

Abstract
We report the synthesis and biological evaluation of a series of (−)‐englerin A analogues obtained along our previously reported synthetic route based on a stereoselective gold(I) cycloaddition process. This synthetic route is a convenient platform to access analogues with broad structural diversity and has led us to the discovery of unprecedented and easier‐to‐synthesize derivatives with an unsaturation in the cyclopentyl ring between C4 and C5. We also introduce novel analogues in which the original isopropyl motif has been substituted with cyclohexyl, phenyl, and cyclopropyl moieties. The high selectivity and growth‐inhibitory activity shown by these new derivatives in renal cancer cell lines opens new ways toward the final goal of finding effective drugs for the treatment of renal cell carcinoma (RCC).
Keywords: enantioselective synthesis, englerin A, gold catalysis, natural products, renal cancer, tumor growth inhibition
Introduction
Since it was isolated from the stem bark of the east African plant Phyllanthus engleri by scientists at the US National Cancer Institute,1, 2 englerin A (1) (Scheme 1), a complex guaiane sesquiterpene, has attracted a lot of attention among the scientific community due to its selective inhibition of renal cancer cell line growth.3 Several total syntheses of englerin A4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 and analogues18, 19, 20, 21, 22, 23, 24, 25 have been reported to date. Recently, two distinct mechanisms for englerin A′s anticancer activity have been proposed, agonism of protein kinase C theta (PKCθ)26 and agonism of short transient receptor potential channels 4 and 5 (TRPC4/5).27
Scheme 1.
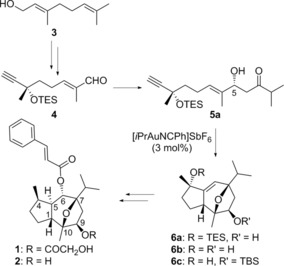
Enantioselective synthesis of (−)‐englerin A (1) based on a stereoselective gold(I)‐catalyzed [2+2+2] alkyne/alkene/carbonyl cycloaddition.6
We completed the enantioselective synthesis of (−)‐englerin A (1) and B (2)6 in 18 steps and 7 % overall yield from inexpensive geraniol (3) via α,β‐unsaturated aldehyde 4 (Scheme 1), following a similar approach to that used previously for the synthesis of (+)‐orientalol F and pubinernoid B.28 Thus, the highly stereoselective intramolecular gold(I)‐catalyzed [2+2+2] alkyne/alkene/carbonyl cycloaddition29 of 1,6‐enyne 5 a led to advanced intermediates 6 a–c with two different OR groups, which allow for the facile preparation of many derivatives.
Herein we report the synthesis and biological evaluation of a series of englerin A analogues prepared by following our synthetic route which allowed us to identify very active compounds with a double bond between C4 and C5. Interestingly, as part of the optimization and scale‐up, we found that only 1,6‐enynes of type 5 a with this specific relative configuration react productively in the gold(I)‐catalyzed cascade reaction to form oxatricyclic compounds.
Results and Discussion
The cinnamate and glycolate moieties are known to be key for englerin′s activity.18, 19, 21, 30, 31 Therefore, to take advantage of the different advanced intermediates 6 a–c generated through our synthetic route, our strategy consisted of adding these substituents to the oxatricyclic intermediates with various levels of unsaturation. In this way, we could have direct access to a set of analogues with great structural diversity.
The synthesis of an initial library of analogues started from the tricyclic structure 6 c (Scheme 2).6 The free hydroxy group at position C4 was esterified under standard conditions to give cinnamate 7 a, which could be further functionalized under Yamaguchi conditions32 to give the glycolate product 7 b in moderate yield. Together with this, the lactate derivative 7 c was also synthesized. The lactate moiety has shown good activities in other analogues.21 Following the total synthesis plan, 6 c can be isomerized to 8 a in two steps by an oxidation/reduction protocol,6, 28 allowing the synthesis of englerin A and B analogues with an extra unsaturation in the cyclopentyl ring (between C4 and C5). The cinnamate substituent was added by following the usual esterification method with cinnamoyl chloride, and the C9 alcohol was deprotected to give analogue 9 a (45 % yield, two steps). Esterification under Yamaguchi conditions with the TBDPS‐protected glycolic acid followed by deprotection afforded analogue 9 b (63 %, two steps).
Scheme 2.

Synthesis of (−)‐englerin derivatives from 6 c. Reagents and conditions: a) 1. cinnamoyl chloride, DMAP, Et3N, CH2Cl2, 45 °C, 2. TBAF, THF, 23 °C, 6 h: 7 a 85 %, 9 a, 45 %, 12 a 66 %; b) 1. esterification conditions: RCOOH, DMAP, NEt3, 2,4,6‐trichlorobenzoyl chloride, toluene, 23 °C, 1 h, 2. deprotection TBDPS conditions (when needed): TBAF, AcOH, THF, 4 h, 23 °C: 7 b 58 %, 7 c 76 %, 9 b 63 %, 11 b, 72 %, 12 b 60 %, 12 c 39 %, 12 d 78 %, 12 e 58 %, 12 f 50 %; c) CrO3(2,5‐dimethylpyrazole) (3 equiv), CH2Cl2, 23 °C, 73 %; d) WCl6 (2 equiv), nBuLi (4 equiv), THF, 0→50 °C, 2 h, 82 %; e) [Ir(py)(PCy3)(COD)][BArF] (30 mol %), H2 (80 bar), CH2Cl2, 23 °C, 4 days, quant. (1:1 d.r.).
Continuing with intermediate 8 a, a hydrogenation of the sterically hindered tetra‐substituted alkene was performed using Pfaltz′s Ir catalyst,33 leading to a separable 1:1 mixture of diastereomers 10 a–b. Cinnamate derivatives 11 a–b, 12 a–f of both diastereomers were prepared under standard conditions. For the diastereomer with the same configuration as the naturally occurring englerin A, 11 a, we wanted to synthesize a new derivative with an amine instead of a free hydroxy group. Thus, esterification with commercially available d‐Boc‐alanine led to 11 b. Unfortunately, removal of the Boc group could not be achieved under the usual reaction conditions. For the other diastereomer 10 b, the glycolate residue was also introduced to give 12 b (60 %). Other ester derivatives 12 c–e and carbonate 12 f were similarly prepared.
This first library of compounds was tested against renal cancer cell lines A498 and UO‐31. The results are summarized in Table 1. The first compounds synthesized from 6 c with the cinnamate moiety at C4 and a double bond between C5 and C6 showed minimal cell growth inhibition of the two renal cancer cell lines. As already observed with englerin B, new compounds lacking the glycolate residue (R=H) showed much lower activity than those containing this ester. Compounds 9 b and 12 b showed interesting inhibition of growth against the A498 cell line. Only lactate 12 c has a similar IC50 value to that of its glycolate counterpart, whereas the three other analogues 12 d–f show much lower activity. The basic conclusion after this initial screening was that the C4−C5 unsaturated series and the cis ring fusion series are active and worthy of further exploration, whereas the C5−C6 unsaturated series was of no biological interest.
Table 1.
Biological activity of the first library of compounds.
| Cell line | GI50 [μm][a] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 a | 7 b | 7 c | 9 b | 9 k | 11 b | 12 a | 12 b | 12 c | 12 d | 12 e | 12 f | |
| A498 | >40 | 32 | 33 | <2.5 | >40 | >40 | 18 | 1.5 | 3.1 | >40 | 40 | >40 |
| UO‐31 | >40 | 31 | 32 | 10 | >40 | >40 | 22 | 8.2 | 16 | >40 | 40 | >40 |
[a] Results of preliminary screening to identify any activity present in the structures. Values were obtained by NCI′s internal two‐cell renal assays (see Supporting Information) by interpolation; these are rough estimates using five concentration points and duplicate wells per point at each concentration. The usual cutoff for cell growth inhibition in 48 h assays is 10 μm. Note that compound 9 k (Scheme 3) was also tested using the internal two‐cell renal assay. The compounds with activity (9 b, 12 b, 12 c) were then tested in the NCI 60 screen (Table 2).
The good activity results observed with 9 b are especially important, because early intermediates with this carbon skeleton are readily available by our gold(I)‐catalyzed synthetic route, in contrast to those requiring the non‐stereoselective hydrogenation of 8 a (Scheme 2). Therefore, we decided to focus our synthetic efforts on a second library of compounds maintaining the double bond between C4 and C5.
Scale‐up of our original synthesis6 to the multigram scale (up to 50 g of geraniol) could be performed with minor changes in the first steps and with minor loss in enantioselectivity (Sharpless epoxidation 9:1 e.r., see Supporting Information). To further simplify the synthesis and to access new diastereomeric structures, a simple aldol reaction with 3‐methyl‐2‐butanone (instead of the stereoselective Denmark reaction) was performed to obtain an approximate 1:1 mixture of diastereomeric β‐hydroxy ketones 5 a/a’ (Scheme 3). This mixture was directly submitted to the gold(I)‐catalyzed cycloaddition reaction. Surprisingly, instead of the expected 1:1 mixture of diastereomers at C9, 6 a was the only product that could be isolated by column chromatography from the reaction mixture (30 % yield, 60 % based on 5 a with 5R,10S configuration). Presumably the gold(I)‐catalyzed reaction 5 a’ with 5S,10S configuration is interrupted at one of the final cyclization reactions and fails to form the tricyclic skeleton. Although not atom‐economical, this procedure opened the door for quick access to derivatives of englerin with various substituents at the C7 position by the use of readily available ketones in the aldol reaction with aldehyde 4. Thus, aldols 5 b–e with cyclohexyl, phenyl, cyclopropyl, and tert‐butyl substituents were obtained as an approximate 1:1 mixture of diastereomers.
Scheme 3.

Synthesis of derivatives 9 c–s. Reagents and conditions: a) LDA, RCOMe, THF, −78 °C, 15 h: 5 a/a’ 78 %, 5 b/b’ 78 %, 5 c/c’ 70 %, 5 d/d’ 91 %, 5 e/e’ 80 %; b) [iPrAuNCPh]SbF6 (3 mol %), CH2Cl2, 23 °C, 5 h: 6 a 30 %, 6 d 32 %, 6 e 24 %, 6 f 19 %, 6 g, 11 %; c) TBAF, THF, 23 °C, 12 h, 30–82 %; d) DMAP, imidazole, TBDMSCl, 23 °C, 69–84 %; e) 1. CrO3, pyridine, CH2Cl2, 23 °C, 1 h, 2. CeCl3(H2O)7, NaBH4, MeOH, 23 °C, 5 min, 43–70 %; f) WCl6 (2 equiv), nBuLi (4 equiv), THF, 0→50 °C, 2 h: 8 a 82 %, 8 b 74 %, 8 c and 8 d not isolated; g) 1. cinnamoyl chloride (or RCOCl), DMAP, Et3N, CH2Cl2, 45 °C, 4–12 h, 2. TBAF, THF, 23 °C, 12 h: 9 n 48 %, 9 o 58 % (3 steps), 9 p 40 % (3 steps), 9 e 43 %; h) 1. RCOOH, DMAP, NEt3, 2,4,6‐trichlorobenzoyl chloride, toluene, 23 °C, 1 h, and 2. TBAF, AcOH, THF, 4 h, 23 °C (deprotection of TBDPS group), 9 c 27 %, 9 d quant., 9 f 81 %, 9 g 80 %, 9 h 53 %, 9 i 41 %, 9 q 66 %, 9 r 38 %, 9 s 78 %. With slight modifications (see Supporting Information): 9 j 94 %, 9 k 95 %, 9 l 31 %, 9 m 26 %. PhcPr=1,2‐trans‐cyclopropylphenyl.
In all cases the gold(I)‐catalyzed reaction gave a single diastereomer, albeit in 11–32 % yield. Interestingly, the mixture of phenyl derivatives 5 c/c’ could be separated by semi‐preparative chiral chromatography (Supporting Information), and the two main stereoisomers were independently submitted to the gold(I)‐catalyzed reaction using [iPrAuNCPh]SbF6 under the standard reaction conditions. As predicted, only one of them led to oxatricyclic 6 e, whereas the other stereoisomer gave an inseparable complex mixture of compounds from which no other cyclic structure could be detected.
With the oxatricyclic structures in hand (6 g was not studied in further detail because of the very low yield), the synthetic route was followed to get the corresponding englerin analogues with the unsaturation in the cyclopentyl ring, between C4 and C5, and isopropyl, cyclohexyl, phenyl, and cyclopropyl substituents at position C7 (Scheme 3). Alcohol deprotection and protection strategies followed by the oxidation/reduction isomerization protocol28 led to the key intermediates 8 a–d, which were converted into a second library of compounds 9 c–s by selective esterification reactions.
This second library of compounds was tested in the NCI 60 one‐dose screen (except 9 k (Table 1), 9 e and 9 o (not tested)), along with compounds 9 b, 12 b, and 12 c. Compounds with apparent renal selectivity34 were then tested in the NCI 60 five‐dose protocol at the high concentration of 10−4 m (Tables 2 and 3, see Supporting Information for complete data). As expected, analogues with a free hydroxy group at position C9, namely englerin B derivatives 9 c, 9 d, 9 n, and 9 p displayed very low growth inhibitory activity. For the different esters of the alcohol at C6, acetate 9 h had no activity against renal cancer cells, whereas the introduction of a longer chain in 9 f resulted also in poor activity. On the other hand, the introduction of the cyclopropyl ring in 9 g resulted in moderate GI50 values for the renal cancer cell lines, although an interesting selectivity for the RXF393 line could be observed.
Table 2.
Activity results for compounds with apparent renal selectivity.
| Cell line[a] | GI50 [μm][b] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 9 b | 9 g | 9 i | 9 j | 9 q | 9 r | 9 s | 12 b | 12 c | |
| 786‐0 | 1.1 | 35 | 15 | 3.2 | 85 | 1.5 | 5.2 | 3.2 | 37 | 35 |
| A498 | 0.010 | 0.034 | 0.24 | 0.23 | 2.2 | 0.021 | 0.095 | 0.10 | 0.74 | 2.2 |
| ACHN | 0.017 | 0.022 | 3.0 | 0.14 | 4.5 | 0.033 | 0.045 | 0.034 | 1.4 | 6.6 |
| CAKI‐1 | 0.39 | 1.1 | 11 | 6.2 | 13 | 5.9 | 13 | 11 | 13 | 23 |
| RXF 393 | 0.059 | 0.13 | 0.041 | 0.011 | 8.9 | 0.029 | 0.039 | 0.022 | 35 | 29 |
| SN12C | 1.0 | 20 | 9.3 | 1.5 | 10 | 0.32 | 1.1 | 0.74 | 19 | 28 |
| TK‐10 | 1.9 | 100 | 17 | 16 | 93 | 12 | 22 | 19 | 100 | 100 |
| UO‐31 | 0.015 | 0.040 | 9.5 | 1.0 | 20 | 0.35 | 0.62 | 0.63 | 1.8 | 10 |
[a] The eight cell lines in the renal cancer panel characterized at the outset of the NCI 60 screening project are distinct in molecular, cytological, and clinical parameters, and are intended to be representative of different kidney cancers.35 [b] Positive control standard adriamycin (NSC #123127; see Supporting Information). Data are the average values of n=2 assays except 9 j (n=1) and 1 (n=3); (−)‐englerin A as reference.
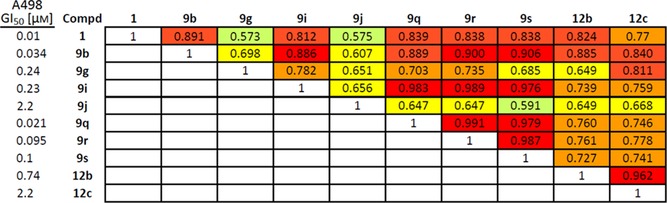
Table 3.
Matrix COMPARE at the GI50 level of response between selected englerin A analogues and englerin A (1).[a]
|
[a] Heat map colors indicate increments of 0.1 in the Pearson correlation; in the NCI 60 data, the statistical significance of differences in patterns has generally been drawn at a coefficient of 0.5.
Substitution of the glycolate appendix by the alanine and glycine derivatives 9 k–m did not give good inhibition results. Poor potency and activity was observed in these three cases. The acetyl derivative 9 j displayed better results, as it showed good selectivity for renal cancer cell lines, but with moderate GI50 values. The lactate analogue 9 i gave an interesting selectivity profile and very good growth inhibition potency in some of the lines.
Englerin analogues with substitution of the isopropyl motif at C7 are rare. Previous analogues in which the isopropyl group is substituted by ethyl or methyl groups have been described by Christmann and co‐workers18 and showed lower growth inhibition. Here, in the guaiane structure with a double bond in the cyclopentyl ring, we found that substituting the isopropyl group with a bulkier cyclohexyl in 9 q was beneficial for the selectivity and growth inhibitory activity for most of the lines, relative to 9 b. Adding the phenyl group at C7 in 9 r also shows similar benefits and even lower GI50 values in some cases. In addition, the cyclopropyl analogue 9 s shows good potency and selectivity against the growth of renal cancer cell lines.
The selectivity of the compounds listed in Table 2 was examined by using the COMPARE algorithm.36 All of the compounds had meaningful Pearson correlation coefficients at the GI50 level of response with each other and with englerin A (Table 3). Although 9 j had somewhat lower coefficients than the rest, this can be attributed to its low potency. In fact, the correlation for NCI 60 tests of englerin A and englerin B acetate at the GI50 level, previously reported, is only 0.491, with englerin B acetate being 400‐fold weaker than englerin A.1
Conclusions
In summary, we synthesized new englerin analogues by following our previously reported enantioselective synthesis of (−)‐englerin A. We found that our synthetic route is a good platform to access varied analogues, which allowed us to discover an unprecedented and easier way to synthesize a family of derivatives with a cyclopentene ring. In addition, novel analogues with substituents distinct from isopropyl could be prepared by a simple non‐stereoselective aldol reaction followed by a very selective gold(I)‐catalyzed cycloaddition reaction that furnished oxatricyclic products 6 a and 6 d–g as a single diastereomer and in good enantiomeric excess (minimum e.r. 9:1) from an epimeric mixture. The structure–activity relationship findings of this study are being used in the design of more potent and bioavailable drug candidates.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Acknowledgements
We thank MINECO Severo Ochoa Excellence Accreditation 2014–2018 (SEV‐2013‐0319), project (CTQ2013‐42106‐P), the European Research Council (Advanced Grant No. 321066), the AGAUR (projects 2014 SGR 818 and 2010 VALOR 00015), and the ICIQ Foundation. We also thank Imma Escofet and the Chiraltech and Cromatography unit ICIQ for technical support. This research was supported in part with funds from the Intramural Research Program, National Cancer Institute (project 1 ZIA BC01147 003, to J.A.B. and T.T.R.). We thank the Developmental Therapeutics Program of NCI for NCI 60 testing, and E. Goncharova, J. Evans, and T. Bostaph for their implementation of the COMPARE algorithm.
L. López-Suárez, L. Riesgo, F. Bravo, T. T. Ransom, J. A. Beutler, A. M. Echavarren, ChemMedChem 2016, 11, 1003.
Contributor Information
Dr. John A. Beutler, Email: beutlerj@mail.nih.gov.
Prof. Antonio M. Echavarren, Email: aechavarren@iciq.es.
References
- 1. Ratnayake R., Covell D. G., Ransom T. T., Gustafson K. R., Beutler J. A., Org. Lett. 2009, 11, 57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beutler J. A., Ratnayake R., Covell D., Johnson T. R. (US National Institutes of Health), WO 2009088854 A1, 2009.
- 3.Views and reviews:
- 3a. Willot M., Christmann M., Nat. Chem. 2010, 2, 519–520; [DOI] [PubMed] [Google Scholar]
- 3b. Chain W. J., Synlett 2011, 2605–2608; [Google Scholar]
- 3c. Pouwer R. H., Richard J.-A-, Tseng C.-C., Chen D. Y.-K., Chem. Asian J. 2012, 7, 22–35; [DOI] [PubMed] [Google Scholar]
- 3d. Lu Y., Yao H.-Q., Sun B. F., Chin. J. Org. Chem. 2012, 32, 1–12. [Google Scholar]
- 4. Willot M., Radtke L., Könning D., Fröhlich R., Gesser V. H., Strohmann C., Christmann M., Angew. Chem. Int. Ed. 2009, 48, 9105–9108; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9269–9272. [Google Scholar]
- 5. Zhou Q., Chen X., Ma D., Angew. Chem. Int. Ed. 2010, 49, 3513–3516; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3591–3594. [Google Scholar]
- 6. Molawi K., Delpont N., Echavarren A. M., Angew. Chem. Int. Ed. 2010, 49, 3517–3519; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3595–3597. [Google Scholar]
- 7. Nicolaou K. C., Kang Q., Ng S. Y., Chen D. Y.-K., J. Am. Chem. Soc. 2010, 132, 8219–8222. [DOI] [PubMed] [Google Scholar]
- 8. Xu J., Caro-Diaz E. J. E., Theodorakis E. A., Org. Lett. 2010, 12, 3708–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Wang C.-L., Sun B.-F., Chen S.-G., Ding R., Lin G. Q., Xu J.-Y., Shang Y.-J., Synlett 2012, 263–266; [Google Scholar]
- 9b. Wang J., Chen S.-G., Sun B.-F., Lin G.-Q., Shang Y.-J., Chem. Eur. J. 2013, 19, 2539–2547. [DOI] [PubMed] [Google Scholar]
- 10. Li Z., Nakashige M., Chain W. J., J. Am. Chem. Soc. 2011, 133, 6553–6556. [DOI] [PubMed] [Google Scholar]
- 11. Takahashi K., Komine K., Yokoi Y., Ishihara J., Hatakeyama S., A. J. Org. Chem. 2012, 77, 7364–7370. [DOI] [PubMed] [Google Scholar]
- 12. Lee J., Parker K. A., Org. Lett. 2012, 14, 2682–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zahel M., Keßberg A., Metz P. A., Angew. Chem. Int. Ed. 2013, 52, 5390–5392; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5500–5502. [Google Scholar]
- 14. Zhang J., Zheng S., Peng W., Shen Z., Tetrahedron Lett. 2014, 55, 1339–1341. [Google Scholar]
- 15. Hanari I., Shimada N., Kurosaki Y., Thrimurtulu N., Nambu H., Anada M., Hashimoto S., Chem. Eur. J. 2015, 21, 11671–11676. [DOI] [PubMed] [Google Scholar]
- 16. Gao P., Cook S. P., Org. Lett. 2012, 14, 3340–3343. [DOI] [PubMed] [Google Scholar]
- 17. Kusama H., Tazawa A., Ishida K., Iwasawa N., Chem. Asian J. 2015, 11, 64–67. [DOI] [PubMed] [Google Scholar]
- 18. Radtke L., Willot M., Sun H., Ziegler S., Sauerland S., Strohmann C., Fröhlich R., Habenberger P., Waldmann H., Christmann M., Angew. Chem. Int. Ed. 2011, 50, 3998–4002; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4084–4088. [Google Scholar]
- 19. Chan K. P., Chen D. Y.-K., ChemMedChem 2011, 6, 420–423. [DOI] [PubMed] [Google Scholar]
- 20. Sun B.-F., Wang C.-L., Ding R., Xu J.-Y., Lin G.-Q., Tetrahedron Lett. 2011, 52, 2155–2158. [Google Scholar]
- 21.
- 21a. Navickas V., Ushakov D. B., Maier M. E., Ströbele M., Meyer H.-J., Org. Lett. 2010, 12, 3418–3421; [DOI] [PubMed] [Google Scholar]
- 21b. Ushakov D. B., Navickas V., Ströbele M., Maichle-Mössmer C., Sasse F., Maier M. E., Org. Lett. 2011, 13, 2090–2093. [DOI] [PubMed] [Google Scholar]
- 22. Dong L., Jiao X.-Z., Liu X.-Y., Tian C-S., Li X-Y., Yao Y-Y., Xie P., J. Asian Nat. Prod. Res. 2014, 16, 629–639. [DOI] [PubMed] [Google Scholar]
- 23. Akee R. K., Ransom I., Ratnayake R., McMahon J. B., Beutler J. A., J. Nat. Prod. 2012, 75, 459–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abe H., Tezuka A., Kobayashi T., Ito H., Heterocycles 2014, 88, 651–662. [Google Scholar]
- 25. Acerson M. J., Bingham B. S., Allread C. A., Andrus M. B., Tetrahedron Lett. 2015, 56, 3277–3280. [Google Scholar]
- 26. Sourbier C., Scroggins B. I., Ratnayake R., Prince I. L., Lee S., Lee M.-J., Nagy P. L., Lee Y. H., Trepel J. B., Beutler J. A., Linehan W. M., Neckers L., Cancer Cell 2013, 23, 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Carson C., Raman P., Tullai J., Xu L., Henault M., Thomas E., Yeola S., Lao J., McPate M., Verkuyl J. M., Marsh G., Sarber J., Amaral A., Bailey S., Lubicka D., Pham H., Miranda N., Ding J., Tang H.-M., Ju H., Tranter P., Ji N., Krastel P., Jain R. K., Schumacher A. M., Loureiro J. J., George E., Berellini G., Ross N. I., Bushell S. M., Erdemli G., Solomon J. M., PLoS One 2015, 10, e0127498; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Akbulut Y., Gaunt H. J., Muraki K., Ludlow M. J., Amer M. S., Bruns A., Vasudev N. S., Radtke L., Willot M., Hahn S., Seitz T., Ziegler S., Christmann M., Beech D. J., Waldmann H., Angew. Chem. Int. Ed. 2015, 54, 3787–3791; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3858–3862. [Google Scholar]
- 28. Jiménez-Núñez E., Molawi K., Echavarren A. M., Chem. Commun. 2009, 7327–7329. [DOI] [PubMed] [Google Scholar]
- 29. Jiménez-Núñez E., Claverie C. K., Nieto-Oberhuber C., Echavarren A. M., Angew. Chem. Int. Ed. 2006, 45, 5452–5455; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5578–5581. [Google Scholar]
- 30. Sulzmaier F. J., Li Z., Nakashige M. L., Fash D. M., Chain W. J., Ramos J. W., PLoS One 2012, 7, e48032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Williams R. T., Yu A. L., Dicciani M. B., Theodorakis E. A., Batova A., J. Exp. Clin. Cancer Res. 2013, 32, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Inanaga J., Hirata K., Saeki H., Katsuki T., Yamaguchi M. A., Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar]
- 33. Wüstenberg B., Pfaltz A., Adv. Synth. Catal. 2008, 350, 174–178. [Google Scholar]
- 34.Selectivity in the NCI 60 screen has been defined as differential potency among the human cancer cells in the screen. Non-selectivity means that the 60 cells respond similarly (potency within 10-fold).
- 35. Shoemaker R. H., Nat. Rev. Cancer 2006, 6, 813–823. [DOI] [PubMed] [Google Scholar]
- 36. Paull K. D., Shoemaker R. H., Hodes L., Monks A., Scudiero D. A., Rubinstein L., Plowman J., Boyd M. R., J. Natl. Cancer Inst. 1989, 81, 1088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary