

Abstract
Background: As targeted cancer therapies and molecular profiling become widespread, the era of “precision oncology” is at hand. However, cancer genomes are complex, making mutation-specific outcomes difficult to track. We created a proof-of-principle, CUSTOM-SEQ: Continuously Updating System for Tracking Outcome by Mutation, to Support Evidence-based Querying, to automatically calculate and display mutation-specific survival statistics from electronic health record data.
Methods: Patients with cancer genotyping were included, and clinical data was extracted through a variety of algorithms. Results were refreshed regularly and injected into a standard reporting platform. Significant results were highlighted for visual cueing. A subset was additionally stratified by stage, smoking status, and treatment exposure.
Results: By August 2015, 4310 patients with a median follow-up of 17 months had sufficient data for survival calculation. As expected, epidermal growth factor receptor (EGFR) mutations in lung cancer were associated with superior overall survival, hazard ratio (HR) = 0.53 (P < .001), validating the approach. Guanine nucleotide binding protein (G protein), q polypeptide (GNAQ) mutations in melanoma were associated with inferior overall survival, a novel finding (HR = 3.42, P < .001). Smoking status was not prognostic for epidermal growth factor receptor–mutated lung cancer patients, who also lived significantly longer than their counterparts, even with advanced disease (HR = 0.54, P = .001).
Interpretation: CUSTOM-SEQ represents a novel rapid learning system for a precision oncology environment. Retrospective studies are often limited by study of specific time periods and can lead to incomplete conclusions. Because data is continuously updated in CUSTOM-SEQ, the evidence base is constantly growing. Future work will allow users to interactively explore populations by demographics and treatment exposure, in order to further investigate significant mutation-specific signals.
Keywords: health information management, electronic health records, genomics, information science, precision medicine, neoplasms
BACKGROUND AND SIGNIFICANCE
Cancer is a heterogeneous set of more than 120 diseases with widely varying prognoses and treatments. It represents the second most common cause of death in the United States and has increasing impact worldwide.1 Due to rapid changes in technology and treatment as well as the growing complexity of the healthcare delivery ecosystem, the National Academy of Medicine has described cancer care as a “system in crisis.”2 The future of cost-effective and high-quality cancer care depends on rapid learning systems that optimize the utility of routine observational data gathered from the clinic, including outcomes.3 Adding significantly to the challenge of cancer care, the explosion in knowledge of somatic cancer genomic alterations has continued apace.4–6 It has been recognized for some time that certain “driver” mutations are central to the pathogenesis and virulence of cancer, whereas “passenger” mutations may be nothing more than red herrings.7–9 Large-scale efforts such as the Cancer Genome Atlas have begun to reveal the “genomic landscape” of a variety of common and deadly tumor types, e.g., melanoma,10 breast cancer,11 and lung cancer.12 Targeted agents, such as vemurafenib, a v-raf murine sarcoma viral oncogene homolog B (BRAF) inhibitor, and crizotinib, an anaplastic lymphoma receptor tyrosine kinase inhibitor,13,14 have ushered in the era of precision oncology, as evidenced by the focus on oncology in President Obama’s recently announced Precision Medicine Initiative.15 The Cancer Genome Atlas Pan-Cancer analysis project16 has demonstrated that recurring driver mutations, such as BRAF p.V600E, are found across diverse cancer types.17 Many of these recurrent mutations are predicted to lead to tumor cell susceptibility to currently approved medications and/or therapies undergoing clinical trial evaluations, such that many newer antineoplastics have been approved only in the context of a specific mutation or set of mutations. However, recent results have clearly demonstrated that similarly mutated cancers do not all respond to the same targeted agents.18
Currently, oncologists are expected either to subspecialize to the point where they can manage a tractable number of genomic alterations and their associated prognostic and treatment implications within working memory, or to rely on external knowledge bases such as those provided by third-party laboratories (e.g., Illumina Inc., Foundation Medicine Inc.) or other knowledge management systems (e.g., the Syapse Precision Medicine Platform, My Cancer Genome). While knowledge bases are highly valuable, they have rarely integrated clinical information such as outcomes and treatment exposure. To our knowledge, a system of prospectively monitoring mutation-specific outcomes has not been developed for routine clinical care and secondary data analysis.
OBJECTIVE
We sought to develop a tool that would automatically extract mutation-specific outcome data, in near real-time, from various electronic health record (EHR) data sources across our single large academic institution, and synthesize the results for visual analysis. The intent of this system, named CUSTOM-SEQ: a Continuously Updating System for Tracking Outcome by Mutation, to Support Evidence-based Querying, is to (1) provide continuously updating feedback on outcomes observed in the clinical domain and (2) enable the identification of potential covariates and confounders that have been difficult to extract from medical records without manual abstraction.
As a proof-of-concept, we implemented the system to evaluate all genotyped cancer patients seen at Vanderbilt University Medical Center (VUMC) for mutation-specific survival. We also developed automated methods to stratify a subpopulation of lung cancer patients for tobacco-specific survival, stage-specific survival, and treatment exposure as a function of mutation status.
MATERIALS AND METHODS
Constructing the patient cohort
The data source used for this analysis is the VUMC Research Derivative (RD), an identifiable database of clinical and related data derived from VUMC’s clinical information systems and restructured for research and quality programs.19 As of August 2015, the RD contained information on >3 million patients dating back to 1992. The RD contains diagnosis, treatment, demographic, and outcome data recorded in structured, semi-structured, or free text fields. While the dates and titles of documents scanned from outside institutions, e.g., outside pathology reports, are retained, the scanned images are not included in the RD.
The eligible patient cohort included any patient with SNaPshot tumor genotyping data in the RD. SNaPshot is a fast, high-throughput, multiplex mutational profiling method based on the Applied Biosystems SNaPshot platform.20–23 Test results are reported as predicted protein alterations based on the observed genetic variant(s). SNaPshot testing has been performed at VUMC since 2010, initially on lung cancer and melanoma specimens, with disease-specific panels currently available for acute myeloid leukemia, breast cancer, colorectal cancer, glioma, lung cancer, and melanoma.
Automated algorithms for mutation-specific survival data extraction
In order to be included in the baseline analysis, patients had to have sufficient data in the EHR to automatically extract or calculate the following data elements: (1) SNaPshot test results, (2) tumor type, (3) date of diagnosis, and (4) date of death or last contact. We also required that patients had at least one Current Procedural Terminology (CPT), fourth edition (copyright 2013, American Medical Association) code for a billable clinical encounter within ± 365 days of SNaPshot testing, in order to exclude patients who were not seen at VUMC for their cancer diagnosis (see Supplemental Table 1). SNaPshot test results were structured laboratory values that did not require any algorithm development beyond custom mapping of internal laboratory codes to Human Gene Nomenclature Committee names.24,25 Date of death or last contact was also available in structured format from multiple sources. The other data elements, however, required the development of heuristic algorithms for accurate data extraction. The tumor type was classified using International Classification of Diseases, ninth edition, Clinical Modification (ICD-9-CM) administrative codes with a “winner take all” adjudication when multiple eligible ICD-9-CM codes were present (see Supplemental Table 2). Date of diagnosis, which can be challenging to determine for patients who were originally diagnosed outside of VUMC, was determined through a combination of structured data analysis and natural language processing, as previously described.26 Further details are available in the Supplemental Methods; the data analysis workflow is summarized in Figure 1.
Figure 1:

Simplified schematic of identification of analyzable patients as of January 2014, followed by algorithm extraction of tumor type, date of diagnosis, and date of death or last contact using a variety of structured and unstructured data sources. Binary digits icon represents metadata including date stamps of scanned documents; text icon represents searchable electronic text data; PDF icon represents scanned images; Social Security icon represents the Social Security Death Index. CPT: Current Procedural Terminology, 4th edition code (copyright 2013 American Medical Association); ICD-9-CM: International Classification of Diseases, 9th Edition, Clinical Modification; PDF: Portable Document Format.
Patients were stratified by their SNaPshot test results, with assignment to the category “None” if no mutations were detected. For a given tumor type classification, gene mutations with fewer than 10 occurrences were grouped into an “Other” category so as to minimize the risk of re-identification, per common practice.27 If a specimen was found to have more than one mutation in the same gene, the patient was only counted once. If a specimen was found to have mutations in more than one gene, the patient was counted once for each mutation category. If a patient had more than one SNaPshot test, the earliest test was used for the analysis.
Evaluation of data extraction algorithms
In order to evaluate the accuracy of the automated classification and date extraction algorithms, a stepwise quality assurance approach was undertaken, with iterative improvements undertaken until a prespecified level of performance was achieved. By the end of this process, ∼2% of identified charts had been randomly selected for quality assurance review. All reviewed charts underwent manual abstraction by at least two abstractors with clinical subject matter expertise, and interannotator agreement (IAA) was calculated by Cohen’s kappa (κ).28 Any disagreements in manual abstraction were adjudicated by a third abstractor, with persistent discrepancies resolved by discussion among the three abstractors.
Exploration of covariates extracted from EHR data
The baseline analysis of mutation-specific survival revealed a potential signal for improved survival in EGFR-mutated lung cancer patients (see Results section, below). We therefore explored the addition of several covariates for lung cancer patients diagnosed prior to April 30, 2014: (1) smoking status, (2) stage at diagnosis, and (3) treatment exposure, including the oral EGFR tyrosine kinase inhibitor erlotinib (Tarceva, Genentech Inc., South San Francisco, CA, USA). Smoking status and intravenous treatment exposure status were available in structured format. Stage was extracted from clinical notes, as previously described.29 The methods to extract each of these covariates are further detailed in the Supplemental Methods.
Survival analysis
Overall survival was plotted according to the Kaplan–Meier method. We then employed a Cox proportional hazards regression model to estimate the hazard ratio (HR) for overall survival as a function of mutational status. For each mutation category, the Wald statistic for that category versus all other categories combined was calculated. If the P-value for this comparison was ≤0.05 divided by the number of mutation categories (the Bonferroni correction for multiple hypothesis testing),30 this category was labeled as significant, as denoted by an asterisk in the figure legend. If any mutation category was significant, the background of the figure was colored pale green; otherwise the background was colored pale pink. Except for the baseline analysis, we combined covariates so as to generate no more than four survival curves at once, to preserve visualization quality. All statistical tests were two-sided.
General methods
Structured Query Language (SQL) queries to Netezza and Oracle databases were built into Extract, Transform, and Load (ETL) processes using Talend Studio (Talend Inc.). The dashboard for visual analytics was constructed and displayed using JasperReports Server version 4.5.1 (Jaspersoft Corporation, San Francisco, CA, USA). Survival analyses were performed using R version 3.0.2 and the R package Survival (http://www.r-project.org). Mutation-specific survival curves were calculated, graphed, and injected into the dashboard. The dashboard was updated on a weekly basis as part of a scheduled ETL process. The development version of the dashboard was made available to members of the Vanderbilt-Ingram Cancer Center and was accessed securely using direct Lightweight Directory Access Protocol (LDAP) authentication. All the components of the general tool were seamlessly integrated; the expanded stratification analysis was performed manually during the pilot project period using Aginity Workbench (Aginity Inc.). The general tool was determined to be non-human-subject research by the VUMC Institutional Review Board (IRB), per 45 CFR §46.102(d) (VUMC IRB #131613). The expanded stratification analysis was determined to be exempt (VUMC IRB #140697); all authors with access to data had the appropriate HIPAA training. This study was performed in accordance with the STROBE Statement, Version Four.31
RESULTS
Population
A total of 4310 patients were identified as having undergone SNaPshot testing and meeting the Current Procedural Terminology inclusion criteria as of August 21, 2015; the first specimen was tested in July 2010.21,22 The majority of specimens had zero or one cancer mutation detected; a minority (N = 327) had two or more simultaneous mutations detected, as shown in Table 1. Median follow-up from the date of diagnosis to the date of death or last contact was 17 months (interquartile range [IQR] = 6–40 months). Example screenshots from the live dashboard (as of November 25, 2015) are shown in Supplemental Figures 1 and 2.
Table 1:
Cancer patients and specimens by primary anatomic site or histology
| Cancer type | Number of patients (%)b | Specimens with no mutation detected | Specimens with one mutation detected | Specimens with two or more mutations detected |
|---|---|---|---|---|
| Acute myeloid leukemia | 504 (12) | 316 | 121 | 67 |
| Breast cancera | 466 (11) | 343 | 118 | 5 |
| Colorectal cancera | 469 (11) | 252 | 180 | 37 |
| Glioma | 158 (4) | 97 | 54 | 7 |
| Lung cancera | 1364 (32) | 767 | 530 | 67 |
| Melanoma | 1200 (28) | 422 | 711 | 67 |
| Other diagnoses | 166 (4) | 128 | 35 | 3 |
| Total | 4310 (100) | 2325 | 1749 | 253 |
aFor these cancers, classification is by anatomic site of origin.
bNumber of individual patients adds up to >100%, because some patients had more than one cancer diagnosis.
Evaluation of algorithm accuracy
The algorithms for determining cancer type, date of diagnosis, and date of death were manually evaluated on 75 charts each. IAA for date of diagnosis was κ = 0.79; all but one discrepancy were resolved by adjudication. The median absolute discrepancy between the manually and algorithmically determined dates of diagnosis was 2 days (IQR, 0–260 days). IAA was κ = 1.0 for date of death and tumor type classification. The median absolute discrepancy between the manually and algorithmically determined dates of death was 0 days (IQR, 0–10 days).
Baseline survival analysis
As of August 21, 2015, EGFR mutation was associated with improved overall survival in lung cancer patients, with an estimated median overall survival of 10.1 years for a patient with any EGFR mutation, versus 4.1 years in patients with no detected EGFR mutations (HR 0.53, 95% CI (Confidence Interval), 0.38-0.73, P < .001 ; Figure 2). In melanoma, guanine nucleotide binding protein (G protein), q polypeptide (GNAQ) mutation was associated with decreased overall survival, with an estimated median overall survival of 1.3 years for a patient with any GNAQ mutation, versus 9.4 years in patients with no detected GNAQ mutations (HR 3.42, 95% CI, 2.10-5.58, P < .001; Figure 3). BRAF mutation in melanoma did not appear to confer a survival advantage (P = .11).
Figure 2:

Lung cancer gene mutation-specific survival analysis at different time points; SNaPshot mutation panel testing began in July 2010. (A) survival curves as of March 2011, 3 months prior to the first significant finding; (B) in June 2012, EGFR mutation was statistically associated with superior survival, whereas KRAS mutation was statistically associated with inferior survival; (C) in December 2013, KRAS mutation is no longer significant; (D) at the most recent analysis (August 2015), EGFR mutation remains a significant predictor for survival. BRAF: v-raf murine sarcoma viral oncogene homolog B; EGFR: epidermal growth factor receptor; ERBB2: v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2; KRAS: Kirsten rat sarcoma viral oncogene homolog; PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha.
Figure 3:

Melanoma gene mutation-specific survival analysis at different time points; SNaPshot mutation panel testing began in July 2010. (A) survival curves as of June 2012, 6 months prior to first significant finding; (B) first significant finding, in December 2012, of inferior survival associated with GNAQ mutation; (C) similar results are seen in December 2013; and (D) the most recent analysis, August 2015. BRAF: v-raf murine sarcoma viral oncogene homolog B; CTNNB1: catenin (cadherin-associated protein), beta 1, 88 kDa; GNA11: guanine nucleotide binding protein (G protein), alpha 11 (Gq class); GNAQ: guanine nucleotide binding protein (G protein), q polypeptide; KIT: v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; NRAS: neuroblastoma RAS viral (v-ras) oncogene homolog.
Survival by smoking status
Lung cancer survival was clearly influenced by smoking status (Table 2). On average, patients who had never smoked lived more than 3 years longer than their counterparts who had ever smoked (HR 1.96, 95% CI, 1.39-2.75, P < .001; Figure 4A). When the population was stratified by smoking and EGFR status, EGFR-mutated patients lived longer than their counterparts who had no EGFR mutation detected, regardless of their personal smoking status, and tobacco use was no longer a significant driver of mortality in the EGFR-mutated patients (HR 1.43, 95% CI, 0.69-2.98, P = .339; Figure 4B).
Table 2:
Stratification by various clinical variables combined with mutation information provides further insights into the outcomes of patients
| Clinical variable | Genomic variable | Number of cases | Median OS, years |
|---|---|---|---|
| Tobacco Use = Ever | – | 806 | 2.83 |
| Tobacco Use = Never | – | 133 | 6.21 |
| Tobacco Use = Ever | EGFR mutated | 47 | 14.19 |
| Tobacco Use = Ever | EGFR wild type | 759 | 2.73 |
| Tobacco Use = Never | EGFR mutated | 57 | 8.73 |
| Tobacco Use = Never | EGFR wild type | 76 | 3.44 |
| Stage = I | – | 182 | 5.57 |
| Stage = II | – | 73 | 2.47 |
| Stage = III | – | 179 | 2.83 |
| Stage = IV | – | 343 | 1.07 |
| Stage = I or II | EGFR mutated | 30 | Not reached |
| Stage = I or II | EGFR wild type | 260 | 6.01 |
| Stage = III or IV | EGFR mutated | 73 | 2.20 |
| Stage = III or IV | EGFR wild type | 452 | 1.24 |
Note that the numbers do not always add up to the total number of lung cancer cases, because some clinical variables were missing or unknown.
Figure 4:
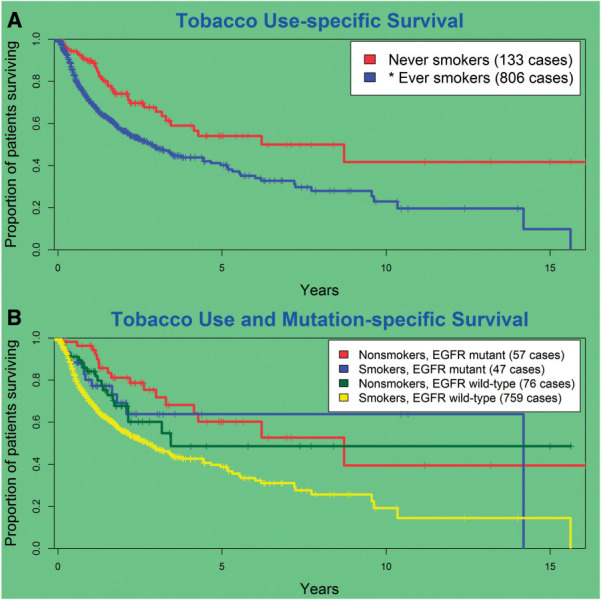
(A) Smoking is a strong overall predictor for inferior outcome but (B) the apparent effect is no longer significant when stratifying by EGFR mutation status.
Survival by stage at diagnosis
As shown in Figure 5A and Table 2, survival generally worsens as a function of advancing stage, with stage IV patients (metastatic disease present at the time of diagnosis) having the worst overall survival. However, when stratifying by stage and EGFR mutation status (Figure 5B), EGFR-mutated patients with advanced-stage disease (stages III and IV) appeared to have a better prognosis than similar patients without a detected EGFR mutation (HR 0.54, 95% CI, 0.37-0.78, P = .001).
Figure 5:
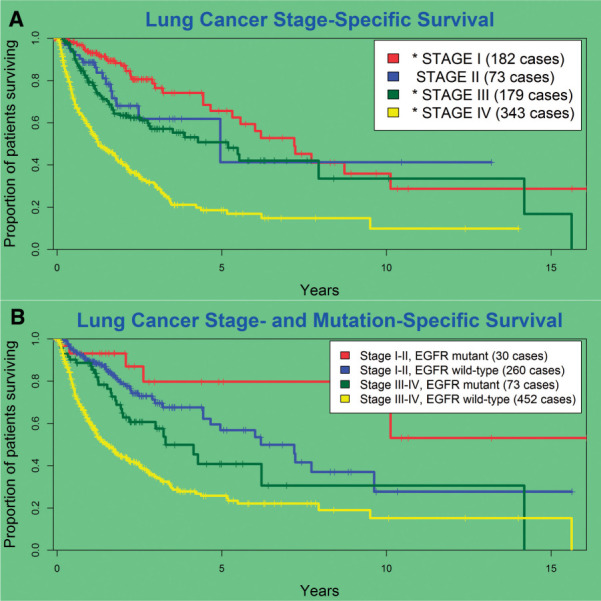
(A) Prognosis generally worsens with advancing stage; (B) EGFR mutation status allows for further stratification into distinct prognostic subgroups.
Survival by stage and treatment exposure
For the N = 77 advanced-stage (stages III and IV) lung cancer patients with an EGFR mutation, 91% (N = 70) received erlotinib, whereas only 35% (N = 27) were administered a platinum drug, as shown in Table 3. As compared to the EGFR wild type group, EGFR-mutated patients were much more likely to receive erlotinib, (Odds Ratio) OR 23.3 (95% CI, 10.4-61.3, P < .0001). Conversely, EGFR-mutated patients were less likely to receive conventional platinum chemotherapy, OR 0.57 (95% CI, 0.33-0.96, P = .0275). For the EGFR-mutated patients, survival was not statistically significantly different based on treatment exposure, but the sample size was small.
Table 3:
EGFR-mutated lung cancer patients were more likely to have ever received erlotinib and less likely to have ever received a platinum-based chemotherapy.
| Lung cancer genotype | +erlotinib exposure | No erlotinib exposure | +platinum exposure | No platinum exposure |
|---|---|---|---|---|
| EGFR-mutated (N = 77) | 70 | 7 | 27 | 50 |
| EGFR wild type (N = 511) | 153 | 358 | 249 | 262 |
DISCUSSION
We have demonstrated a tool that can generate tumor mutation–specific survival curves in near-real time at the institutional level. The tool also enables an exploratory analysis into characteristics that may drive lung cancer–specific survival, based on underlying automated extraction algorithms. While these algorithms introduce some inaccuracies, this should be acceptable given that the primary purpose of this tool is hypothesis generation; further scientific investigations would likely require some degree of manual chart review under separate IRB approval. One particular use case that has generated initial enthusiasm by institutional users is the identification of “exceptional responders,” i.e., patients or patient populations who appear to do particularly better or worse than other similar groups.32 Other possible uses of CUSTOM-SEQ are descriptive statistics for operational and reporting needs and identification of potential signals to target for quality improvement.
Our exploratory analysis built off a possible exceptional response signal detected in EGFR-mutated lung cancer. EGFR is a transmembrane glycoprotein that is a receptor for proteins from the epidermal growth factor family. When mutated in lung cancer, the kinase activity of EGFR is increased, which leads to upregulation of pro-survival signaling pathways in the tumor cell.33 EGFR mutations in lung cancer have been shown in the clinical trial population to confer a survival advantage34,35; our results appear to confirm this finding in an unselected clinical population. From these same initial studies on patients with EGFR mutations, it is known that they are more likely to be female, of Asian ethnicity, and nonsmokers. They are also much more likely to be treated with EGFR tyrosine kinase inhibitors, given the strong evidence that this class of agents provides an apparent overall survival advantage when used as first-line treatment.36 Thus, we introduced the ability to stratify by various clinical factors in CUSTOM-SEQ, which enhances the utility considerably. For example, ever having smoked was a clear negative prognostic factor for lung cancer survival, although this finding did not appear to extend to the EGFR-mutated subgroup. Stage also modified survival as expected, but stage-for-stage the EGFR-mutated subgroup had better overall survival than their counterparts. Finally, as expected, EGFR-mutated lung cancer patients were much more likely to receive tyrosine kinase inhibitor therapy. Interestingly, they were also less likely to receive conventional chemotherapy, and this finding could be the basis for a pragmatic clinical trial of therapeutic approaches using targeted agents.37 For example, emerging data suggests that use of both the Bruton’s tyrosine kinase inhibitor ibrutinib in chronic lymphocytic leukemia and the PI(3)Kα inhibitor alpelisib in breast cancer is accompanied by very rapid progression once resistance develops, in contrast to the gradual relapses historically seen in these diseases.38,39
There are several important limitations to our approach. Our findings are limited by the single-institution nature of the study; the customized extraction algorithms developed here may require further customization to work with other EHR systems and in other localities. Due to this limitation, multi-level stratification, e.g., examining the impact of race on the population of EGFR-mutated nonsmoking lung cancer patients, will often be underpowered. Large national efforts such as the American Society of Clinical Oncology’s CancerLinQ may address this by aggregating and normalizing large amounts of information across many practices.40,41 Certain genotyping tests, e.g., anaplastic lymphoma receptor tyrosine kinase rearrangement testing, were not included in our analysis because the results of this fluorescence in situ hybridization test were not available in a computable format at the time. Accurate assignment of the date of death requires an ongoing feed from third-party data sources, e.g., the National Death Index, due to the fact that many cancer patients pass away at home or in hospice settings. Using our algorithms, we identified ∼30% of the patients as having been deceased; this number may substantially underestimate the actual value, and efforts to capture evidence of death using natural language processing are ongoing. In general, we chose not to rely heavily on cancer registry data, since typically only the first course of treatment is recorded, and many cases seen at a tertiary care center such as VUMC have progressed beyond initial treatment and would be considered nonanalytic.
We have made the assumption that a mutation found by SNaPshot testing was present at diagnosis, even if the date of diagnosis preceded the date of SNaPshot testing by years or decades. This assumption is likely to be valid, as the majority of patients would not have been expected to have received targeted therapies prior to mutation testing. However, a subset is likely to have experienced tumor genomic evolution42; this may be especially true for patients with an exceptionally long disease-free interval, e.g., certain breast cancer and melanoma patients. Acquired resistance will also become more common as patients undergo treatment with targeted therapies in the community.43,44 For the minority of patients with 2 or more mutations, we counted them twice, once for each mutated gene. As the complexity of the clinically relevant mutation landscape expands, this approach will need to be readdressed. For example, somatic cancer mutation analysis platforms such as FoundationOne™ (Foundation Medicine Inc., Cambridge, MA, USA) have reported a median clinically relevant mutation count of approximately 3 per sample analyzed.45 We also considered mutation only at the gene level and did not take into consideration that some mutations are known to confer resistance (e.g., EGFR p.T790M in lung adenocarcinoma) and some to confer sensitivity (e.g., EGFR p.L858R in lung adenocarcinoma), based on preclinical and clinical trial data.46 Future work will incorporate this information, especially as findings of in vitro resistance are confirmed in clinical settings or in large collaborative efforts such as ClinGen.47,48
The median survival estimate based on the Kaplan–Meier survival curve is less reliable in this analysis given the high percentage of censoring; however, the hazard ratio remains a reliable estimator of differential survival.49 We intentionally used an unadjusted Cox proportional hazards model, with the understanding that significance may be reduced substantially after adjusting for confounders. There is also a risk that unobserved Type II errors are occurring. Future iterations of the tool will allow users to interactively adjust for demographics and treatment exposure; these steps were carried out manually during this pilot evaluation.50
Patients presenting at a tertiary-care oncology clinic represent a unique population. Aside from being generally more frail and having more co-morbidities, they have often received part of their cancer care elsewhere. The true population prevalence of clinically relevant mutations in such patients is unknown, although an overall 46% mutation rate found through SNaPshot testing is similar to that found through similar “hotspot” tests. More expansive panels such as FoundationOne have detection rates as high as 76%.45 For lung cancer, the estimated prevalence of the 2 most common (and mutually exclusive51) mutated genes, EGFR and KRAS, is 10–35% and 15–25%, respectively.52 During the development of the tool described here, we came across many challenging scenarios, e.g., patients with 2 or more synchronous or metachronous cancer diagnoses, sometimes occurring in the same organ, and patients with very long disease-free intervals, on the order of years to decades. With the caveat that we were unable to account for all outlier conditions, we found that our informatics extraction algorithms performed well for the purposes of this exploratory tool. It is likely that, with some local tweaks, these algorithms could be adapted to other clinical sites with rich EHR data, making this method potentially generalizable.
We had several significant findings in lung cancer and melanoma, some of which persisted through stratification, as discussed above. While the effect of EGFR mutation on lung cancer prognosis is known, GNAQ mutations have not previously been shown to be an independent predictor for melanoma survival. However, somatic mutations in GNAQ have been found in ∼50% of primary uveal melanomas and up to 28% of uveal melanoma metastases, whereas they are rare in other melanomas.53 Therefore, the observation of inferior survival could be a function of anatomic site rather than mutation-specific; others have suggested that GNAQ mutations do not alter disease-free survival for uveal melanomas.54 This again illustrates that the purpose of an exploratory tool such as CUSTOM-SEQ is for hypothesis generation; ICD-9-CM codes may not accurately distinguish site of disease, and additional research would be required to determine if this finding persists after stratifying by primary site of melanoma. An interesting finding with quality and cost-control implications is that 30% of EGFR wild-type lung cancer patients received erlotinib at some point. While this seems to contradict the lack of sensitivity expected in this situation,55 it must be noted that erlotinib was approved in 2004 “for the treatment of locally advanced or metastatic NSCLC after failure of at least one prior chemotherapy regimen,” and did not have a mutation-specific label until 2013.56 Thus, it would be expected that wild-type patients who had progressed on conventional chemotherapy may have been treated with erlotinib, although the response rate may have been very low. When changes in labeling such as this do occur, CUSTOM-SEQ could be used to look for changes in patterns of care.
One observation is particularly noteworthy, and this is the circumstance where KRAS mutation was statistically significantly associated with inferior survival in lung cancer for a period of almost 12 months (Figure 2B) before losing statistical significance (Figure 2C). There are many possible explanations for such a finding. The likelihood of a Type I error is small; at one point the P-value for the association was .0003. In general, this finding and a similar finding for BRAF mutation in melanoma for one 2-week interval demonstrates the importance of cautiously interpreting findings of statistical significance.57 Unlike in a randomized clinical trial, where the survival analyses are often conducted only at predetermined interim analysis time points,58 the method described herein allows for continuously updated survival analyses. As systems are developed that have the ability to display data at any time point, investigators are going to have to create rules to prevent over-sampling and possibly over-interpreting their results.
CONCLUSION
In conclusion, we have developed a tool that generates cancer mutation–specific survival statistics in near real-time, while also enabling a historic look-back to identify transition points of statistical significance. Signals can be investigated further using stratification, which will be interactive in future iterations. CUSTOM-SEQ can be used for a variety of purposes, including quality assessment, operational needs, and hypothesis generation. CUSTOM-SEQ is a promising new addition to a precision oncology environment.
ACKNOWLEDGEMENTS
We would like to acknowledge Qingxia Chen of the Vanderbilt Department of Biostatistics for her advice regarding statistical considerations and Joseph Burden and Scott Sobecki of the VICC Research Informatics Core for their programmatic support.
SUPPLEMENTARY MATERIAL
Supplementary material is available online at http://jamia.oxfordjournals.org/.
CONTRIBUTORS
J.L.W., W.P., and M.A.L. conceived CUSTOM-SEQ. L.W. provided the data. J.L.W., R.A., and P.C. participated in QA and suggested algorithm improvements. W.P. and J.A.S. provided feedback on the look and feel of the product. All authors contributed to the initial draft manuscript and approved the final manuscript.
FUNDING
This study was supported in part by donations by the Robert J. Kleberg Jr and Helen C. Kleberg Foundation, the T.J. Martell Foundation, and an anonymous foundation. Additional support was provided by grant funding through the Vanderbilt Institute for Clinical and Translational Research [ULTR000445 from National Center for Advancing Translational Sciences of the National Institutes of Health (NCATS/NIH)] and the Vanderbilt-Ingram Cancer Center Core Grant [P30-CA68485-18 from National Cancer Institute (NCI)]. The funders had no role in study design; in the collection, analysis, and interpretation of data; in the writing of the report, or in the decision to submit the paper for publication.
COMPETING INTERESTS
The authors declare that no competing interests exist.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. [DOI] [PubMed] [Google Scholar]
- 2. Delivering High-Quality Cancer Care: Charting a New Course for a System in Crisis. Washington, DC: The National Academies Press; 2013. [PubMed]
- 3.Abernethy AP, Etheredge LM, Ganz PA, et al. Rapid-learning system for cancer care. J Clin Oncol. 2010;28(27):4268–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Allen EM, Wagle N, Levy MA. Clinical analysis and interpretation of cancer genome data. J Clin Oncol. 2013;31(15):1825–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. [DOI] [PubMed] [Google Scholar]
- 8.Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haber DA, Settleman J. Cancer: drivers and passengers. Nature. 2007;446(7132):145–146. [DOI] [PubMed] [Google Scholar]
- 10.Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stephens PJ, Tarpey PS, Davies H, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang R, Pan Y, Li C, et al. Analysis of major known driver mutations and prognosis in resected adenosquamous lung carcinomas. J Thorac Oncol. 2014;9(6):760–768. [DOI] [PubMed] [Google Scholar]
- 13.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–16703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med. 2015;373(8):726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Danciu I, Cowan JD, Basford M, et al. Secondary use of clinical data: The Vanderbilt approach. J Biomed Inform. 2014;52:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dias-Santagata D, Akhavanfard S, David SS, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2(5):146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su Z, Dias-Santagata D, Duke M, et al. A platform for rapid detection of multiple oncogenic mutations with relevance to targeted therapy in non-small-cell lung cancer. J Mol Diagn. 2011;13(1):74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS One. 2012;7(4):e35309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan E, Goff L, Cardin D, et al. Routine multiplexed mutational analysis of colorectal cancers — a single institution experience. ECOG-ACRIN Young Investigators’ Symposium. Fort Lauderdale, FL; 2012. [Google Scholar]
- 24.Levy MA, Lovly CM, Pao W. Translating genomic information into clinical medicine: lung cancer as a paradigm. Genome Res. 2012;22(11):2101–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. Genenames.org: the HGNC resources in 2015. Nucleic Acids Res. 2015;43(Database issue):D1079–D1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warner JL, Wang L, Atreya R, Carney P, Burden J, Levy MA. Automated extraction of date of cancer diagnosis from EMR data sources. AMIA Workshop on Data Mining for Medical Informatics 2014 cited: http://www.dmmh.org/dmmi2014_submission_1.pdf Accessed 22 November 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weber GM, Murphy SN, McMurry AJ, et al. The Shared Health Research Information Network (SHRINE): a prototype federated query tool for clinical data repositories. J Am Med Inform Assoc. 2009;16(5):624–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hripcsak G, Rothschild AS. Agreement, the f-measure, and reliability in information retrieval. J Am Med Inform Assoc. 2005;12(3):296–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warner JL, Levy MA, Neuss MN. Feasibility and accuracy of extracting cancer stage information from narrative electronic health record data. J Oncol Pract. 2015. [DOI] [PubMed] [Google Scholar]
- 30.Rice TK, Schork NJ, Rao DC. Methods for handling multiple testing. Adv Genet. 2008;60:293–308. [DOI] [PubMed] [Google Scholar]
- 31.von Elm E, Altman DG, Egger M, et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. J Clin Epidemiol. 2008;61(4):344–349. [DOI] [PubMed] [Google Scholar]
- 32.Hellmann MD, Kris MG, Rudin CM. Medians and milestones in describing the path to cancer cures: telling “Tails”. JAMA Oncol. 2015:1–3. [DOI] [PubMed] [Google Scholar]
- 33.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305(5687):1163–1167. [DOI] [PubMed] [Google Scholar]
- 34.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. [DOI] [PubMed] [Google Scholar]
- 35.Clinical Lung Cancer Genome P, Network Genomic M. A genomics-based classification of human lung tumors. Sci Transl Med. 2013;5(209):209ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou C, Wu YL, Chen G, et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer (OPTIMAL, CTONG-0802). Ann Oncol. 2015;26(9):1877–1883. [DOI] [PubMed] [Google Scholar]
- 37.Lurie JD, Morgan TS. Pros and cons of pragmatic clinical trials. J Comp Effectiveness Res. 2013;2(1):53–58. [DOI] [PubMed] [Google Scholar]
- 38.Jain P, Keating M, Wierda W, et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood. 2015;125(13):2062–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juric D, Castel P, Griffith M, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature. 2015;518(7538):240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sledge GW, Jr, Miller RS, Hauser R. CancerLinQ and the future of cancer care. American Society of Clinical Oncology educational book /ASCO American Society of Clinical Oncology Meeting. 2013;33:430–434. [DOI] [PubMed] [Google Scholar]
- 41.Kantarjian H, Yu PP. Artificial intelligence, big data, and cancer. JAMA Oncol. 2015;1(5):573–574. [DOI] [PubMed] [Google Scholar]
- 42.Meador CB, Micheel CM, Levy MA, et al. Beyond histology: translating tumor genotypes into clinically effective targeted therapies. Clin Cancer Res. 2014;20(9):2264–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10(11):760–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rehm HL, Berg JS, Brooks LD, et al. ClinGen—the Clinical Genome Resource. N Engl J Med. 2015;372(23):2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aronson SJ, Rehm HL. Building the foundation for genomics in precision medicine. Nature. 2015;526(7573):336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee ET, Go OT. Survival analysis in public health research. Ann Rev Public Health. 1997;18:105–134. [DOI] [PubMed] [Google Scholar]
- 50.Purchase HC, Andrienko N, Jankun-Kelly T, Ward M. Theoretical Foundations of Information Visualization. Information Visualization: Springer; 2008: 46–64. [Google Scholar]
- 51.Jang TW, Oak CH, Chang HK, Suo SJ, Jung MH. EGFR and KRAS mutations in patients with adenocarcinoma of the lung. Korean J Int Med. 2009;24(1):48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lovly CM, Horn L, Pao W. Molecular Profiling of Lung Cancer. 2015 [Accessed November 22, 2015]; http://www.mycancergenome.org/content/disease/lung-cancer/ [Google Scholar]
- 53.Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bauer J, Kilic E, Vaarwater J, Bastian BC, Garbe C, de Klein A. Oncogenic GNAQ mutations are not correlated with disease-free survival in uveal melanoma. Br J Cancer. 2009;101(5):813–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. U.S. Food and Drug Administration. FDA Approves First Companion Diagnostic to Detect Gene Mutation Associated with a Type of Lung Cancer. 2013 [Accessed November 22, 2015]; http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm352230.htm .
- 57.Ioannidis JP. Why most published research findings are false. PLoS Med. 2005;2(8):e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lewis RJ. An introduction to the use of interim data analyses in clinical trials. Ann Emerg Med. 1993;22(9):1463–1469. [DOI] [PubMed] [Google Scholar]