there are as yet no studies exploring the developmental changes in the amounts of endocannabinoids or their synthesizing enzymes.
We show that t-LTP is dependent on NMDA receptors (Fig. 1C,E). Although we have not performed the testing, NMDA receptors may also be involved in the induction of t-LTD (Sjöström et al., 2003; Bender et al., 2006). Are there changes in the activation of NMDA receptors that may be relevant to a developmental switch or an extinction of STDP? The dominant subunit components of NMDA receptors change from NR2B to NR2A at TCA-L4 synapses during the first postnatal week (Barth and Malenka, 2001; Lu et al., 2001; Yanagisawa et al., 2004). Although there are currently no studies regarding such a subunit change at TCA-L2/3 synapses, it was shown that suppressing the emergence of the NR2A subunit is unable to block the extinction of pairing-induced LTP at TCA-L4 synapses in NR2A-knock-out mice (Lu et al., 2001). This change in the properties of NMDA receptors may not be relevant to the developmental changes of STDP.
CB1R signaling and disruption of the neural network
We have demonstrated that endogenous CB1R signaling is required for the formation of normal thalamocortical projections. We also showed that exogenously applied CB1R ligands disrupt the normal formation of thalamocortical projections. CB1R is one of the most abundant G-protein-coupled receptors in the brain and is prevalent throughout the CNS. Our findings predict that CB1Rs may play an important role in network formation in other areas of the brain. This hypothesis should be explored in more detail in future work, as the abuse of substances that change cannabinoid signaling may cause serious disruptions in neural projections. Considering that most CB1R ligands are highly lipophilic and easily pass the blood–brain barrier, the use of cannabinoids and their analogs needs to be studied with respect to their effects on brain plasticity.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research on Innovative Area Mesoscopic Neurocircuitry 23115515 and 25115721 to F.K., MEXT26430022 to C.I., United States-Japan Brain Research Cooperative Program to C.I. and F.K., Takeda Science Foundation to C.I. and F.K., National Institutes of Health NS048884 to H.-C.L., and Baylor IRDDRC core facility National Institutes of Health/National Institute of Child Health and Human Development P30-HD024064 to H.-C.L We thank Drs. Masanobu Kano, Yuki Hashimotodani, Michael P. Stryker, Yoshio Hata, and Ken Mackie for reading the early version of the manuscript and providing comments and suggestions; Drs. Beat Lutz and Giovanni Marsicano for CB1R global KO and conditional allele mice; Drs. Klaus-Armin Nave and Sandra Goebbels for NEX-Cre mouse line; and Ximin Zhou and Chris P. Jew for technical assistance.
The authors declare no competing financial interests.
References
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-J. [DOI] [PubMed] [Google Scholar]
- Agmon A, O'Dowd DK. NMDA receptor-mediated currents are prominent in the thalamocortical synaptic response before maturation of inhibition. J Neurophysiol. 1992;68:345–349. doi: 10.1152/jn.1992.68.1.345. [DOI] [PubMed] [Google Scholar]
- Agmon A, Yang LT, O'Dowd DK, Jones EG. Organized growth of thalamocortical axons from the deep tier of terminations into layer IV of developing mouse barrel cortex. J Neurosci. 1993;13:5365–5382. doi: 10.1523/JNEUROSCI.13-12-05365.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw A, Duff G, Zabouri N, Cécyre B, Chaine N, Cherif H, Tea N, Lutz B, Ptito M, Bouchard JF. Concerted action of CB1 cannabinoid receptor and deleted in colorectal cancer in axon guidance. J Neurosci. 2011;31:1489–1499. doi: 10.1523/JNEUROSCI.4134-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester-Rosado CJ, Albright MJ, Wu CS, Liao CC, Zhu J, Xu J, Lee LJ, Lu HC. mGluR5 in cortical excitatory neurons exerts both cell-autonomous and -nonautonomous influences on cortical somatosensory circuit formation. J Neurosci. 2010;30:16896–16909. doi: 10.1523/JNEUROSCI.2462-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AL, Malenka RC. NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses. Nat Neurosci. 2001;4:235–236. doi: 10.1038/85070. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Connors BW. Short-term dynamics of thalamocortical and intracortical synapses onto layer 6 neurons in neocortex. J Neurophysiol. 2002;88:1924–1932. doi: 10.1152/jn.2002.88.4.1924. [DOI] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urbán GM, Monory K, Marsicano G, Matteoli M, Canty A, Irving AJ, Katona I, Yanagawa Y, Rakic P, Lutz B, Mackie K, Harkany T. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science. 2007;316:1212–1216. doi: 10.1126/science.1137406. [DOI] [PubMed] [Google Scholar]
- Bernardo KL, Woolsey TA. Axonal trajectories between mouse somatosensory thalamus and cortex. J Comp Neurol. 1987;258:542–564. doi: 10.1002/cne.902580406. [DOI] [PubMed] [Google Scholar]
- Bodor AL, Katona I, Nyíri G, Mackie K, Ledent C, Hájos N, Freund TF. Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J Neurosci. 2005;25:6845–6856. doi: 10.1523/JNEUROSCI.0442-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano SM, Shatz CJ. Activity-dependent cortical target selection by thalamic axons. Science. 1998;281:559–562. doi: 10.1126/science.281.5376.559. [DOI] [PubMed] [Google Scholar]
- Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- Dent EW, Barnes AM, Tang F, Kalil K. Netrin-1 and semaphorin 3A promote or inhibit cortical axon branching, respectively, by reorganization of the cytoskeleton. J Neurosci. 2004;24:3002–3012. doi: 10.1523/JNEUROSCI.4963-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foeller E, Feldman DE. Synaptic basis for developmental plasticity in somatosensory cortex. Curr Opin Neurobiol. 2004;14:89–95. doi: 10.1016/j.conb.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/S0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- Fujiyama F, Furuta T, Kaneko T. Immunocytochemical localization of candidates for vesicular glutamate transporters in the rat cerebral cortex. J Comp Neurol. 2001;435:379–387. doi: 10.1002/cne.1037. [DOI] [PubMed] [Google Scholar]
- Fukudome Y, Ohno-Shosaku T, Matsui M, Omori Y, Fukaya M, Tsubokawa H, Taketo MM, Watanabe M, Manabe T, Kano M. Two distinct classes of muscarinic action on hippocampal inhibitory synapses: M2-mediated direct suppression and M1/M3-mediated indirect suppression through endocannabinoid signalling. Eur J Neurosci. 2004;19:2682–2692. doi: 10.1111/j.0953-816X.2004.03384.x. [DOI] [PubMed] [Google Scholar]
- Fukunishi A, Maruyama T, Zhao H, Tiwari M, Kang S, Kumanogoh A, Yamamoto N. The action of Semaphorin7A on thalamocortical axon branching. J Neurochem. 2011;118:1008–1015. doi: 10.1111/j.1471-4159.2011.07390.x. [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44:611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/S0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- Itami C, Kimura F. Developmental switch in spike timing-dependent plasticity at layers 4–2/3 in the rodent barrel cortex. J Neurosci. 2012;32:15000–15011. doi: 10.1523/JNEUROSCI.2506-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itami C, Samejima K, Nakamura S. Improved data processing for optical imaging of developing neuronal connectivity in the neonatal mouse barrel cortex. Brain Res Protoc. 2001;7:103–114. doi: 10.1016/S1385-299X(01)00048-4. [DOI] [PubMed] [Google Scholar]
- Itami C, Kimura F, Kohno T, Matsuoka M, Ichikawa M, Tsumoto T, Nakamura S. Brain-derived neurotrophic factor-dependent unmasking of “silent” synapses in the developing mouse barrel cortex. Proc Natl Acad Sci U S A. 2003;100:13069–13074. doi: 10.1073/pnas.2131948100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kawamura Y, Fukaya M, Maejima T, Yoshida T, Miura E, Watanabe M, Ohno-Shosaku T, Kano M. The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J Neurosci. 2006;26:2991–3001. doi: 10.1523/JNEUROSCI.4872-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura F, Baughman RW. Distinct muscarinic receptor subtypes suppress excitatory and inhibitory synaptic responses in cortical neurons. J Neurophysiol. 1997;77:709–716. doi: 10.1152/jn.1997.77.2.709. [DOI] [PubMed] [Google Scholar]
- Kimura F, Nishigori A, Shirokawa T, Tsumoto T. Long-term potentiation and N-methyl-D-aspartate receptors in the visual cortex of young rats. J Physiol Lond. 1989;414:125–144. doi: 10.1113/jphysiol.1989.sp017680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura F, Itami C, Ikezoe K, Tamura H, Fujita I, Yanagawa Y, Obata K, Ohshima M. Fast activation of feedforward inhibitory neurons from thalamic input and its relevance to the regulation of spike sequences in the barrel cortex. J Physiol. 2010;588:2769–2787. doi: 10.1113/jphysiol.2010.188177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HC, Gonzalez E, Crair MC. Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron. 2001;32:619–634. doi: 10.1016/S0896-6273(01)00501-3. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutiérrez SO, van der Stelt M, López-Rodriguez ML, Casanova E, Schütz G, Zieglgänsberger W, Di Marzo V, Behl C, Lutz B. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–88. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- Min R, Nevian T. Astrocyte signaling controls spike timing-dependent depression at neocortical synapses. Nat Neurosci. 2012;15:746–753. doi: 10.1038/nn.3075. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Fukaya M, Shimizu H, Watanabe M. Subtype switching of vesicular glutamate transporters at parallel fibre-Purkinje cell synapses in developing mouse cerebellum. Eur J Neurosci. 2003;17:2563–2572. doi: 10.1046/j.1460-9568.2003.02698.x. [DOI] [PubMed] [Google Scholar]
- Mulder J, Aguado T, Keimpema E, Barabás K, Ballester Rosado CJ, Nguyen L, Monory K, Marsicano G, Di Marzo V, Hurd YL, Guillemot F, Mackie K, Lutz B, Guzmán M, Lu HC, Galve-Roperh I, Harkany T. Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc Natl Acad Sci U S A. 2008;105:8760–8765. doi: 10.1073/pnas.0803545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa Y, O'Leary DD. Dynamic patterned expression of orphan nuclear receptor genes RORalpha and RORbeta in developing mouse forebrain. Dev Neurosci. 2003;25:234–244. doi: 10.1159/000072271. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Watakabe A, Hioki H, Fujiyama F, Tanaka Y, Yamamori T, Kaneko T. Transiently increased colocalization of vesicular glutamate transporters 1 and 2 at single axon terminals during postnatal development of mouse neocortex: a quantitative analysis with correlation coefficient. Eur J Neurosci. 2007;26:3054–3067. doi: 10.1111/j.1460-9568.2007.05868.x. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsam A, Seif I, Gaspar P. Refinement of thalamocortical arbors and emergence of barrel domains in the primary somatosensory cortex: a study of normal and monoamine oxidase a knock-out mice. J Neurosci. 2002;22:8541–8552. doi: 10.1523/JNEUROSCI.22-19-08541.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Lujan R, Rozov A, Burnashev N, Somogyi P, Sakmann B. Target-cell specific facilitation and depression in neocortical circuits. Nat Neurosci. 1998;1:279–285. doi: 10.1038/1092. [DOI] [PubMed] [Google Scholar]
- Rice FL, Van der Loos H. Development of the barrels and barrel field in the somatosensory cortex of the mouse. J Comp Neurol. 1977;171:545–560. doi: 10.1002/cne.901710408. [DOI] [PubMed] [Google Scholar]
- Roland AB, Ricobaraza A, Carrel D, Jordan BM, Rico F, Simon A, Humbert-Claude M, Ferrier J, McFadden MH, Scheuring S, Lenkei Z. Cannabinoid-induced actomyosin contractility shapes neuronal morphology and growth. eLife. 2014;3:e03159. doi: 10.7554/eLife.03159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E. Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol. 2001;531:807–826. doi: 10.1111/j.1469-7793.2001.0807h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senft SL, Woolsey TA. Growth of thalamic afferents into mouse barrel cortex. Cereb Cortex. 1991;1:308–335. doi: 10.1093/cercor/1.4.308. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kulik A, Roberts JD, Ohishi H, Nusser Z, Kaneko T, Somogyi P. Target-cell-specific concentration of a metabotropic glutamate receptor in the presynaptic active zone. Nature. 1996;381:523–525. doi: 10.1038/381523a0. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/S0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Sun HY, Dobrunz LE. Presynaptic kainate receptor activation is a novel mechanism for target cell-specific short-term facilitation at Schaffer collateral synapses. J Neurosci. 2006;26:10796–10807. doi: 10.1523/JNEUROSCI.2746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki C, Yamasaki M, Uchigashima M, Konno K, Yanagawa Y, Watanabe M. Cytochemical and cytological properties of perineuronal oligodendrocytes in the mouse cortex. Eur J Neurosci. 2010;32:1326–1336. doi: 10.1111/j.1460-9568.2010.07377.x. [DOI] [PubMed] [Google Scholar]
- Uesaka N, Uchigashima M, Mikuni T, Nakazawa T, Nakao H, Hirai H, Aiba A, Watanabe M, Kano M. Retrograde semaphorin signaling regulates synapse elimination in the developing mouse brain. Science. 2014;344:1020–1023. doi: 10.1126/science.1252514. [DOI] [PubMed] [Google Scholar]
- Vitalis T, Lainé J, Simon A, Roland A, Leterrier C, Lenkei Z. The Type 1 cannabinoid receptor is highly expressed in embryonic cortical projection neurons and negatively regulates neurite growth in vitro. Eur J Neurosci. 2008;28:1705–1718. doi: 10.1111/j.1460-9568.2008.06484.x. [DOI] [PubMed] [Google Scholar]
- Watson S, Chambers D, Hobbs C, Doherty P, Graham A. The endocannabinoid receptor, CB1, is required for normal axonal growth and fasciculation. Mol Cell Neurosci. 2008;38:89–97. doi: 10.1016/j.mcn.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Yanagisawa T, Tsumoto T, Kimura F. Transiently higher release probability during critical period at thalamocortical synapses in the mouse barrel cortex: relevance to differential short-term plasticity of AMPA and NMDA EPSCs and possible involvement of silent synapses. Eur J Neurosci. 2004;20:3006–3018. doi: 10.1111/j.1460-9568.2004.03756.x. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Kameyama K, Esumi K, Daimyo Y, Watanabe M, Hata Y. Developmental and visual input-dependent regulation of the CB1 cannabinoid receptor in the mounse visual cortex. PLoS One. 2013;8:e53082. doi: 10.1371/journal.pone.0053082. [DOI] [PMC free article] [PubMed] [Google Scholar]