

Abstract
Background
Altered metabolism is a hallmark of cancer. However, the role of genomic changes in metabolic genes driving the tumour metabolic shift remains to be elucidated. Here, we have investigated the genomic and transcriptomic changes underlying this shift across ten different cancer types.
Results
A systematic pan-cancer analysis of 6538 tumour/normal samples covering ten major cancer types identified a core metabolic signature of 44 genes that exhibit high frequency somatic copy number gains/amplifications (>20 % cases) associated with increased mRNA expression (ρ > 0.3, q < 10−3). Prognostic classifiers using these genes were confirmed in independent datasets for breast and kidney cancers. Interestingly, this signature is strongly associated with hypoxia, with nine out of ten cancer types showing increased expression and five out of ten cancer types showing increased gain/amplification of these genes in hypoxic tumours (P ≤ 0.01). Further validation in breast and colorectal cancer cell lines highlighted squalene epoxidase, an oxygen-requiring enzyme in cholesterol biosynthesis, as a driver of dysregulated metabolism and a key player in maintaining cell survival under hypoxia.
Conclusions
This study reveals somatic genomic alterations underlying a pan-cancer metabolic shift and suggests genomic adaptation of these genes as a survival mechanism in hypoxic tumours.
Electronic supplementary material
The online version of this article (doi:10.1186/s13059-016-0999-8) contains supplementary material, which is available to authorized users.
Keywords: Cancer, Integrative biology, Metabolism, Tumour microenvironment
Background
The role of the microenvironment in driving tumour progression is increasingly recognised. Hypoxia is one of the key physiological and microenvironmental differences between tumour and normal tissues; it induces DNA amplification and damage, whilst reducing repair [1]. Moreover, hypoxia along with acidosis increases clonal selection, resulting in aggressive cancer phenotypes. A crucial mechanism underlying such evolution in a highly dynamic tumour microenvironment is metabolic reprogramming of tumour cells [2–5]. Metabolic adaptation is an emerging hallmark of cancer [6]. It has also been recognised that altered gene regulatory networks in cancer drive the activity of metabolic pathways (reviewed in [4]). Although there are some well-described mutations, e.g. of IDH1 and IDH2 [7–10], this is not the case for the vast majority of such genes. Thus, the complex landscape of oncogenic activity of metabolic pathways is far from understood. Therefore, there is an urgent need for understanding the genetic basis of regulation of metabolic genes, characterising their role in driving tumour growth and assessing their potential as new therapeutic targets.
Recent studies have examined transcriptional deregulation in metabolic pathways across different cancers [11–13], identifying candidate drivers of cancer metabolic adaptation. Although these studies highlight the molecular underpinnings of altered metabolism, the mechanisms driving the metabolic shift and clinical potential of metabolic drivers remain obscure. For instance, changes in mRNA abundance can arise from either altered transcriptional machinery or genomic aberrations. Therefore, in order to elucidate mechanistic insights into the dysregulated metabolism of cancer cells and identify potential drivers of this hallmark, it is crucial to study genomic aberrations alongside transcriptional changes. Here, we hypothesise that metabolic requirements in the hypoxic tumour microenvironment are driving selection of genomic alterations leading to genetic heterogeneity as well as transcriptional deregulation. To test this hypothesis high-resolution genomic and transcriptomic profiles of metabolic transporters and enzymes in tumour and normal tissue samples are necessary, as well as associated clinical annotations. Courtesy of projects such as The Cancer Genome Atlas (TCGA), the International Cancer Genome Consortium [14] (ICGC) and Metabric [15], genomic, transcriptomic, epigenomic and proteomic profiles across multiple tumour types have been generated in thousands of well-annotated human cancer samples. Thus, using these profiles, we conducted a pan-cancer integrated analysis of 6538 matched genomic and transcriptomic profiles covering ten tumour types in a comprehensive collection of 2752 metabolic enzyme and transporter genes. Specifically, we asked whether (1) somatically acquired mutations are independent drivers of metabolic shift, (2) over-expression of metabolic genes [13, 16] is induced by somatically acquired copy-number alterations, (3) metabolic genes are associated with previously reported cancer drivers [17–19] and (4) metabolic genes are associated with tumour hypoxia [20] and (5) are prognostic. We then evaluated the top-ranked key rate-limiting enzyme SQLE in a panel of aggressive breast cancers and colorectal cancer cell lines.
Results
Pan-cancer metabolic landscape and increased dysregulation in hypoxic tumours
A previously curated list of 2752 metabolic enzyme and transporter genes extracted from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Additional file 1 and Additional file 2: Table S1) was considered, offering coverage of over-expressed metabolic genes in cancer tissues and association with stemness as well as aggressive breast cancers [13]. To systematically identify pan-cancer metabolic gene signatures characterising multi-tissue cancer profiles, genomic (DNA somatic mutations and copy number aberrations) and transcriptomic (mRNA abundance) data from 6538 samples spanning ten cancer types [15, 21–29] were analysed (Additional file 1 and Additional file 2: Table S2).
We first asked whether genomic alterations were globally enriched in metabolic genes. We found no difference in the mean frequency of somatic copy number alterations (SCNAs) across all cancers between the 2752 metabolic genes and other genes (P = 0.85, two-sided proportion test) and a slight but not significant decrease in the mean frequency of somatic mutations (P = 0.06, two-sided proportion test).
Given hypoxia is the main microenvironmental factor associated with dysregulated metabolism [2–5] and genomic instability [1, 30], we asked whether genomic alterations in metabolic genes are associated with tumour hypoxia. To achieve this, we applied a previously validated hypoxia signature [20, 31], which we confirmed to be higher in tumour samples compared with normal tissues for the cancer types considered here (Additional file 1: Figure S1a). We observed an increase in genomic alterations in metabolic genes in hypoxic tumours, with a varying degree of significance depending on the type of alteration and cancer type. Specifically, tumour hypoxia was correlated with the extent of SCNAs in metabolic genes in up to seven of the ten cancer types (P < 0.05, one-sided Wilcoxon test; Additional file 1: Figure S1b, c) and with the extent of somatic mutations in three of the ten cancer types (Additional file 1: Figure S1d). This agrees with recent reports of hypoxia-driven transient SCNAs in cancer [32]. We tested this further by looking at the acquisition of early genomic aberrations in a stable diploid human cancer cell line model in vivo, complementing the results obtained from the cancer samples and from models with a higher background of genetic instability. After treatment with the antiangiogenic drug bevacizumab, inducing hypoxia (Additional file 1: Figure S2a), we observed SCNA events exclusive to the treatment group (Additional file 1: Figure S2b, c). Considering the chromosomal stability of this cell line, this represents a significant genetic change. Interestingly, the main alteration was in the copy number of chromosome 12, which is significantly enriched for metabolism genes compared with other chromosomes (Poverlap = 0.03, hypergeometric test; Additional file 1: Figure S2d). Given the variety of cancer types and experimental systems considered, the above results taken together suggest that genomic alterations in metabolic pathways, and particularly in SCNAs, act as a conserved selective pressure in hypoxic tumours.
We then asked if we could identify specific SCNA events linked with over-expression across cancer types; if identified, such events would constitute candidate metabolism drivers. This is based on the commonly accepted hypothesis that a strong correlation between DNA and mRNA levels is indicative of a gain of function. Specifically, we selected the top 10 % over-expressed genes with respect to normal tissue, across all tumour types (log2 fold change >0.5; Additional file 1 and Additional file 2: Table S3), and further limited this group to those which were recurrently gained or amplified (putative) in at least 20 % of samples in each cohort (Additional file 1: Figure S3). Next, we performed a pan-cancer correlation analysis which revealed a total of 277 metabolic genes exhibiting positive correlation across different cancer types (Spearman’s ρ > 0.3, q < 10−3; Additional file 1 and Additional file 2: Tables S4 and S5).
Interestingly, the frequency of these candidate metabolic drivers varied remarkably between cancers, from six genes (kidney renal cell carcinoma (KIRC)) to 143 genes (ovarian cancer (OV)) (Additional file 1: Figure S4a). The number of candidate metabolic drivers was significantly higher than expected by chance in breast carcinoma (BRCA), colorectal adenocarcinoma (COADREAD), glioblastoma multiforme (GBM) and OV (P < 10−4; using 10,000 randomly chosen non-metabolic gene sets; see the “Permutation analysis” section in the “Methods”). To identify metabolic processes essential to multiple tumours, we prioritised 44 core metabolic drivers with significantly correlated DNA and mRNA changes in cancer samples (Spearman’s ρ > 0.3, q < 10−3) in at least three of the ten tumour types (Fig. 1a; Additional file 1: Figure S4b, c; Additional file 1; Additional file 2: Table S6). The top candidate metabolic drivers were PYCRL, ALG3 and NUDT1, which were correlated in at least seven of the ten tumour types. The list also contained TYMS, a well-known chemotherapeutic target [33], and another five genes (GAPDH, PYCR1, TPI1, TSTA3 and TTYH3) for which inhibition of expression has been previously shown to reduce cell viability in in vitro and in vivo functional genomics screens [13]. These 44 genes were significantly enriched in five overlapping metabolic processes (Fig. 1b; Additional file 1; Additional file 2: Table S7), including pyrimidine metabolism (six genes, q = 1.97 × 10−4), drug metabolism (six genes, q = 1.97 × 10−4) and purine metabolism (five genes, q = 0.039).
Fig. 1.
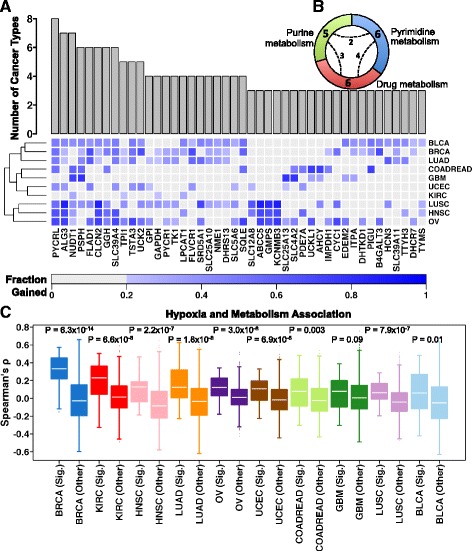
Candidate core pan-cancer drivers of metabolic dysregulation and hypoxia association. a For each candidate driver, the cumulative bar chart shows the inclusion frequencies representing the number of cancer types whereby mRNA and copy number profiles were correlated. The inclusion criterion was arbitrarily set to a minimum of three cancers. The heat map shows the extent of genomic aberrations for the selected candidate drivers as well as the corresponding cancer types. The intensity indicates the fraction of the cohort in which a given gene has either a gain or amplification. The rows (tumour types) were ordered using hierarchical clustering. b The distribution of core candidate metabolic drivers in statistically enriched compartments of metabolic pathways. The number in each segment indicates the metabolic genes present in that compartment. The dashed arcs (with numbers) indicate the number of genes shared between the compartments (Additional file 1 and Additional file 2: Table S7). c Cancer-wise mRNA correlations (Spearman’s correlation coefficients) of core metabolic signature (44 genes, Sig. group) and hypoxia score and compared with the correlations between non-selected metabolic genes (Other) and hypoxia score. For each cancer type, the distribution of both sets of correlations was compared using a one-sided Wilcoxon test and the P values are displayed. Box plots are sorted (high on the left to low on the right) by the median correlation coefficient in the signature (Sig.) set of scores. To avoid bias, three genes common to the metabolism signature and hypoxia signature and another up to 17 genes (depending upon the cancer type) common to the non-selected metabolic group and hypoxia signature were removed. BLCA bladder urothelial carcinoma, BRCA breast invasive carcinoma, LUAD lung adenocarcinoma, COADREAD colorectal adenocarcinoma, GBM glioblastoma multiforme, UCEC uterine corpus endometrial carcinoma, KIRC Kidney renal clear cell carcinoma, LUSC lung squamous cell carcinoma, HNSC head and neck squamous cell carcinoma, OV Ovarian serous cystadenocarcinoma
We then asked if there was an increased association of these 44 genes with hypoxia relative to other metabolic genes. Remarkably, nine of the ten cancer types showed significantly greater positive correlation between the expression of the 44 core metabolic drivers and the hypoxia signature compared with the remainder of the metabolic genes (Fig. 1c; P ≤ 0.01, one-sided Wilcoxon test). A similar pattern was maintained in five of the ten cancer types when considering SCNA profiles (Additional file 1: Figure S5a). The association between hypoxia and these 44 genes was also reflected in a significantly elevated fraction of copy number gains for these genes in patients with higher hypoxia in four of the ten cancer types (Additional file 1: Figure S5b; P < 0.05, one-sided Wilcoxon test). Conversely, hypoxia was not associated with the extent of somatic mutations in these 44 genes (Additional file 1: Figures S5c). This reflects the lower frequency of somatic mutations in these genes, with an overall mutation rate ranging from 0.056 % (PYCRL) to 2.212 % for (ABCC5) (Additional file 1 and Additional file 2: Table S8a). Evaluation of all metabolic genes revealed only 12 out of 2752 genes somatically mutated at a mean frequency of >5 % (Additional file 1 and Additional file 2: Table S8b). These results support the hypothesis that a core group of metabolic genes are preferentially selected through genomic gains/amplifications, with rare exceptions such as IDH1/2 [7–10]. Equivalent analysis focussing on down-regulated genes with genomic losses resulted in only three recurrent candidates of metabolism (Additional file 1 and Additional file 2: Table S9).
In summary, we identified a core set of 44 metabolic genes which are over-expressed in tumour samples through genomic gains/amplification, exhibit low rates of somatic mutations and demonstrate correlation with tumour hypoxia.
Association of candidate metabolic drivers with previously reported cancer drivers
We next asked whether previously reported validated/putative cancer drivers could also account for metabolic dysregulation of the 44 core metabolic drivers or could be linked with the selection of SCNAs. To answer this, we compiled a comprehensive list of previously reported cancer drivers from three established sources [17–19] and tested whether our core metabolism signature was associated with the somatic mutational events underlying these drivers. Significant overlap (71 genes, P < 10−4; permutation analysis) was observed amongst the three sources of cancer drivers and a smaller but still significant overlap of four genes (B4GALT3, CYC1, GMPS and SLC25A10) was observed between the combined list from the three databases and the metabolic signature (P = 0.045, hypergeometric test) (Fig. 2a).
Fig. 2.
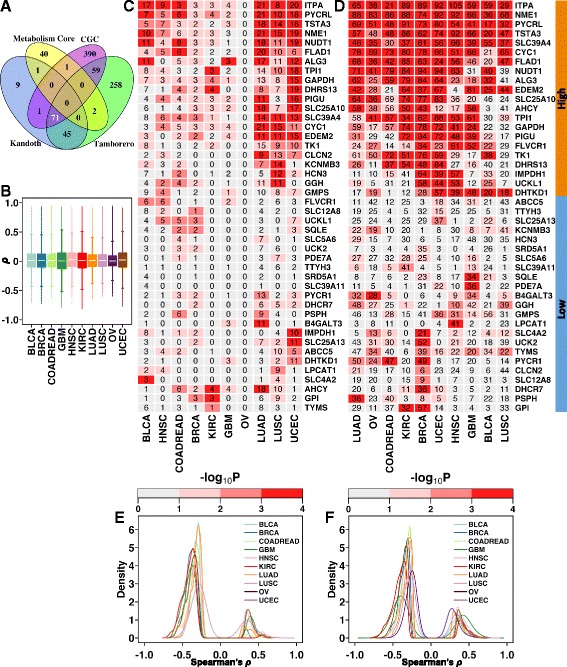
Metabolic compartmental enrichment and candidate drivers of tumourigenesis. a Overlap of genes between the core candidate metabolic drivers (yellow) and three lists (blue, purple and green) of previously reported cancer drivers [17–19]. The number in white (71) indicates overlap between the previously reported cancer drivers. b Pan-cancer null distributions of Spearman’s correlation coefficients between mRNA profiles of one million random (matched across tumour types) gene pairs. c, d Pan-cancer enrichment analysis of previously reported cancer-specific drivers (c) and the global (combined) list of candidate cancer drivers (d). The numbers in the cells indicate the total number of drivers correlated or anti-correlated with the corresponding metabolic gene (rows). The cutoffs for correlation were derived from the null distribution (95th and 5th percentiles) as shown in Fig. 2b. The cell colour indicates the probability values of expecting the observed correlation by random chance alone, with high intensities suggesting higher enrichment of previously reported cancer drivers in genes correlated with the candidate metabolic drivers. Tumour types (columns) were grouped by hierarchical clustering. The gene-wise (rows) clustering groups for the merged list of cancer drivers (d) are highlighted in orange (high correlation group) and blue (low correlation group). e, f Density plots of Spearman’s correlation coefficients of candidate metabolic drivers exhibiting significant correlation with the cancer-specific (e) and global list of previously reported cancer drivers (f). BLCA bladder urothelial carcinoma, BRCA breast invasive carcinoma, COADREAD colorectal adenocarcinoma, GBM glioblastoma multiforme, HNSC head and neck squamous cell carcinoma, KIRC kidney renal clear cell carcinoma, LUAD lung adenocarcinoma, LUSC lung squamous cell carcinoma, OV Ovarian serous cystadenocarcinoma, UCEC uterine corpus endometrial carcinoma
We further tested the likelihood of observing these previously reported cancer drivers (somatic mutations driven) in any of the 2752 metabolism genes and observed that metabolism genes were significantly enriched in the non-mutated component of the genome (the overlap with CGC drivers was 26 genes, P < 1 × 10−5; with Kandoth drivers it was seven genes, P = 1.4 × 10−4; with Tamborero drivers it was six genes, P = 4 × 10−5; 105permutations; see the “Permutation analysis” section in the “Methods”). Since the cancer drivers in these three databases [17–19] were catalogued mainly based on somatic point mutations and indels (with the exception of CGC, which also contained amplifications, translocations and germline mutations), we assessed whether core metabolic signature genes exhibit SCNAs preferentially. By comparing the proportions of samples that had only SCNAs or only mutations, we conclude that core metabolic signature genes indeed have a tendency to harbour SCNAs as opposed to somatic mutations (Additional file 1: Figure S5d). This finding agrees with the previously illustrated cancer genome hyperbola model which depicts, on a whole genomic scale, SCNAs to be inversely correlated with mutational events [34].
Owing to this limited overlap between the metabolic signature and previously reported cancer drivers, we asked whether any association exists between the mRNA profiles of these cancer drivers and the metabolic signature (“Methods”). First, the background distribution of mRNA–mRNA correlation between random gene pairs was established for each cancer type (one million permutations; see the “Permutation analysis” section in the “Methods”; Fig. 2b; Additional file 1; Additional file 2: Table S10). Next, correlation analysis was performed between mRNA profiles of the core metabolic signature and mRNA profiles of (1) cancer-type specific candidate drivers [18] mutated in at least 5 % of the cohort and (2) a merged global high-confidence cancer gene consensus list [18, 19] (Additional file 1 and Additional file 2: Table S11). Cancer-type specific analysis revealed modest to strong correlation between the expression of previously reported cancer drivers and at least some of the 44 core metabolic drivers, except for OV, where no significant correlation was identified (Fig. 2c). Conversely, the analysis of the global list of 180 previously reported cancer drivers showed a correlation between a large proportion of these 44 genes and at least the expression of one cancer driver gene (Fig. 2d) and revealed a subgroup of these genes (Fig. 2d, orange covariate) significantly associated with expression of several previously reported cancer drivers across cancers (P < 0.05; 10,000 random permutations; see the “Permutation analysis” section in the “Methods”). These genes were enriched in drug, pyrimidine and purine metabolism (q values 8.34 × 10−8 to 0.0001, minimum overlap of three genes). In contrast, a second subgroup showed a weak correlation with expression of previously reported cancer drivers (Fig. 2d, blue covariate) and no enrichment in any biological pathway. In both analyses, inspection of the correlation values revealed a strong overall trend of inverse correlation between the expression of the 44 core metabolic signature genes and that of previously reported cancer drivers (Fig. 2e, f).
Finally, we compared the mRNA profiles of metabolic genes by stratifying them into mutant and wild-type groups using each of the cancer type-specific drivers. Significant differences (|log2 fold change| > 1, q value < 0.1) in mRNA abundance for eight metabolic genes (with sparse overlap between five cancer types) were observed, demonstrating limited dependence on the mutational status of previously reported cancer drivers (TP53, ATRX, IDH1, NFE2L2, BAP1, USP9X and KEAP1) (Additional file 1 and Additional file 2: Table S12).
In summary, these results indicate that SCNAs are the main events underlying over-expression of the 44 core candidate metabolic drivers, although they also suggest that previously reported (non-metabolic) cancer drivers may contribute to a dysregulated metabolism via indirect regulation of metabolic genes.
Clinical relevance of the newly identified candidate metabolic drivers
We asked whether the mRNA abundance profile of the 44 candidate metabolic drivers is a prognostic indicator, further substantiating gain of function. Univariate survival analysis of cancer type-specific candidate metabolic drivers revealed a number of genes that were prognostic (11 BRCA, two COADREAD, one GBM, three KIRC, two lung squamous cell carcinoma (LUSC) and six OV; P < 0.05, log-rank test; Additional file 1 and Additional file 2: Table S13). By pre-selecting prognostic genes (P < 0.1, log-rank test) over 1000 cross-validation iterations, we trained multivariate prognostic models including clinical variables for breast cancer and tested them on held-out subsets of the respective cohorts (see the “Survival analysis” section in the “Methods”). The BRCA (median concordance index = 0.63) and KIRC (median concordance index = 0.62) classifiers were consistently prognostic (1000 classifiers on randomly generated training sets) (Fig. 3a).
Fig. 3.
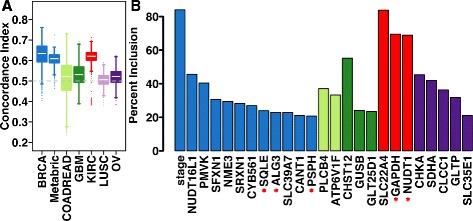
Prognostic significance of candidate metabolic drivers. a Distribution of concordance indices in held-out validation set/s using 1000 prognostic classifiers for each cancer type (see the “Survival analysis” section in the “Methods” ). The dashed grey line (concordance index = 0.5) represents a model with no prognostic ability. For breast cancer, models were trained using half of the TCGA cohort and tested on the other half as well as independently tested on the Metabric cohort. b Percentage inclusion of the most frequently chosen genes in each cancer type (>20 % of models) following univariate and LASSO feature selection performed on the training cohort. Red asterisks indicate genes present in the core metabolic signature
Importantly, BRCA prognostic classifiers were validated on an independent Metabric breast cancer cohort [15] (median concordance index = 0.61), indicating the robustness of the breast cancer classifiers. The BRCA classifiers were most frequently composed of clinical stage and three core metabolic signature genes (SQLE, ALG3 and PSPH) whilst KIRC classifiers predominantly constituted two core metabolism genes (NUDT1 and GAPDH) (Fig. 3b). COADREAD, GBM, LUSC and OV prognostic classifiers did not validate on their respective validation sets (Fig. 3a). Using the most frequently selected metabolism genes in breast cancer, a multivariate molecular classifier was created using the entire TCGA BRCA cohort and was successfully validated in the Metabric cohort (hazard ratio (HR)Q4 vs Q1 = 2.49, P = 1.11 × 10−16; Additional file 1: Figure S6a). Since clinical stage demonstrated a strong tendency of selection in the permutation analysis, we adjusted the molecular classifier for clinical stage and it remained an independent predictor of patient outcome (HRQ4 vs Q1 = 1.89, P = 6.45 × 10−7; Additional file 1: Figure S6b).
Subtype-specificity and hypoxia association of metabolic drivers
Cancer types and subtypes are heterogeneous [21, 24, 28]. We focused on breast cancer, where the presence of many different subtypes has been extensively reported [15], and also observed the strong association between hypoxia, genomic alterations and metabolism (Fig. 1; Additional file 1: Figure S1). We asked if genomic and clinical heterogeneity in breast cancer could be associated with metabolic differences. We used an independent cohort (Metabric) [15] further stratifying by well-known breast cancer subtypes (PAM50) [35]. Of the 103 (97 mapped to Metabric) candidate metabolic genes identified in the TCGA BRCA series (Additional file 1 and Additional file 2: Table S4), 49 maintained a positive correlation between mRNA abundance and copy number gains in the Metabric cohort (compared with 63 in the BRCA cohort) (ρ > 0.3, q < 10−3; Additional file 1 and Additional file 2: Table S14). The subtype-specific analysis revealed 19 core breast cancer metabolic genes present in all subtypes, as well as genes exclusive to aggressive subtypes (Basal-like, Her2-enriched and Luminal B; Fig. 4a; Additional file 1 and Additional file 2: Table S14). The number of genes showing positive mRNA–DNA correlations within each subtype was variable, with the highest in the aggressive subtypes: basal-like (50/97, P = 2 × 10−4) and Her2-enriched (52/97, P < 10−4) diseases (Additional file 1 and Additional file 2: Table S15).
Fig. 4.
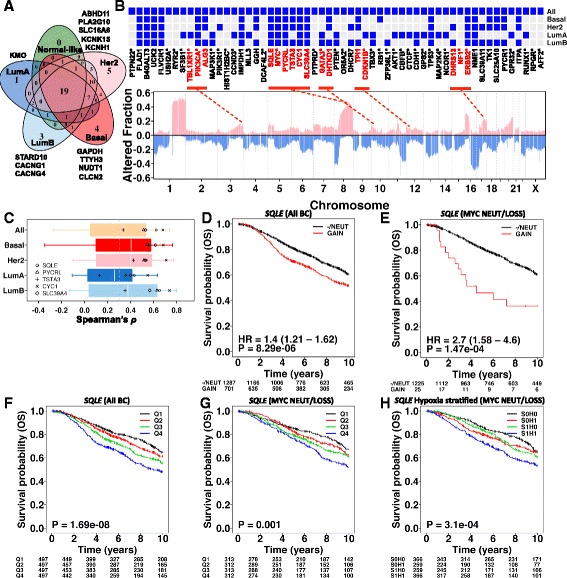
Independent validation of candidate metabolic drivers in the Metabric breast cancer cohort (n = 1991). a The overlap of candidate metabolic drivers identified in the TCGA BRCA cohort and tested for the correlation between mRNA and gene copy number data (log2 ratio) in the Metabric cohort. The numbers indicate correlated genes in the intrinsic subtypes of breast cancer (PAM50). Due to absence of subtype-specific candidate metabolic genes in the PAM50 subtype ‘Normal-like’ breast cancer, it was not considered in subsequent analyses. b Genome-wide altered copy number fraction in the complete Metabric cohort (n = 1991). Bottom: pink peaks indicate fractional copy number gains/amplifications; blue peaks indicate fractional homozygous or heterozygous deletions. The gene symbols of candidate metabolic genes are located at the top in the order of their genomic location (left to right: chromosome 1 to X). Gene symbols with asterisks indicate significantly altered known breast cancer genes [25], MYC and ERBB2. Genes in red represent clusters of metabolic and significantly altered breast cancer genes that are in genomic proximity of each other. The red dashed lines show their approximate loci. Top: heat map showing the presence (blue) and absence (grey) of candidate metabolic genes across breast cancer intrinsic subtypes (PAM50) (as summarised in Fig. 4a). The subtype-specific significantly altered known breast cancer genes [25] are also highlighted with asterisks. c Correlation between mRNA and copy number data for genes in chromosome 8q24 amplicon using the complete Metabric cohort (All) and intrinsic subtypes of breast cancer. The candidate metabolic drivers are highlighted with unique symbols to show their mRNA dependence on gene dosage. d Copy number-based Kaplan–Meier analysis of SQLE in the Metabric breast cancer cohort. There were only five cases for which copy number state = heterozygous loss and, therefore, these were merged with the copy number diploid (NEUT, n = 1282) group. Genomic gains and amplifications were collapsed into one group (GAIN). e Same as (d) using the MYC diploid/loss subset of the Metabric breast cancer cohort. f mRNA-based Kaplan–Meier analysis of SQLE in the Metabric breast cancer cohort. Samples were split into four groups based on 75th percentile, median and 25th percentile of log2 mRNA abundance of SQLE (lowest = Q1, highest = Q4). g mRNA-based Kaplan–Meier analysis of SQLE in the MYC diploid/loss subset of the Metabric breast cancer cohort. h Median-dichotomised mRNA-based Kaplan–Meier analysis of SQLE further stratified into hypoxia high and low risk groups (S0H0 = low SQLE and low hypoxia, S0H1 = low SQLE and high hypoxia, S1H0 = high SQLE and low hypoxia, S1H1 = high SQLE and high hypoxia). BC breast cancer, OS overall survival
The subtype-specific candidates showed some, although limited, overlap of five genes with previously reported cancer drivers (Additional file 1: Figure S7a–e). Next, we inspected the genomic distribution of breast cancer candidate metabolic drivers and previously reported cancer drivers. Examination of co-occurrence of genes in proximal chromosome bands revealed five clusters (which included nine metabolic genes) whereby genomic gains were significantly collocated. For instance, gain in chromosome 8q24 of MYC was significantly associated with SQLE, PYCRL, TSTA3, CYC1 and SLC39A4 (P = 0 to 3 × 10−283, Fishers exact test; Fig. 4b; Additional file 1 and Additional file 2: Table S16).
A similar trend was maintained in basal-like, Her2-enriched and luminal B breast cancers (PBasal = 6.56 × 10−53 to 1.96 × 10−38, PHer2 = 5.48 × 10−54 to 4.37 × 10−27, PLumB = 1.34 × 10−106 to 1.81 × 10−78; Fishers exact test; Fig. 4b; Additional file 1 and Additional file 2: Table S16), in line with previous studies demonstrating a link between MYC co-amplification and these genes as well as association with poor prognosis [36, 37]. Analysis of mRNA and DNA profiles on the chromosome 8q24 locus emphasised the importance of these five metabolism genes as they all demonstrated greater than average concordance between mRNA and DNA profiles (Fig. 4c).
As hypoxia is a driver of genomic gains (Additional file 1: Figure S1b, c) [32], we tested hypoxic association of SCNAs in these five genes, confirming significant positive correlation between SCNAs and the hypoxia signature [20] (Spearman’s ρ: SQLE = 0.31, PYCRL = 0.25, TSTA3 = 0.25, CYC1 = 0.25, SLC39A4 = 0.24; P < 10−3).
Of the five MYC co-amplified candidate metabolic drivers, CYC1 and TSTA3 demonstrated significant association with poor survival using both DNA- and mRNA-based groupings (CYC1SCNA: HR = 1.26, 95 % confidence interval (CI) = 1.08–1.47, P = 0.003; CYC1mRNA: Pquartiles = 3.9 × 10−5; TSTA3SCNA: HR = 1.28, 95 % CI = 1.1–1.49, P = 0.002; TSTA3mRNA: Pquartiles = 0.037) but SQLE demonstrated the highest prognostic ability on both mRNA- and DNA-derived risk groups (Fig. 4d–g). Since we observed a modest increase in SQLE mRNA abundance in the TP53 mutant group in breast cancer (Additional file 1 and Additional file 2: Table S12), we further interrogated this association with TP53 mutation status in the TCGA breast cancer cohort (where all data modalities were available) and found that SQLE’s prognostic ability was decreased when adjusting for TP53 mutation status (P = 0.03 to 0.18) (Additional file 1: Figure S8). However, SQLE’s mRNA association with prognosis was independent of MYC gain/amplification as we could recapitulate it in MYC neutral cases. Furthermore, following adjustment for clinical covariates (age, stage and grade), SQLE genomic classification remained an independent prognostic factor (HR = 3.2, 95 % CI = 1.64–6.26, P = 6.64 × 10−4) (Additional file 1 and Additional file 2: Table S17). Importantly, stratification of SQLE’s median-dichotomised (mRNA) risk groups by the hypoxia signature [20] indicated a worse prognosis group associated with SQLE over-expression in hypoxic tumours (Fig. 4h).
Next, we extended our breast cancer candidate metabolic drivers by repeating the analyses on each of the PAM50 subtypes in a large breast cancer series having sufficient representation of all subtypes (Additional file 1 and Additional file 2: Table S18). These data indicated 12 core genes common across all subtypes, including SQLE and SLC39A4 (Additional file 1: Figure S7F) as core metabolic drivers of all breast cancers. We also tested these subtype-specific candidate metabolic genes for the presence of subtype-specific mutational events using the TCGA BRCA study [25]. Consistent with our previous observation of lack of mutational events in candidate metabolic drivers, none of the subtype-specific metabolic genes were significantly mutated.
In summary, our results indicate that the heterogeneity in breast cancer subtypes can be partially recapitulated by underlying metabolic signatures. These subtype-specific breast cancer metabolism signatures contained a common set of five genes (SQLE, PYCRL, TSTA3, CYC1 and SLC39A4) which were co-amplified with MYC on chromosome 8q24 and showed a positive correlation with hypoxia. Of these genes, SQLE SCNA as well as mRNA profiles were associated with poor clinical outcome in an independent breast cancer cohort, with worse prognosis in hypoxic tumours.
The top ranked, MYC co-amplified, metabolic driver SQLE is key for cell survival under hypoxic conditions
Focussing on the MYC locus (chromosome 8q24), we sought to elucidate the role of five candidate metabolism drivers (SQLE, PYCRL, TSTA3, CYC1 and SLC39A4). All of these genes have shown varying but significant levels of essentiality in previous cell line shRNA screens [38] (Additional file 1 and Additional file 2: Table S19). Of these, SQLE was not only ranked amongst the top candidates in our analysis but also displayed putative gains/amplifications in breast cancer cell lines from the Cancer Cell Line Encyclopaedia (CCLE) [39] (Additional file 1: Figure S9a–d). We further verified SQLE copy number gains and mRNA correlation in both breast and colorectal cancer cell lines (Fig. 5a, b). In general, our findings from the clinical studies were corroborated in the CCLE by the correlation patterns between the DNA and mRNA profiles of candidate metabolism drivers (Additional file 1: Figure S10a–d). Given the positive correlation of five candidate metabolic genes located at MYC amplicon genes with the hypoxia gene expression signature in breast cancer (Spearman’s ρ: SQLE = 0.31, PYCRL = 0.25, TSTA3 = 0.25, CYC1 = 0.25, SLC39A4 = 0.24; P < 10−3), we tested whether these five genes are directly induced by hypoxia and/or, when expressed, confer survival advantage under hypoxia. In vitro mRNA abundance analysis under hypoxia and normoxia revealed strong hypoxic dependence for SQLE in HCC1569 and HCC1806 breast cancer cells and HCT116 colorectal cancer cells (P < 0.05; unpaired t-test) (Fig. 5c, d). Analysis of SQLE protein expression confirmed hypoxic induction of SQLE in HCC1806 and, to a lesser extent, in HCC1569. None of the other chromosome 8q24 genes demonstrated hypoxic regulation (Additional file 1: Figure S11a–d). We validated the copy-number of SQLE and MYC in the breast and colorectal cell lines relative to the genome ploidy with quantitative PCR (Additional file 1: Figure S12). With the exception of HCC1954 and MDA-MB-231, all other cell lines had a significantly increased number of copies of SQLE (P < 0.05, unpaired t-test). The MYC copy number was also significantly increased (P < 0.05, unpaired t-test) in all cell lines except HCC1569 cells.
Fig. 5.
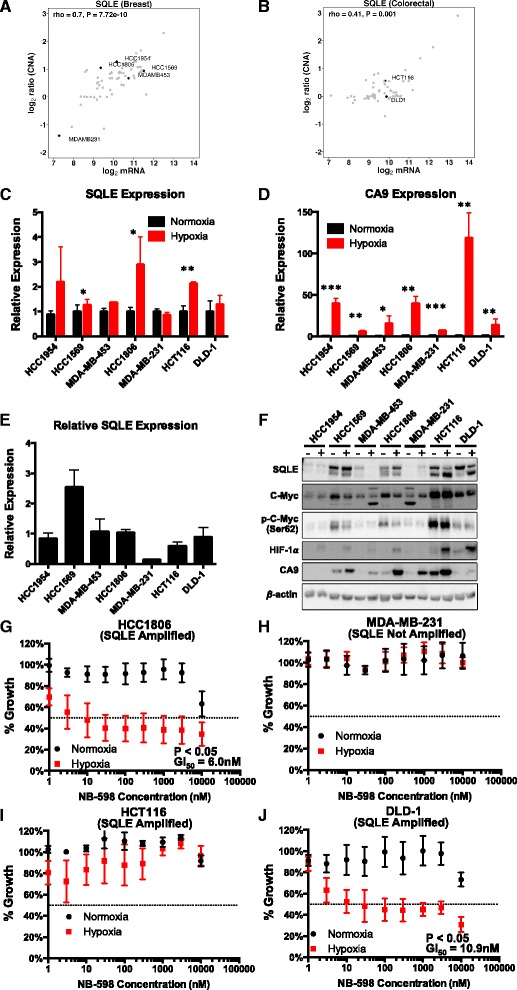
In vitro validation of SQLE is normoxia and hypoxia. a SQLE expression and copy number correlation in CCLE breast cancer lines. Cell lines used for in vitro validation are labelled. b SQLE expression and copy number correlation in CCLE colorectal cancer lines. Cell lines used for in vitro validation are labelled. c SQLE expression in cancer cell lines under 24-h normoxia and hypoxia (1 % O2). d CA9 expression (hypoxia control) in cancer cell lines under 24-h normoxia and hypoxia (1 % O2). e SQLE expression in cancer cell lines relative to one another. f Western blot analysis showing the levels of SQLE, MYC, phospho-MYC (Ser62), HIF-1α and CA9 after 24 h of normoxia and hypoxia (1 % O2). The two SQLE bands correspond to different isoforms, which were further confirmed by siRNA knockdown western blots (Additional file 1: Figure S11q). g–j SQLE inhibition with NB-598 reduces cell viability at a range of concentrations with a GI50 of 6 nM in HCC1806 (g) and GI of 10.9 nM in DLD-1 (j) under hypoxia (1 % O2) but not normoxia. The effect of the compound was not inhibitory enough to calculate a GI50 value in MDA-MB-231 (h) or HCT116 (i) cells under normoxia or hypoxia (1 % O2). HCC1954 and HCC1569 are HER2+ breast cancer cell lines. MDA-MB-453, HCC1806 and MDA-MB-231 are triple receptor negative breast cancer cell lines. HCT116 and DLD-1 are colorectal cancer cell lines. Error bars are standard deviations. ***P < 0.001, **P < 0.01, *P < 0.05, n = 3
SQLE requires oxygen as a co-factor for its enzymatic reaction; thus, we hypothesised that it may show decreased activity in hypoxia and the selection of early genes in the cholesterol biosynthesis pathway for amplification in hypoxia, such as HMG-CoA reductase [40], may contribute to overcome the block. The increased amount of SQLE may compensate for decreased activity in hypoxia. The relative expression of SQLE at the mRNA (Fig. 5e) or protein (Fig. 5f) levels did not show any significant relationship to DNA copy number. This suggests other mechanisms such as epigenetic regulation, as shown previously [37], may also play a role in increasing SQLE expression in tumours.
Given SQLE’s MYC-independent prognostic ability (Fig. 4e, g), association with hypoxia in breast and colorectal cancer samples (Spearman’s ρ: Breast-Metabric = 0.31, Breast-BRCA = 0.42, P < 10−3; COADREAD = 0.17, P = 0.003) and hypoxic induction in three cell lines amongst those tested (Fig. 5c), we focussed on functional analysis of SQLE. Breast and colorectal cancer cells were treated with SQLE inhibitor (NB-598) under hypoxia and normoxia (Fig. 5g–j). There was no effect on cell viability in GI50 assays under normoxic conditions, but the highly amplified triple negative breast cancer cell line HCC1806 and modestly amplified colorectal cancer cell line DLD-1 showed clear sensitivity to NB-598 under hypoxia (1 % O2; Fig. 5g, j). We further investigated the functional significance of SQLE by clonogenic assay (Fig. 6a–g). One cell line showed reduced survival in hypoxia alone (P < 0.001); however, in six of the seven cell lines tested, there was a significant synergistic effect of SQLE inhibition and hypoxia. Equivalent knockdown analyses on TSTA3 and PYCRL, two of the other four top candidate metabolism genes, followed by cell viability and clonogenic assays indicated a smaller yet significant effect for PYCRL knockdown; in particular, PYCRL knockdown reduced the number of colonies in HCC1806 and DLD-1 cells only under hypoxia (P < 0.05; Additional file 1: Figure S11e–p).
Fig. 6.
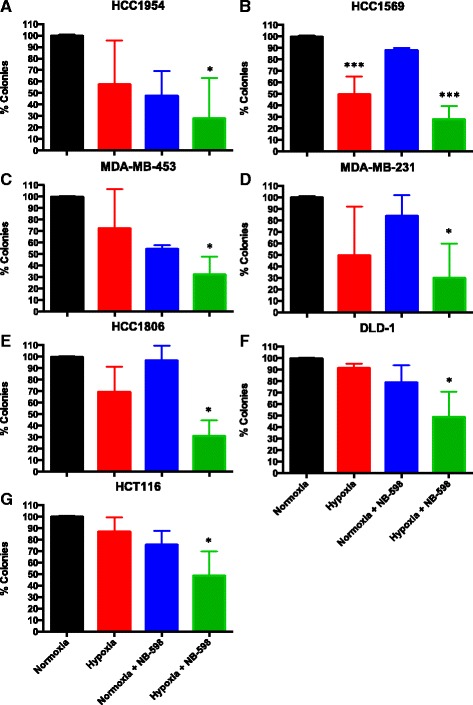
Clonogenic analysis of SQLE. a–g Clonogenic analysis of SQLE inhibition by NB-598 under normoxia and hypoxia (1 % O2) in HCC1954 (a), HCC1569 (b), MDA-MB-453 (c), MDA-MB-231 (d), HCC1806 (e), DLD-1 (f) and HCT116 (g) cells. ***P < 0.001, *P < 0.05, n = 3. Black, red, blue and green bars correspond to the percentage of colonies under normoxia and hypoxia and NB-598 cells under normoxia and hypoxia, respectively. Error bars are standard deviations
In summary, SQLE expression in vitro demonstrated significant increases under hypoxia in three of the seven breast and colorectal cancer cell lines. SQLE was also confirmed as a driver of colorectal and triple receptor negative breast cancers and a key gene in maintaining cell survival under hypoxia.
Discussion
The metabolism of rapidly dividing tumour cells is shifted towards increased anabolic processes and redox homeostasis compared with normal cells. Further, the hypoxic microenvironment adds additional metabolic stress, to which tumour cells have to adapt. Indeed, much research over the past decade has shown increased expression of and dependence on many genes involved in the tumour hypoxic microenvironment [41]. Moreover, complex interactions between oncogenic signalling pathways such as MYC and metabolism have been identified [2, 4, 6, 42]. Essential metabolic pathways hold potential to offer new druggable targets [43] and have served as therapeutic targets in a number of cancer types. In order to systematically identify new potential targets, we devised and exploited a novel in silico approach revealing metabolic dysregulation as an underlying source of selective genomic heterogeneity in a large pan-cancer study.
In a comprehensive analysis of 6538 samples from ten different cancer types, considering both genotypic (DNA) and phenotypic (mRNA and clinical prognosis) data , we show that over-expression of metabolic genes is substantially enriched with somatically acquired gains and amplifications. Our results suggest that these genomic alterations form a core part of the metabolic reprogramming that is conserved across different cancer types. Specifically, we identified a core signature of 44 metabolic drivers and revealed tumour site-specific metabolic genomic landscapes. We present evidence that differential metabolic requirements contribute to genomic heterogeneity amongst tumours and aggressiveness specifically in kidney and breast cancers. Core metabolic prognostic signatures are identified, comprising NUDT1 and GAPDH in kidney renal cell carcinoma and SQLE, ALG3 and PSPH in two independent breast carcinoma cohorts. Importantly, this identifies patients who might benefit from treatment with inhibitors of these genes, which in some cases are already being tested pre-clinically (e.g. GAPDH [44] and NUDT1 [45] inhibitors).
Importantly, we show that both copy number amplification and expression of the identified metabolic driver genes are significantly associated with tumour hypoxia, suggesting a mechanism of metabolic reprogramming via adaptation within a hypoxic tumour microenvironment. We complement this analysis with evidence from in vitro and in vivo models supporting the hypothesis of selection pressure under hypoxia.
Amongst the top ranked candidates, we show SQLE is a metabolic driver in multiple cancers and demonstrate its association with prognosis and tumour hypoxia (Figs. 1 and 3). Importantly, high SQLE expression in hypoxic tumours was significantly associated with poor survival. SQLE is often co-amplified with the cancer driver MYC but SQLE expression is prognostic independent of MYC status. Furthermore, functional characterisation demonstrates SQLE’s specific role in maintaining proliferative integrity and regulating cell survival exclusively under hypoxia in a panel of cancer cell lines and independently of MYC amplification, confirming SQLE as a potential driver of selection under hypoxia. Interestingly, SQLE protein has also been detected in exosomes of breast cancer cells (MDA-MB-231) [46]. A recent study has also demonstrated that over-expression of SQLE promotes cell proliferation and migration and acts as a positive regulator of ERK signalling in hepatocellular carcinoma cells [47]. A second study, published while this manuscript was in revision, independently confirmed SQLE as bona fide oncogene by amplification, further supporting our findings [48]. SQLE catalyses a rate-limiting oxygenation step in sterol biosynthesis. Cholesterol is vital to cell membrane structure and also functions as a precursor of fat-soluble vitamins and steroid hormones [49]. It also forms part of lipid rafts which co-ordinate receptor-mediated signal transduction pathways [50]. Of the pan-cancer metabolic signature, SQLE and DHCR7, which catalyses cholesterol synthesis, were recently identified as the mediators of radiotherapy resistance in pancreatic cancer [51]. Hypoxic accumulation of squalene, the substrate of SQLE, occurs in glioblastoma, likely due to the dependence on oxygen as a co-factor for SQLE catalysis [52].
In summary, our study highlights a key role for hypoxia as a driver of genomic adaptation, including metabolic genes not directly induced by hypoxia and not previously identified as cancer drivers, such as SQLE. Importantly, drug inhibitors exist for several of the identified genes, allowing prompt translation and testing of these findings in clinical trials, with appropriate patient selection.
Conclusions
Our pan-cancer analysis of genomic and transcriptomic profiles reveals a conserved landscape of metabolic dysregulation in human cancers linked with tumour hypoxia. We identified a core group of candidate metabolic driver genes as amplified and over-expressed across cancer types and strongly associated with tumour hypoxia in terms of both expression and DNA copy number. We also show that these metabolic drivers had not been previously identified as tumour suppressor and/or oncogenes and their over-expression in cancer cannot be explained by genomic alterations in known cancer drivers. Amongst the top candidate metabolic drivers for which drug inhibitors exist, we show that SQLE is independently prognostic and plays a key role in maintaining cell survival under hypoxia.
Methods
Pre-processing and filtering
mRNA abundance, DNA copy number and somatic mutation profiles for TCGA datasets were downloaded from TCGA DCC (gdac), release 2014-01-15. For mRNA abundance, Illumina HiSeq rnaseqv2 level 3 RSEM normalised profiles were used. Genes whereby >75 % of samples had zero reads were removed from the respective dataset. GISTIC v2 [53] level 4 data were used for copy number analysis and log2 ratios were processed to call SCNA states (human genome assembly Hg19). Briefly, SCNA gains/amplifications were based on 0: t < 0.2; 1: 0.2 ≤ t ≤ 1; 2: t > 1 and losses were based on 0: t > −0.2; −1: −0.2 ≥ t ≥ −1; −2: t < −1, where t denotes the log2 ratio. Level 1 clinical data were used for survival analysis. TCGA GBM and OV cancer microarray data were used for mRNA differential expression analysis as normal samples were not available on the Illumina HiSeq platform. mRNA data were converted to log2 scale for subsequent analyses. Analysis of differential mRNA abundance of candidate metabolic drivers stratified by mutational status of cancer type-specific drivers was based on TCGA DCC-reported MutSig v2.0 calls.
The Metabric breast cancer dataset was pre-processed, summarised and quantile-normalised from the raw expression files generated by Illumina BeadStudio (R packages: beadarray v2.4.2 and illuminaHuman v3.db_1.12.2). Raw Metabric files were downloaded from the European Genome-phenome Archive (EGA; study ID EGAS00000000083). Data files of one Metabric sample were not available at the time of our analysis and were therefore excluded. Probe to gene-level mapping was performed by keeping the most variable (standard deviation) probe. Log2 scaled data were used for differential expression analysis. Metabric copy number segmented genomic region data as published in the original study [15] (human genome assembly Hg18) were processed to create gene by patient log2 ratio and call profiles.
Processing of xenograft samples
Mice were housed at BMS, University of Oxford, UK and procedures were carried out under a Home Office license. Six- to seven-week-old female BALB/c NuNu mice were injected subcutaneously in the lower flank with 100 μl Matrigel (BD Bioscience) and 1 × 107 HCT116 cells suspended in 100 μl of serum-free medium. Tumour growth was monitored three times per week measuring the length (L), width (W) and height (H) of each tumour using calipers. Volumes were calculated from the formula 1/6 × π × L × W × H. For bevacizumab treatment mice were injected intraperitoneally every 3 days (10 mg/kg) until sacrifice once randomised groups had xenografts which had reached an average of 150 mm3 in size. When tumours reached 1.44 cm3 the mice were sacrificed by cervical dislocation.
Pre-processing of Affymetrix CytoScan HD data
Affymetrix CytoScan HD array profiling was performed on three untreated HCT116-derived xenografts and three bevacizumab-treated samples. Data were normalised to a large collection of reference human normals as packaged with R package rawcopy (v1.0). Resulting logR ratio data were segmented using rawcopy’s default segmentation algorithm, PSCBS (Additional file 1 and Additional file 2: Table S20). Gene mapping to segmented regions was performed by rawcopy using RefSeq annotation. Since xenografts were exposed to bevacizumab for a limited period of time, we used relatively lenient logR ratio thresholds of ±0.1 for copy number gains and losses (Additional file 1 and Additional file 2: Table S21). Genes which were split in regions of gain as well as loss in the bevacizumab-treated groups were filtered out from subsequent analysis. Raw Affymetrix CEL files are available through the EBI Array Express portal (http://www.ebi.ac.uk/arrayexpress/) under accession number E-MTAB-4200.
Fraction of transcriptome altered (genomic instability marker)
Genome instability in different gene sets (core metabolism signature, non-core metabolism, all metabolism and rest of the genome) was estimated at the gene level. For each gene, the fraction of patients with copy number calls 1,2 (gain/amplified) were regarded as the gained fraction and copy number calls −1,−2 were classed as the loss fraction. Across all gene sets, the fractions altered per gene were averaged to estimate an overall measure of the fraction of the cohort altered. This was estimated separately for gains and losses.
Differential gene expression analysis
Differentially expressed genes were identified using Welch’s t-test. P values were adjusted for multiple comparisons using the Benjamini–Hochberg method. Genes with log2 fold change >0.5 and q < 0.05 (Benjamini–Hochberg method) were regarded as over-expressed, except for bladder urothelial carcinoma (BLCA), ovarian serous cystadenocarcinoma (OV) and uterine corpus endometrial carcinoma (UCEC), for which only a log2 fold-change >0.5 threshold was applied to keep the initial feature selection sets (top 10 % over-expressed metabolic genes) a similar size.
Correlation analysis
All correlation analyses (mRNA–mRNA and mRNA–log2 copy number ratio) were performed using Spearman’s rank correlation.
Pathway analysis
Pathway enrichment analysis for human samples was conducted using GeneCodis [54] against the KEGG database.
Permutation analysis
The null distribution to estimate the enrichment of highly correlated metabolic genes at the mRNA and DNA levels was estimated by randomly selecting 10,000 genes from the non-metabolic section of the transcriptome. The gene set size was matched to the cancer type-specific size of genes exhibiting over-expression and gains/amplification (>20 %) in the metabolic genome.
Null distributions for mRNA–mRNA correlation for each cancer type were established by randomly sampling two distinct sets of 1000 genes, resulting in one million random correlations matched across cancer types. The reference distribution for mRNA–SCNA log2 profiles was established on common genes across all ten cancer types (14,923 genes).
Cancer-specific lists of candidate drivers were extracted from Kandoth et al. [18], further limiting the list to genes which showed >5 % mutation frequency in the respective cohorts. A global list of previously reported cancer drivers was compiled as a merged list of all Kandoth et al. genes and Tamborero et al. [19] putative drivers annotated as ‘High Confidence Driver’ and also present in the Cancer Gene Consensus [17]. For each cancer type, mRNA–mRNA correlations were estimated between core candidate metabolic drivers and the above-mentioned known candidate driver lists. Using the 95th and 5th percentile thresholds of null distributions for mRNA–mRNA correlations, correlation counts tables (Fig. 2c, d) were established. The probability values for each cell (Fig. 2c, d) were estimated by randomly sampling 10,000 subsets of genes and counting the number of times core candidate metabolic drivers showed correlation counts greater than observed. The same procedure was repeated for both cancer-specific candidate drivers (Fig. 2c) and the global list of candidate drivers (Fig. 2d).
Null distributions for overlap between metabolism genes and CGC, Kandoth et al. and Tamborero et al. drivers (as described previously) were estimated by generating 100,000 random subsets of genes from the non-metabolism part of the genome. Probabilities of observing an overlap with metabolism genes as low as 26 (CGC), 7 (Kandoth) and 6 (Tamborero) were subsequently estimated using the randomly generated counts.
Co-occurrence analysis
We established a 2 × 2 counts table for each pair of genes using copy number call data. For each pairwise comparison, the table contained counts for when both genes had amplification, counts exclusive to one gene and counts for when neither had amplification. Due to different copy number-calling algorithms, for the Metabric dataset, gains and amplifications were treated as one category (amplifications). Fisher’s exact test was used to estimate the co-occurrence of copy number amplifications between a given pair of genes.
Gene essentiality estimation
Gene essentiality estimates were generated in a panel of breast cancer cell lines using the COLT cancer database [38] (http://dpsc.ccbr.utoronto.ca/cancer). Data were filtered using default parameters, limited to breast cancer cell lines and a GARP (Gene Activity Ranking Profile) score P value <0.1.
Cancer cell line encyclopaedia analysis
mRNA abundance and gene copy number log2 ratio data from the CCLE were downloaded from the Broad Institute (http://www.broadinstitute.org/ccle/). For gene copy number status, a threshold of 0.2 and –0.2 (consistent with thresholds used for patient data) was applied to log2 ratio profiles to determine putative gains/amplifications and losses, respectively. Breast cancer cell line subtype classifications were inferred from previously published data [55].
Survival analysis
Survival modelling was limited to cancers with available survival data: BRCA, COADREAD, GBM, KIRC, LUSC, OV and Metabric. A Cox proportional hazards model was used to estimate the hazard ratio and a Wald test was used to test the significance of outcome difference between the risk groups. For univariate analysis, mRNA abundance profiles were median-dichotomised to established high-risk (high-abundance) and low-risk (low-abundance) groups. Overall survival time was truncated at 10 years for all cancers except for aggressive tumours (GBM, Metabric’s basal-like and Her2-enriched subtypes), where the truncation was done at 5 years.
For multivariate analysis, genes with correlated mRNA and copy-number profiles were selected by cancer type (BRCA, COADREAD, GBM, KIRC, LUSC and OV; Additional file 1 and Additional file 2: Table S5). Cohorts were randomly split into two halves (training and validation cohorts), except GBM, which was split 2:1 due to the small number of samples (n = 154). For each cancer type, genes were pre-selected through a univariate Cox proportional hazards model followed by log-rank test (P < 0.1; training cohort only). Selected genes were taken forward for multivariate Cox modelling with LASSO feature selection (L1 penalty) in a tenfold cross-validation setting. The model with the smallest mean cross-validation error was selected and tested on the held-out validation cohort. For BRCA, all models contained clinical covariates of age (dichotomised at 50 years) and stage (1 = baseline, 2, 3, 4). These were also tested on the fully independent Metabric breast cancer cohort. Predicted response scores and predicted risk were evaluated for survival association in validation set/s using the concordance index.
Survival modelling was performed in the R statistical environment (v3.0.1) using the R packages survival v2.37-4, glmnet v1.9-5, glmpath v0.95, survcomp v1.10.0 and randomForestSRC v1.4.
Cell culture, GI50 and clonogenic analysis
Cells were maintained in a humidified incubator at 5 % CO2 and 37 °C. For hypoxic exposure, cells were grown at 1 % O2, 5 % CO2 and 37 °C in an INVIVO2 400 (Baker Ruskinn, USA).
MDA-MB-231, MDA-MB-453 and HCT116 cells were maintained in DMEM whilst HCC1806, HCC1954, HCC1569 and DLD-1 were maintained in RPMI, both supplemented with 10 % fetal bovine serum. SQLE inhibitor, NB-598, was available from MedChemexpress (USA).
For GI50 analysis, cells were plated in 96-well plates (1000 cells/well) and, 24 h later, NB-598 was added at a range of concentrations from 1 nM to 10 μM. The 96-well plates were then placed under normoxia or hypoxia for 72 h for the colorectal cancer cell lines (HCT116 and DLD-1) or 120 h for the breast cancer cell lines (HCC1954, HCC1569, MDA-MB-453, HCC1806 and MDA-MB-231). Cell viability at the start of treatment or at the endpoint was assessed using CyQuant (C7026, Life Technologies, UK) according to the manufacturer’s instructions. GI50 was calculated using PRISM (GraphPad, USA).
For clonogenic analysis, cells were plated in 100-mm dishes at 1000 cells/dish and 8 h later, once cells were attached, NB-598 was added at a concentration of 30 nM. Treated and untreated plates were incubated in normoxia or hypoxia for 72 h. After 72 h, cell culture medium was replaced with fresh medium followed by incubation in normoxia for 7–14 days until colonies were clear. The medium was removed and replaced with 0.5 % methylene blue dissolved in 50 % ethanol. Plates were incubated for 1 h at room temperature before washing in tap water. Colonies were visualised and counted utilising the GelCount™ system (Oxford Optronix, UK).
Quantitative PCR
mRNA was extracted with Trizol (Invitrogen, CA, USA) according to the manufacturer’s instructions. DNA was extracted using DNAzol (Invitrogen, CA, USA) according to thr manufacturer’s instruction. Quantitative PCR was carried out as described previously [56] utilising the Applied Biosystems 7900HT Real-Time PCR System. Expression of genes was normalised against the expression of the control genes RPL11 and β-Actin. The primer sequences for mRNA analysis were: CA9 forward, CTTGGAAGAAATCGCTGAGG; CA9 reverse, TGGAAGTAGCGGCTGAAGTC; SQLE forward, AAACGGGAGGCCTCTAAATC; SQLE reverse, AGATGGCCTCGGACTCAAG; RPL11 forward, CTTTGGCATCCGGAGAAAT; RPL11 reverse, TCCAGATTTCTTCTGCCTTG; β-Actin forward, CCAACCGCGAGAAGATGA; β-Actin reverse, CCAGAGGCGTACAGGGATAC; PYCRL forward GACAGGAACCTATGTCACTTTCAA; PYCRL reverse, TGGCAAAGATGACGAGCA; TSTA3 forward, GAAGATGAGGTCTCCATCAAGG; TSTA3 reverse, CCCATCCGACTTGGTTGTAT; CYC1 forward, CTTCGCGGGGTAGTGTTG; CYC1 reverse TTCGACGACAAGGCCACT; SLC39A4 forward, CCTCTTCCTGCTGCACAAC; SLC39A4 reverse, CATCCTCGTACAGGGACAGC. As chromosome 8, on which SQLE and MYC are located, is highly aberrant in breast cancer, control primers were designed from alternative chromosome regions for DNA copy number analysis. We identified regions which generally represented the genome ploidy of patient/cell line DNA. These were 2q11.1 (TEKT4) and 14q11.2 (POTEG). The primer sequences for DNA analysis were: SQLE forward, GCGTGCGACGGTTACTCT; SQLE reverse, GCTTCCTCACCAGCATCC; MYC forward, CGGTTTTCGGGGCTTTAT; MYC reverse, GGCTCTTCCACCCTAGCC; PYCRL forward, GGCACCACCATCTATGGACT; PYCRL reverse, CCTACTTTCTGCTGAGCTCCTT; TSTA3 forward, AGCTGGAAGACAGGATCAGG; TSTA3 reverse, AGAGCGGATGGAATGCAG; POTEG forward, GCGATCTGCTGGCTACTACC; POTEG reverse, CCAAATGGCTTCTTCACAGAG; TEKT4 forward, GCTGACCACACACAGTCCTC; TEKT4 reverse, GGGCCCACGTCGTCTTT.
Western blots
Cell lysates were separated on 10 % SDS-PAGE and transferred to a PVDF membrane. Primary antibodies were used at 1:1000 unless otherwise stated. Antibodies against CA9 (gift from J. Pastorek, Institute of Virology, Slovak Republic), HIF1 (BD Biosciences, USA), β-Actin (A3854, Sigma, UK), SQLE (12544-1-AP, Proteintech, USA), MYC (9402, Cell Signalling, USA), TSTA3 (HPA023301, Sigma, UK) and PYCRL (H00065263-M01) were utilised. Appropriate secondary horseradish peroxidase-linked antibodies were used (Dako, UK). Immunoreactivity was detected with chemiluminescence (Amersham, UK) and visualised using Image Quant LS4000 mini (GE Healthcare, UK).
Visualisation
All visualisations were prepared in the R statistical environment (v3.0.1).
Acknowledgements
This study makes use of data generated by the Molecular Taxonomy of Breast Cancer International Consortium, which was funded by Cancer Research UK and the British Columbia Cancer Agency Branch. The results published here are based, in part, upon data generated by The Cancer Genome Atlas pilot project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions who constitute the TCGA research network can be found at http://cancergenome.nih.gov/.
Funding
This study was funded by an EU framework 7 EURECA grant to FMB (funding SH, RVS, LW), an EU framework 7 p-medicine grant to FMB and ALH (funding SH, LW) and Cancer Research UK grants to FMB and ALH (funding SH, AM, SW) and the Breast Cancer Research Foundation (ALH) and was also supported by the NIHR Oxford Biomedical Research Centre.
Availability of data and materials
An open source R package (iDOS) implementing the methodology described here to identify candidate drivers is being made available through CRAN (http://cran.r-project.org). The iDOS package and associated R scripts are also available as Additional file 3 and on github (https://github.com/shazi828/iDOS/releases) with associated GNU General Public License v2 (https://github.com/shazi828/iDOS). A citable version of the source code and data are available through Zenodo with doi: 10.5281/zenodo.51787 (https://zenodo.org/record/51787).
Authors’ contributions
SH, AM, ALH and FMB conceived the idea. ALH and FMB supervised the study. SH performed computational analyses with contributions from RVS, LMW and FMB. AM performed experimental work with contributions from SW. SH, AM, ALH and FMB co-wrote the manuscript, which all authors approved.
Competing interests
The authors declare that they have no competing interests.
Ethics approval
Mice were housed at BMS, University of Oxford, UK and appropriate procedures were carried out under a Home Office license.
Additional files
Figures S1 to S12 including figure legends and supplementary references. (DOCX 35712 kb)
Tables S1 to S21. (ZIP 10219 kb)
iDOS R package and associated R scripts used in this study. (ZIP (379183 kb)
Footnotes
Syed Haider and Alan McIntyre are co-first authors.
Adrian L. Harris and Francesca M. Buffa are co-senior authors.
Contributor Information
Adrian L. Harris, Email: Adrian.Harris@oncology.ox.ac.uk
Francesca M. Buffa, Email: Francesca.Buffa@oncology.ox.ac.uk
References
- 1.Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180–92. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 2.Benjamin DI, Cravatt BF, Nomura DK. Global profiling strategies for mapping dysregulated metabolic pathways in cancer. Cell Metab. 2012;16:565–77. doi: 10.1016/j.cmet.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Favaro E, Bensaad K, Chong MG, Tennant DA, Ferguson DJ, Snell C, Steers G, Turley H, Li JL, Gunther UL, et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. 2012;16:751–64. [DOI] [PubMed]
- 4.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364–73. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 5.Green DR, Galluzzi L, Kroemer G. Metabolic control of cell death. Science. 2014;345:1250256. doi: 10.1126/science.1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Chen C, Liu Y, Lu C, Cross JR, Morris JP, Shroff AS, Ward PS, Bradner JE, Thompson C, Lowe SW. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev. 2013;27:1974–85. [DOI] [PMC free article] [PubMed]
- 8.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. [DOI] [PMC free article] [PubMed]
- 9.Lu C, Venneti S, Akalin A, Fang F, Ward PS, Dematteo RG, Intlekofer AM, Chen C, Ye J, Hameed M, et al. Induction of sarcomas by mutant IDH2. Genes Dev. 2013;27:1986–98. [DOI] [PMC free article] [PubMed]
- 10.Reitman ZJ, Duncan CG, Poteet E, Winters A, Yan LJ, Gooden DM, Spasojevic I, Boros LG, Yang SH, Yan H. Cancer-associated isocitrate dehydrogenase 1 (IDH1) R132H mutation and d-2-hydroxyglutarate stimulate glutamine metabolism under hypoxia. J Biol Chem. 2014;289:23318–28. [DOI] [PMC free article] [PubMed]
- 11.Hu J, Locasale JW, Bielas JH, O'Sullivan J, Sheahan K, Cantley LC, Vander Heiden MG, Vitkup D. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol. 2013;31:522–9. [DOI] [PMC free article] [PubMed]
- 12.Nilsson R, Jain M, Madhusudhan N, Sheppard NG, Strittmatter L, Kampf C, Huang J, Asplund A, Mootha VK. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat Commun. 2014;5:3128. [DOI] [PMC free article] [PubMed]
- 13.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–50. [DOI] [PMC free article] [PubMed]
- 14.International Cancer Genome C, Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, Bhan MK, Calvo F, Eerola I, et al. International network of cancer genome projects. Nature. 2010;464:993–8. [DOI] [PMC free article] [PubMed]
- 15.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52. [DOI] [PMC free article] [PubMed]
- 16.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–50. [DOI] [PMC free article] [PubMed]
- 18.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9. [DOI] [PMC free article] [PubMed]
- 19.Tamborero D, Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Kandoth C, Reimand J, Lawrence MS, Getz G, Bader GD, Ding L, Lopez-Bigas N. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep. 2013;3:2650. [DOI] [PMC free article] [PubMed]
- 20.Buffa FM, Harris AL, West CM, Miller CJ. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br J Cancer. 2010;102:428–35. doi: 10.1038/sj.bjc.6605450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–37. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cancer Genome Atlas Research Network Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas Research Network Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–22. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas Research N. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luoto KR, Kumareswaran R, Bristow RG. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013;4:5. doi: 10.1186/2041-9414-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fox NS, Starmans MH, Haider S, Lambin P, Boutros PC. Ensemble analyses improve signatures of tumour hypoxia and reveal inter-platform differences. BMC Bioinformatics. 2014;15:170. doi: 10.1186/1471-2105-15-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Black JC, Atabakhsh E, Kim J, Biette KM, Van Rechem C, Ladd B, Burrowes PD, Donado C, Mattoo H, Kleinstiver BP, et al. Hypoxia drives transient site-specific copy gain and drug-resistant gene expression. Genes Dev. 2015;29:1018–31. [DOI] [PMC free article] [PubMed]
- 33.Danenberg PV. Thymidylate synthetase--a target enzyme in cancer chemotherapy. Biochim Biophys Acta. 1977;473:73–92. doi: 10.1016/0304-419x(77)90001-4. [DOI] [PubMed] [Google Scholar]
- 34.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–33. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27:1160–7. [DOI] [PMC free article] [PubMed]
- 36.Nair R, Roden DL, Teo WS, McFarland A, Junankar S, Ye S, Nguyen A, Yang J, Nikolic I, Hui M, et al. c-Myc and Her2 cooperate to drive a stem-like phenotype with poor prognosis in breast cancer. Oncogene. 2014;33:3992–4002. [DOI] [PubMed]
- 37.Parris TZ, Kovacs A, Hajizadeh S, Nemes S, Semaan M, Levin M, Karlsson P, Helou K. Frequent MYC coamplification and DNA hypomethylation of multiple genes on 8q in 8p11-p12-amplified breast carcinomas. Oncogenesis. 2014;3, e95. [DOI] [PMC free article] [PubMed]
- 38.Koh JL, Brown KR, Sayad A, Kasimer D, Ketela T, Moffat J. COLT-Cancer: functional genetic screening resource for essential genes in human cancer cell lines. Nucleic Acids Res. 2012;40:D957–963. doi: 10.1093/nar/gkr959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. [DOI] [PMC free article] [PubMed]
- 40.Pallottini V, Guantario B, Martini C, Totta P, Filippi I, Carraro F, Trentalance A. Regulation of HMG-CoA reductase expression by hypoxia. J Cell Biochem. 2008;104:701–9. [DOI] [PubMed]
- 41.McIntyre A, Harris AL. Metabolic and hypoxic adaptation to anti-angiogenic therapy: a target for induced essentiality. EMBO Mol Med. 2015;7(4): 368–379 [DOI] [PMC free article] [PubMed]
- 42.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782–7. [DOI] [PMC free article] [PubMed]
- 43.Tennant DA, Duran RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10:267–77. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- 44.Chapiro J, Sur S, Savic LJ, Ganapathy-Kanniappan S, Reyes J, Duran R, Thiruganasambandam SC, Moats CR, Lin M, Luo W, et al. Systemic delivery of microencapsulated 3-bromopyruvate for the therapy of pancreatic cancer. Clin Cancer Res. 2014;20:6406–17. [DOI] [PMC free article] [PubMed]
- 45.Gad H, Koolmeister T, Jemth AS, Eshtad S, Jacques SA, Strom CE, Svensson LM, Schultz N, Lundback T, Einarsdottir BO, et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature. 2014;508:215–21. [DOI] [PubMed]
- 46.Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, LeBleu VS, Mittendorf EA, Weitz J, Rahbari N, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177–82. [DOI] [PMC free article] [PubMed]
- 47.Sui Z, Zhou J, Cheng Z, Lu P. Squalene epoxidase (SQLE) promotes the growth and migration of the hepatocellular carcinoma cells. Tumour Biol. 2015;36(8):6173–9. [DOI] [PubMed]
- 48.Brown DN, Caffa I, Cirmena G, Piras D, Garuti A, Gallo M, Alberti S, Nencioni A, Ballestrero A, Zoppolia G. Squalene epoxidase is a bona fide oncogene by amplification with clinical relevance in breast cancer. Sci Rep. 2016;6:19435. [DOI] [PMC free article] [PubMed]
- 49.Krause MR, Regen SL. The structural role of cholesterol in cell membranes: from condensed bilayers to lipid rafts. Acc Chem Res. 2014;47:3512–21. doi: 10.1021/ar500260t. [DOI] [PubMed] [Google Scholar]
- 50.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 51.Souchek JJ, Baine MJ, Lin C, Rachagani S, Gupta S, Kaur S, Lester K, Zheng D, Chen S, Smith L, et al. Unbiased analysis of pancreatic cancer radiation resistance reveals cholesterol biosynthesis as a novel target for radiosensitisation. Br J Cancer. 2014;111:1139–49. [DOI] [PMC free article] [PubMed]
- 52.Kucharzewska P, Christianson HC, Belting M. Global profiling of metabolic adaptation to hypoxic stress in human glioblastoma cells. PLoS One. 2015;10 doi: 10.1371/journal.pone.0116740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tabas-Madrid D, Nogales-Cadenas R, Pascual-Montano A. GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 2012;40:W478–483. doi: 10.1093/nar/gks402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prat A, Karginova O, Parker JS, Fan C, He X, Bixby L, Harrell JC, Roman E, Adamo B, Troester M, Perou CM. Characterization of cell lines derived from breast cancers and normal mammary tissues for the study of the intrinsic molecular subtypes. Breast Cancer Res Treat. 2013;142:237–55. [DOI] [PMC free article] [PubMed]
- 56.McIntyre A, Patiar S, Wigfield S, Li JL, Ledaki I, Turley H, Leek R, Snell C, Gatter K, Sly WS, et al. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin Cancer Res. 2012;18:3100–11. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
An open source R package (iDOS) implementing the methodology described here to identify candidate drivers is being made available through CRAN (http://cran.r-project.org). The iDOS package and associated R scripts are also available as Additional file 3 and on github (https://github.com/shazi828/iDOS/releases) with associated GNU General Public License v2 (https://github.com/shazi828/iDOS). A citable version of the source code and data are available through Zenodo with doi: 10.5281/zenodo.51787 (https://zenodo.org/record/51787).